

Summary
The cystic fibrosis transmembrane conductance regulator (CFTR) undergoes rapid turnover at the plasma membrane in various cell types. The ubiquitously expressed N-WASP promotes actin polymerization and regulates endocytic trafficking of other proteins in response to signaling molecules such as Rho-GTPases. In the present study we investigated the effects of wiskostatin, an N-WASP inhibitor, on the surface expression and activity of CFTR. We demonstrate, using surface biotinylation methods, that the steady-state surface CFTR pool in stably transfected BHK cells was dramatically decreased following wiskostatin treatment with a corresponding increase in the amount of intracellular CFTR. Similar effects were observed for latrunculin B, a specific actin-disrupting reagent. Both reagents strongly inhibited macroscopic CFTR-mediated Cl− currents in two cell types including HT29-Cl19A colonic epithelial cells. As previously reported, CFTR internalization from the cell surface was strongly inhibited by a cyclic-AMP cocktail. This effect of cyclic-AMP was only partially blunted in the presence of wiskostatin, which raises the possibility that these two factors modulate different steps in CFTR traffic. In kinetic studies wiskostatin appeared to accelerate the initial rate of CFTR endocytosis as well as inhibit its recycling back to the cell surface over longer time periods. Our studies implicate a role for N-WASP-mediated actin polymerization in regulating CFTR surface expression and channel activity.
Keywords: CFTR, actin, N-WASP, wiskostatin, latrunculin B, endocytosis, cyclic-AMP
1. Introduction
The cystic fibrosis transmembrane conductance regulator (CFTR) is a chloride-selective ion channel found in epithelial cells lining the gastrointestinal tract, airways and reproductive organs. Mature, glycosylated CFTR is delivered from post-golgi compartments to the apical plasma membrane where it regulates the vectorial transport of ions and water across the cell membrane. Interestingly, the wild type protein has a relatively long half-life (16–24 hours in most cell types), although it exhibits rapid turnover (within minutes) at the surfaces of many cell types [1, 2]. Biochemical, biophysical and functional studies of T84 colonic epithelial cells and stably transfected chinese hamster ovary cells provided evidence that CFTR is rapidly internalized from the plasma membrane by clathrin-coated vesicles [2, 3]. The C-terminal tail of CFTR contains two structural features that are important for its endocytosis; (i) a dileucine (1430LL) motif and (ii) a tyrosine-based motif (1424YDSI1427). The latter motif interacts with the clathrin adapter protein complex (AP-2), which facilitates entry of the ion channel into coated pits [4, 5]. Internalized CFTR channels appear to be capable of rapidly recycling back to the plasma membrane, presumably via recycling endosomes [6], which could possibly modulate CFTR expression at the cell surface [7]. However, the mechanism and itinerary of CFTR recycling is not fully understood.
Several studies indicate that the components of the clathrin-mediated endocytic machinery directly or indirectly interact with the actin cytoskeleton, which forms a scaffolding structure beneath the plasma membrane [8, 9]. Disruption of the actin filament network has been demonstrated to compromise the endocytic trafficking of some proteins such as the transferrin receptor [10] and H+ -K+- ATPase proton pumps [11]; however, others have reported no effect on clahrin-mediated endocytosis [12, 13]. Recently, Swiatecka-Urban et al., have reported that CFTR interacts with a complex of actin-binding proteins including myosin VI that facilitate CFTR endocytosis and that may regulate CFTR expression at the cell surface [14]. In addition it was reported that CFTR-mediated Cl− currents were insensitive to stimulation by cyclic-AMP in whole cells or to direct addition of PKA to excised patches when the actin cytoskeleton was first completely disrupted [15, 16]. The authors proposed that a direct interaction between the actin filaments and CFTR was a prerequisite for stimulating channel activity.
In the present study we investigated the effects of two actin-disrupting reagents, namely, wiskostatin and latrunculin B, on regulating the surface expression and activity of CFTR. Wiskostatin is a cell-permeant compound that stabilizes N-WASP in its native, autoinhibited conformation thereby preventing actin polymerization [17–19]. The WASP family proteins are emerging as central regulators of actin assembly vis-à-vis their interactions with Rho family GTPases, phosphatidylinositol 4,5 bisphosphate (PIP2) and Arp2/3 complexes. Moreover, several studies indicate that N-WASP regulates endocytosis and exocytosis, presumably via its role in modulating actin assembly [20–22]. Latrunculin B, which causes actin filament disassembly by sequestering actin monomers, was reported to block the recycling of G-protein coupled receptors internalized in response to β-agonists in human carcinoma cells [23].
We show that wiskostatin substantially reduced the steady-state surface pool of CFTR in biochemical studies in which cell surface CFTR was labeled using a cleavable biotinylating reagent (NHS-SS-biotin). Concomitantly, there was a dramatic increase in the amount of CFTR that accumulated within the cells following wiskostatin treatment although the total CFTR content in the lysates was not appreciably altered. Similar results were obtained using latrunculin B. Both wiskostatin and latrunculin B also dramatically decreased CFTR-mediated currents in two cell types including HT29-Cl19A colonic epithelial cells. In detailed kinetic studies we observed that wiskostatin had complex effects on both the time course of CFTR endocytosis and recycling implying that it may affect both processes. Our results indicate that the filamentous actin and/or N-WASP-dependent actin assembly play a modulatory role in the endocytic trafficking of CFTR.
2. Materials and methods
2.1. Cell culture
BHK cells that were stably transfected with full-length wild-type human CFTR were a gift from J.W. Hanrahan and cultured as described [24]. HT29-CL19A human colonic epithelial cells expressing native CFTR were propagated as described previously [25].
2.2. Cell surface biotinylation
Cell surface proteins were biotinylated with sulfosuccinimidyl-2-(biotinamido)ethyl-1,3-dithiopropionate (sulfo-NHS-SS-Biotin) following previously published protocols [2] with minor modifications. Briefly, BHK-CFTR cells grown in 100-mm dishes were incubated with indicated concentrations of wiskostatin (Calbiochem) or DMSO (vehicle) for 2h at 37° C. Further steps were performed at 0° C. Cells were rinsed with ice-cold PBS (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4, pH 7.4) containing 0.1 mM CaCl2 and 1 mM MgCl2 (PBS-CM) and incubated with 1 mg/ml EZ-Link Sulfo-NHS-SS-biotin (Pierce) in PBS (pH adjusted to 8.0) for 30 min. Cell monolayers were rinsed with cold PBS-CM and unreacted biotin molecules were quenched with PBS containing 1% BSA for 10 min before cell lysis and streptavidin-agarose capture.
2.3. CFTR Internalization assay
To assay the internalization of cell surface CFTR, cell surface proteins were biotinylated as above, after which the cells were warmed to 37° C for varying time periods in the presence of wiskostatin, latrunculin B (Calbiochem) or DMSO (vehicle). In some experiments a cyclic-AMP cocktail (100 μM cpt-cAMP, 20 μM forskolin, 1 mM IBMX) was added to the cells at this stage. At the indicated time points, cells were rapidly cooled by rinsing with cold PBS-CM and biotin molecules exposed at the cell surface were cleaved with a membrane impermeant stripping solution (100 mM MESNA (mercaptoethanesulfonic acid; Sigma), 50 mM Tris, 100 mM NaCl, 1 mM EDTA, 0.2% (w/v) BSA, adjusted to pH 8.0). Cells were treated with 3 changes of stripping solution for 15 minutes each on ice to ensure complete removal of surface-exposed biotin. Stripped cells were rinsed with cold PBS-CM and processed for lysis.
2.4. Cell lysis, streptavidin-agarose capture and immunoblotting
Cells were lysed with 1 ml PBS containing 1% (v/v) Triton X-100 and protease inhibitor cocktail (Roche). Lysates were centrifuged (x10,000 g) to remove cell debris and protein concentrations of clarified lysates were estimated by standard micro-BCA protein assay (Pierce). Equivalent amounts of cell lysates (2 mg) were rotated with 50 μl of packed streptavidin-agarose (Novagen) overnight at 4° C. The streptavidin-agarose beads were washed extensively with lysis buffer and bound proteins were eluted with Laemmli buffer and separated by SDS-PAGE on 4–15% gradient gels (Bio-Rad) followed by immunoblotting on polyvinylidene difluoride membranes (Millipore). CFTR protein was detected using monoclonal antibodies against the C-terminal tail (clones 24-1, R&D Research and GA-1, Santa Cruz Biotech.) and protein bands were visualized by enhanced chemiluminiscence (Pierce). Signals were quantified by densitometric scanning followed by analysis with Scion Image (release beta 4.0.2) (Scion Corp.).
2.5. Electrophysiology
BHK-CFTR cells were grown on plastic coverslips for excised inside-out patch clamp recordings. Patch pipettes were pulled to tip resistances of 3–4 mOhm. CFTR channels were activated following patch excision by PKA catalytic subunit (110 U/ml) and 1.5 mM Mg2+-ATP. Macroscopic currents were recorded in symmetrical solutions containing (in mM): 140 N-methyl-D-glucamine-Cl, 3 MgCl2, 1 EGTA and 10 TES (pH 7.3). Experiments were performed at 21–23°C. Currents were evoked using a voltage ramp protocol from +80 to −80 mV with a 10 sec time period. Data acquisition was performed using pCLAMP9 software (Axon Instruments).
Whole-cell recordings were obtained from HT29-Cl19A cells using methods as previously described [26] and solutions in which Cl− was the only permeant species as previously described for epithelial cells [25]. All experiments were conducted at room temperature (22–24° C) using an EPC-9 patch clamp amplifier (HEKA Electronik GmbH, Germany) and using the Pulse + PulseFit V 8.65 acquisition program (HEKA Electronik GmbH, Germany). Patch-clamp pipettes were pulled to resistances of 6–8 MOhms. using quartz glass and a Sutter model P-2000 puller (Sutter Instrument Co., CA). The pipette solution contained (in mM): 140 N-methyl D-glucamine; 40 HCl; 100 L-glutamic acid; 0.2 CaCl2; 2 MgCl2; 1 EGTA; 10 HEPES; and 2 ATP-Mg, pH 7.2. The bath solution contained (in mM): 140 N-methyl D-glucamine; 140 HCl; 2 CaCl2; 1 MgCl2; and 10 HEPES, pH 7.4. The Cl− equilibrium potential in these experiments was −31 mV. CFTR channels were maximally stimulated by the addition of a cyclic-AMP-activating cocktail (200 μM cyclic-AMP, 10 μM forskolin, 1 mM IBMX ) to the pipette solution with voltage steps ranging from −110 to +110 mV in increments of 10 mV from a holding potential of −40 mV. Data were acquired using a Pentium 4 PC and analyzed off-line using the integrated graphics package, IGOR Pro (WaveMetrics, OR).
2.6. Data analysis and statistics
Each experiment was repeated at least 3 times. The data are expressed as the means ± S.E. Statistical analysis was performed using student’s unpaired t test (two-tailed) to compare the means, and significance was determined at p< 0.05
3. Results
3.1. Wiskostatin decreases steady-state levels of surface CFTR
BHK-CFTR cells were incubated at 0° C with NHS-SS-biotin in order to label CFTR at the cell surface. This reagent covalently binds to extracellular residues (most likely lysines) on surface proteins (see methods) and has been used extensively to label surface CFTR [2, 27]. In the initial experiments we determined the dose of wiskostatin that produced a maximal effect on CFTR expression at the cell surface. Previous in vitro studies have reported that wiskostatin binds to WASP fragments with an apparent Kd ~100 μM [17]. We observed that the actin cytoskeleton was disrupted in >90% of cells treated with this dose of wiskostatin for 2 h by staining with phalloidin-alexa-fluor-488 (not shown). Therefore, BHK-CFTR cells were treated with 0–100 μM wiskostatin for 2 hours prior to biotinylation and subsequent cell lysis. Incubating cells for longer time periods or with higher concentrations resulted in loss of cell adherence that interfered with subsequent washing and processing steps. Biotinylated proteins were captured with streptavidin-agarose beads following cell lysis and CFTR was detected using monoclonal antibodies to its C-terminal tail. Total CFTR was detected by directly immunoblotting the cell lysates. As shown in the panel representing the streptavidin-bound fraction of CFTR (Fig. 1A, upper panel), there was a dose-dependent decrease in the surface CFTR signal in the presence of wiskostatin. We determined by densitometry (Fig. 1B) that about 80% of the CFTR signal was lost from the cell surface at the highest dose of wiskostatin (100 μM) as compared to vehicle control. There was, however, no appreciable change in the total CFTR content in the lysates at all doses of wiskostatin (Fig. 1A, lower panel). From these results it is clear that wiskostatin causes a substantial decrease in CFTR levels at the cell surface but does not alter the total CFTR content of cells (at least for the time and doses used in this study).
Fig. 1.
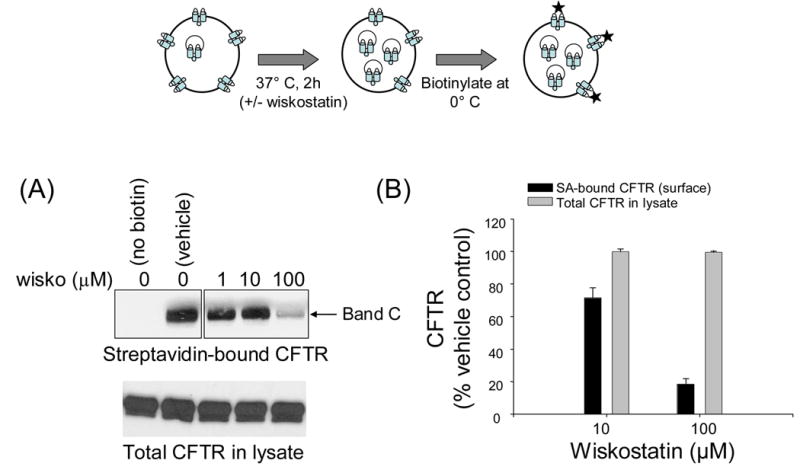
Wiskostatin decreases steady-state surface CFTR expression in a dose-dependent manner. Top panel, schematic representation of labeling cell-surface proteins with NHS-SS-biotin. A, surface-biotinylated CFTR captured with streptavidin-agarose (upper) or total CFTR protein (lower) in lysates of BHK cells treated with vehicle or indicated doses of wiskostatin for 2h. Band C represents mature, fully glycosylated CFTR. The blots in the upper panel represent data from a single experiment in which additional lanes not relevant to this experiment were removed. B, CFTR signals at indicated doses of wiskostatin were quantified by densitometry and normalized to the CFTR signals in vehicle-treated cells. The bars represent means ± S.E. calculated from 3 separate experiments.
3.2. Wiskostatin and latrunculin B cause CFTR to accumulate within cells
Since the preceding results indicated that CFTR was removed from the cell surface following wiskostatin treatment, we next examined the fate of the internalized protein in these cells (Fig. 2). Since N-WASP is known to regulate actin assembly in cells, we also tested the effects of a known actin-disrupting reagent, namely, latrunculin B on the fate of CFTR. In these experiments BHK-CFTR cells were first incubated with NHS-SS-biotin on ice (to inhibit endocytosis) and then incubated in the presence or absence of wiskostatin or latrunculin B for 2 hours at 37° C. During this warming period the cell surface proteins are continuously endocytosed and recycled and reach a new steady-state distribution between the cell surface and intracellular compartments (refer to detailed time-course experiments in Fig 6). Next, the labeled proteins that were surface exposed at steady-state were stripped with a membrane-impermeant reducing reagent, MESNA [2]. The stripping efficiency was determined to be >95% for control cells that were not warmed to 37° C (see Fig. 2B). Biotinylated proteins were captured with streptavidin-agarose and immunoblotted with CFTR antibodies to detect the fraction of initially labeled CFTR that was present inside the cells at steady-state. We observed a dose-dependent increase in the amount of labeled CFTR that accumulated in the cells at steady-state in the presence of wiskostatin; >80% of the surface-labeled CFTR accumulated inside the cells at 100 μM wiskostatin as compared to ~30% in control (vehicle-treated) cells over a period of 2 hours. Similarly, in the presence of 10 μM latrunculin B, 60–70% of surface-biotinylated CFTR accumulated in the cells at steady-state (Fig. 2A, B). The IC50 for latrunculin B is typically in the submicromolar range, and at doses >10 μM was toxic to the cells. Again there was no change in the CFTR content in the lysates at all doses of wiskostatin or latrunculin B. Together with the results shown in Fig 1, these data indicate that wiskostatin and latrunculin caused CFTR to dramatically relocate from the cell surface to intracellular compartments.
Fig. 2.
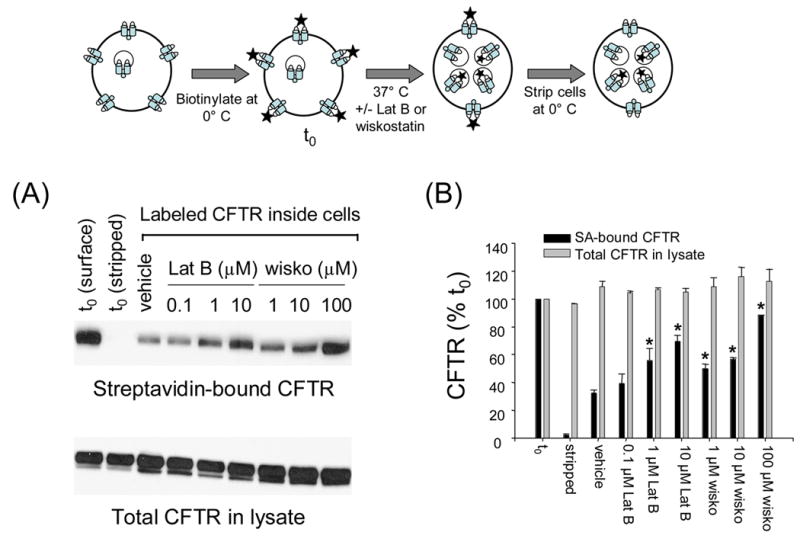
Wiskostatin and latrunculin B increase the fraction of surface-biotinylated CFTR that accumulates within the cells at steady-state. Top panel is a schematic representation of the CFTR internalization assay. t0 represents surface CFTR molecules labeled before wiskostatin/latrunculin B treatment (2h each). A, biotinylated CFTR (upper) or total CFTR protein (lower) in lysates of BHK cells treated with vehicle or indicated doses of latrunculin B or wiskostatin. B, CFTR signals at indicated doses of wiskostatin or latrunculin B were quantified by densitometry and normalized to the CFTR signal at t0. Note that >95% of signal is lost in samples stripped by MESNA at t0.The bars represent means ± S.E. of 3 separate experiments. The asterisks indicate p< 0.05 relative to vehicle control.
Fig. 6.
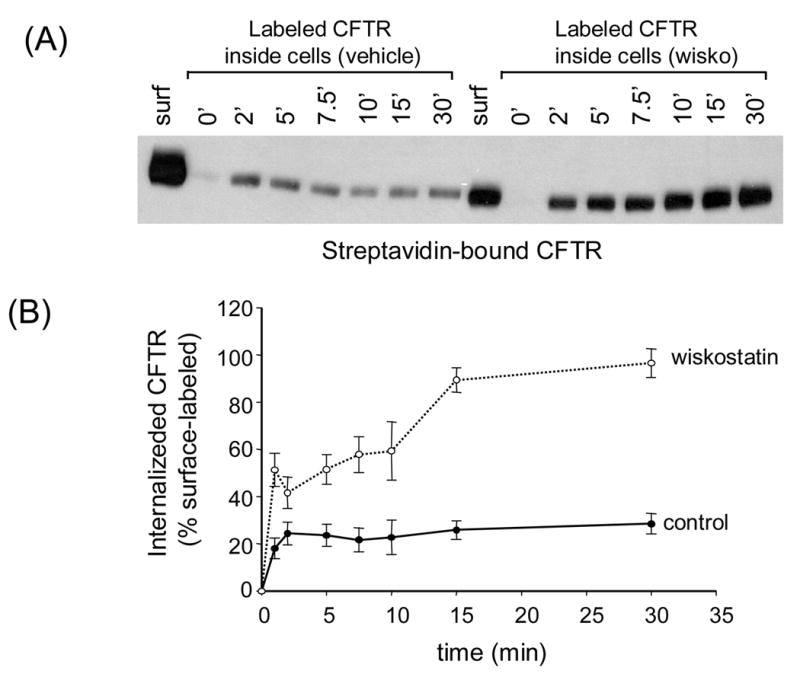
The effects of wiskostatin on the kinetics of CFTR internalization. A, biotinylated CFTR in lysates of BHK cells treated with vehicle or wiskostatin (100 μM) for 2 h at 37° C followed by biotinylation at 0° C. Cells were rewarmed to 37° C for indicated times and the labeled CFTR remaining at the cell surface was stripped by MESNA. Note that the surface CFTR signal was much less for the wiskostatin-treated cells as compared to vehicle control (see also Fig. 1). B, CFTR signals in control or wiskostatin-treated cells were normalized to the respective surface CFTR signals and plotted as a function of time. The data points represent means ± S.E. of 2–7 experiments.
We also observed that latrunculin B showed a tendency to reduce steady-state CFTR expression at the cell surface using the assay of Fig. 1, but the results obtained with this compound were not as reproducible as with wiskostatin (not shown). This disparity could be due to the fact that these reagents have different molecular targets and/or that wiskostatin has a more consistent effect on actin organization.
3.3 Wiskostatin and latrunculin B decrease CFTR-mediated currents in BHK membrane patches and in HT29 colonic epithelial cells
The preceding results indicate that macroscopic CFTR currents should be reduced by wiskostatin if it indeed causes CFTR to redistribute from the cell surface to the cell interior. In order to test this prediction we performed two complementary functional assays in different cell types, (i) macroscopic excised patch clamp experiments in BHK-CFTR cells expressing high levels of recombinant human CFTR and (ii) whole-cell patch clamp studies in HT29-Cl19A human colonic epithelial cells expressing low levels of native CFTR. The BHK-CFTR cells were pre-treated with wiskostatin following which inside-out membrane patches were excised using large tip pipettes. CFTR-mediated currents were activated by adding normally saturating concentrations of PKA and Mg-ATP to the bath. In control patches excised from untreated cells, the CFTR-mediated currents were of typically high magnitude (>400pA; Fig. 3A) due to the presence of several hundred CFTR channels per patch [28]. The fact that the elicited currents were (i) Mg-ATP-dependent (not shown) and (ii) inhibited by addition of the voltage-dependent pore-blocker glibenclamide (see Fig. 3A) confirmed that the currents were CFTR-mediated. By comparison, the PKA-activated and Mg-ATP-dependent currents were consistently much smaller for membrane patches excised from wiskostatin-treated cells (Fig. 3B, C) as also for membrane patches excised from cells pre-treated with latrunculin B (Fig. 3C). The low magnitude of the CFTR-mediated currents indicated that the number of channels in the patch was drastically reduced and/or that the existing channels had very low activity. Because the biochemical results indicated that wiskostatin substantially decreases the number of CFTR molecules at the cell surface, one explanation is that the reduced currents are due to a decrease in the channel density at the plasma membrane. However, it is worth noting that CFTR-mediated currents were inhibited by up to 80% (Fig. 3C) by a dose of wiskostatin (5 μM) that did not produce an equivalent decrease in surface-labeled CFTR (see Fig. 1) (membrane patches could not be reliably obtained at higher doses of wiskostatin). This disparity may be due to the different conditions under which these two assays were performed. In order to test the possibility that wiskostatin could possibly affect the regulatory properties of CFTR as well, wiskostatin (5 μM) was added directly to the cytoplasmic side of excised membrane patches (results not shown). We did not observe an acute reduction of CFTR-mediated currents under these circumstances, indicating that wiskostatin does not direcly affect the gating properties of CFTR channels (see discussion).
Fig. 3.
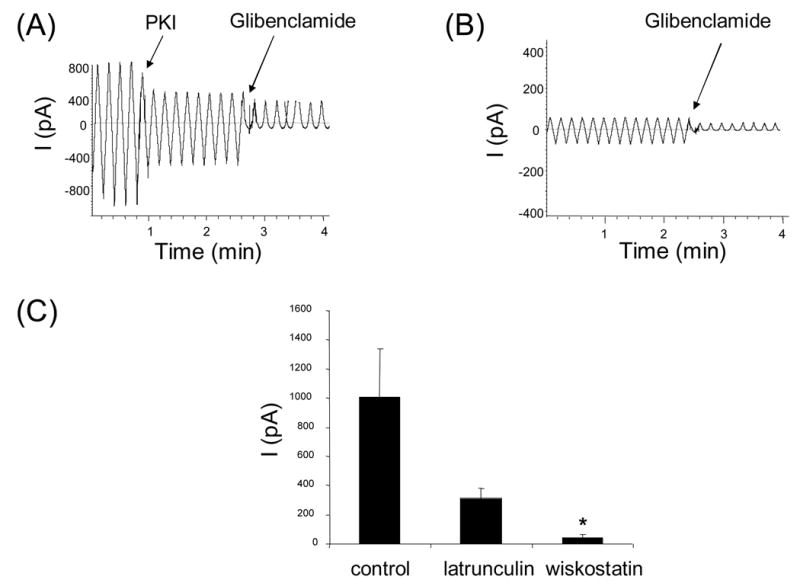
CFTR-mediated chloride currents across excised membrane patches are reduced by pre-treating BHK-CFTR cells with wiskostatin or latrunculin B. A,B. Representative macroscopic currents recordings for inside-out membrane patches excised from a control BHK-CFTR cell (A) or from a cell pre-treated with 5 μM wiskostatin for 2h (B). Inhibitory PKA peptide (PKI; 1.4 μg/ml) ) was added at the first arrow followed by glibenclamide (250 μM), a voltage-dependent pore blocker. Currents were elicited using a voltage ramp protocol (see Methods). C. Mean glibenclamide-sensitive currents (at −80 mV) for membrane patches excised from control cells and from cells pre-treated for 2h with wiskostatin (5 μM) or latrunculin B (1 μM). Data are means ± S.D. calculated from 4–7 experiments. Asterisk indicates p<0.05 relative to control. ( p = 0.09, for latrunculin B-treated cells)
In a complementary set of experiments, CFTR-mediated currents were recorded in HT29-Cl19A epithelial cells under control (untreated) or wiskostatin-treated conditions by whole-cell patch clamp analysis (Fig. 4). These cells express native CFTR and probably represent a more physiologically relevant system for recording CFTR activity. However, these cells express very modest levels of CFTR which makes it difficult to conduct biochemical studies such as surface biotinylation. CFTR-mediated Cl− currents were activated by addition of a cyclic-AMP-cocktail. These currents were validated to be CFTR-mediated on the basis of their dependence on cyclic-AMP activation and block by 1 mM glibenclamide or DPC (not shown). The cyclic-AMP-activated currents were substantially reduced for wiskostatin-treated cells (Fig 4a, b). Likewise, treatment with latrunculin B also inhibited CFTR-mediated currents in the HT-29-Cl19A cells (Fig. 4A, B). The results of the biochemical (surface biotinylation assays) and functional studies indicate that wiskostatin and latrunculin B have striking effects on both the surface expression and macroscopic functional activity of CFTR channels.
Fig. 4.
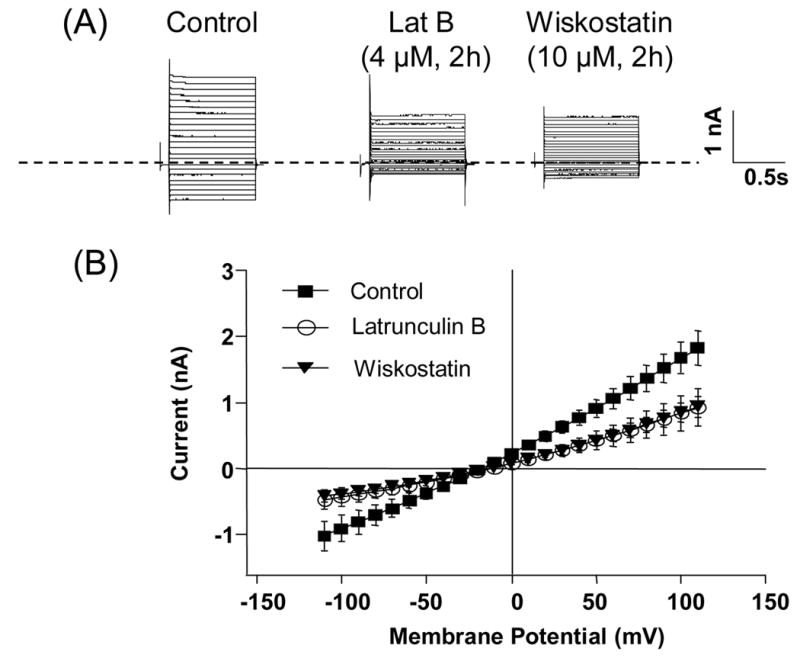
Whole-cell Cl− currents are decreased in HT29-Cl19A cells pre-treated with wiskostatin or latrunculin B. A. Representative cyclic-AMP-activated Cl− currents recorded in control HT29-Cl19A cells and cells pretreated with either latrunculin B (4 μM) or wiskostatin (10 μM) for 2 h. B. Average current-voltage relationships for the cyclic-AMP-dependent currents in control cells (n=5) and cells treated with latrunculin B (n = 5) or wiskostatin (n = 4).
3.4. Cyclic-AMP inhibits CFTR internalization in the presence or absence of wiskostatin
Cyclic-AMP has been reported to inhibit CFTR endocytosis, presumably, because the active channel is not efficiently internalized or that protein phosphorylation alters the recycling of CFTR [1, 2]. In this context we examined the effects of wiskostatin on CFTR accumulation in cells in the presence of a cyclic-AMP cocktail (Fig. 5). BHK-CFTR cells were first biotinylated at 4° C and then exposed to 100 μM wiskostatin in the presence or absence of a cyclic-AMP cocktail for 2 h at 37° C. Following this, the cells were stripped of surface biotin and the fraction of labeled CFTR that accumulated inside the cells at steady-state was assessed by streptavidin capture and immunoblotting. As previously reported, a smaller fraction of labeled CFTR accumulated inside the cells in the presence of cyclic-AMP as compared to control untreated cells (Fig. 5A). Cyclic-AMP also reduced CFTR internalization in the presence of wiskostatin, although a greater fraction of CFTR was internalized than with cyclic-AMP alone (Fig. 5A, B). These data indicate that cyclic-AMP inhibits CFTR internalization even in the presence of wiskostatin, which implies that these two factors may regulate different steps in CFTR traffic.
Fig. 5.
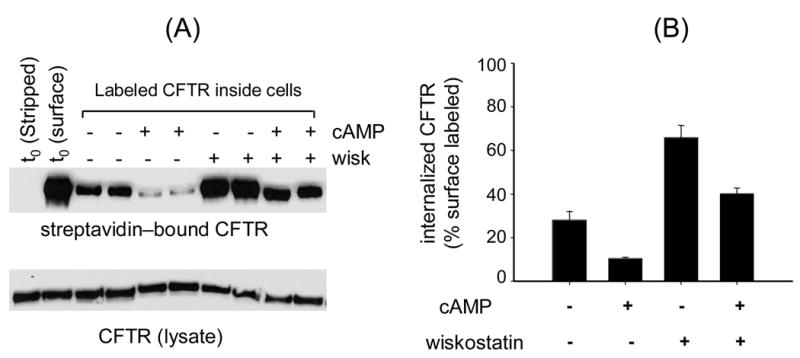
cyclic-AMP inhibits net CFTR internalization from the cell-surface, but to a lesser extent in the presence of wiskostatin. Refer to Fig. 2 for schematic. A, biotinylated CFTR (upper) or total CFTR protein (lower) in lysates of BHK cells harvested at t0 or following treatment with wiskostatin (100 μM) and/or a cyclic-AMP cocktail for 2h (see methods). Duplicate samples for each condition are shown. B, CFTR signals in the presence of wiskostatin and/or cyclic-AMP were quantified by densitometry and normalized to the CFTR signal at t0. The bars represent means ± S.E. of 4 experiments.
3.5. Wiskostatin has complex effects on the kinetics of CFTR internalization and recycling
From the preceding results it is apparent that wiskostatin has pronounced effects on CFTR expression at the cell surface. CFTR channel density at the plasma membrane reflects a balance between the rates of CFTR endocytosis and recycling back to the cell surface. Detailed time course experiments were performed to determine if wiskostatin primarily affected the rate of CFTR endocytosis. BHK-CFTR cells were treated with 100 μM wiskostatin for 2 h following which cells were surface-biotinylated at 0° C and then warmed to 37° C for varying times (0–30 min). At each time point the surface biotin was stripped with MESNA and the biotinylated CFTR inside the cells was detected by streptavidin-capture and immunoblotting as described (Fig. 6A). In the control cells (no wiskostatin) there was a quasi-linear increase in the fraction of CFTR that accumulated in the cells between 0–2 min. At longer times there was no apparent increase in the CFTR signal, presumably because the internalized CFTR is rapidly and efficiently recycled back to the cell surface (Fig. 6B). Two differences were observed for cells treated with wiskostatin: (i) a larger fraction of surface-labeled CFTR was internalized in the first two minutes and (ii) the biotinylated CFTR continued to accumulate in the cells over a longer period and reached steady state more gradually as compared to control (Fig. 6B). These data indicate that wiskostatin may have multiple effects on CFTR endocytosis/recycling: (i) an increase in the initial rate of CFTR internalization (caused either by an increase in the fraction of labeled CFTR molecules that can be internalized, or an increase in the intrinsic rate of endocytosis) and (ii) a reduction in the rate of recycling of internalized CFTR molecules back to the cell surface (evidenced as a slower time-course to reach steady-state).
4. Discussion
CFTR is present largely at the plasma membranes of epithelial cells where it regulates bidirectional transport of ions and water. The carboxy-terminal tail of CFTR contains signals that facilitate its entry into clathrin-coated pits [5] and it is well documented that the channel is constitutively internalized in various cell types [1, 2]. The main goal of this work was to determine if CFTR levels at the cell surface are modulated by actin polymerization and/or N-WASP-mediated actin assembly. We investigated the steady-state distribution of CFTR between the plasma membrane and intracellular compartments in response to latrunculin B, an actin depolymerizing agent and wiskostatin, a cell-permeable allosteric inhibitor of N-WASP that effectively blocks actin assembly. The WASP family proteins together with the Arp2/3 complex are key modulators of actin assembly in various cell types. The WASP family proteins are direct effectors for upstream signaling molecules, namely, Rho-GTPases and PIP2, which regulate many aspects of cell function including vesicle trafficking. Further, the Rho-GTPases are preferred targets for toxins secreted by opportunistic pathogens, including Pseudomonas aeruginosa, that cause life-threatening infections in cystic fibrosis patients. Through modulation of Rho-GTPases activity the pathogens mediate changes in the actin cytoskeleton which may facilitate their entry in cells [29]. In this context it was recently reported that a cell-free filtrate of P. aeruginosa reduced CFTR-mediated transepithelial Cl− secretion by inhibiting the endocytic recycling of CFTR in polarized human airway epithelial cells [30]. Our results raise the possibility that physiologic and pathologic factors that influence WASP activity may modify CFTR expression or functional activity at the plasma membrane. Future studies will be aimed at identifying such factors and determining their role in regulating CFTR surface expression and activity.
4.1. CFTR surface expression and activity are reduced by wiskostatin and latrunculin B
We observed a dose-dependent decrease in CFTR surface expression following wiskostatin treatment of BHK-CFTR cells in biotinylation studies. A large fraction of CFTR (~80%) was lost from the cell surface at 100 μM wiskostatin. There was no change in the total CFTR content in the lysates at all doses of wiskostatin indicating that there was no apparent degradation of CFTR under these conditions. The steady-state surface CFTR expression presumably reflects a balance between rates of internalization and of recycling back to the cell surface. Our results indicate that both wiskostatin and latrunculin B shifted the steady-state distribution towards greater amounts of CFTR inside cells while depleting the ion channel from the cell surface (Fig. 2). Similar results were also obtained with cytochalasin D, a standard actin-disrupting reagent that binds to the growing ends of the actin filaments (unpublished observations). However, the effect of wiskostatin was most consistent and reproducible in these experiments.
The biochemical results were corroborated by functional studies performed in cultured cells. CFTR-mediated Cl− currents were strongly inhibited by wiskostatin and latrunculin B both in BHK fibroblast cells that express high levels of recombinant CFTR and in HT29-CL19A epithelial cells that express low levels of native CFTR. The correlation between the results of the biochemical and functional studies favors the idea that these reagents greatly reduce the number of CFTR channels expressed at the cell surface. However, we observed that the dose of wiskostatin required to inhibit CFTR-mediated currents by >80% in membrane patches excised from BHK-CFTR cells (5 μM; Fig. 2) was less than the dose which resulted in 80% reduction in surface CFTR expression biochemically (100 μM; Fig. 1). One possible explanation is that the readouts for the surface biotinylation assay and the functional (patch clamp) assay cannot be directly compared, because the assays are performed under different conditions (intact cells versus broken cell assays) that may differently affect the actin cytoskeleton. However, we cannot rule out that the reagent could affect the regulation of individual CFTR channels as well. It seems unlikely that wiskostatin directly inhibits CFTR channels given that (i) wiskostatin binds to its specific target (N-WASP) in vitro with an apparent Kd of ~100 μM [17] and (ii) adding wiskostatin directly to the cytoplasmic side of excised membrane patches did not result in an acute reduction of CFTR-mediated currents. This, however, does not rule out other possibilities that have been proposed in the literature such as (i) CFTR activity is modulated by a direct interaction with actin filaments [31] or (ii) wiskostatin indirectly affects CFTR gating within intact cells by releasing a cellular kinase or phosphatase inhibitor that is tethered to F-actin filaments [32].
4.2. Cyclic-AMP inhibits CFTR internalization in the presence or absence of wiskostatin
There is a growing notion that phosphorylation of the CFTR channel and/or its functional activity can modulate its surface expression in some cell types. Prince et al., demonstrated that stimulation with cyclic-AMP decreased the internalization of CFTR from the cell surface by ~60% in T-84 epithelial cells [1]. Lukacs et al., similarly reported that activation of PKA inhibited the rate of CFTR endocytosis in stably transfected chinese hamster ovary cells [2]. Other investigators postulated that cyclic-AMP also stimulated exocytosis/recycling of CFTR to the cell surface [7]. Thus, cyclic-AMP could affect CFTR expression at the cell surface by (i) inhibiting endocytosis or (ii) enhancing recycling. In the present study we observed that cyclic-AMP inhibited CFTR internalization both in the presence and absence of wiskostatin, although this effect was partially attenuated by wiskostatin (Fig. 5). The results indicate that (i) wiskostatin and cyclic-AMP have opposite effects on CFTR expression at the cell surface and (ii) cyclic-AMP can partially override the effects of wiskostatin on CFTR internalization (or recycling; see below). It seems plausible that wiskostatin and cyclic-AMP primarily affect different steps in CFTR trafficking; on the one hand CFTR is endocytosed more slowly from the cell surface (as an effect of cyclic-AMP), whereas wiskostatin inhibits recycling of the internalized CFTR (see below). This could possibly explain the greater accumulation of CFTR inside cells in the presence of both cyclic-AMP and wiskostatin.
4.3. Models to explain effects of wiskostatin on CFTR surface expression
The results of this study support the existence of regulated CFTR trafficking which requires an appropriate balance between (i) internalization of CFTR into endocytic vesicles (endocytosis) and (ii) redistribution of internalized channels back to the plasma membrane (endocytic recycling). Wiskostatin and latrunculin B could affect the distribution of CFTR between these two compartments by (a) enhancing endocytosis from the cell surface, thereby increasing the intracellular pool of CFTR or (b) inhibiting recycling of endocytosed CFTR back to the cell surface (Fig. 7). These possibilities are not mutually exclusive. An effect of wiskostatin on endocytosis is supported by the kinetic data (Fig. 6) which indicated that wiskostatin increased the fraction of surface-labeled CFTR that was endocytosed at early time points. It is possible that some CFTR at the cell surface is tethered to the actin cytoskeleton either directly [31] or indirectly via PDZ-domain interactions with proteins that bind actin (e.g., NHERF1/EBP50, NHERF2/E3KARP and ezrin) [33–35]. Wiskostatin could increase the endocytic retrieval of CFTR by disrupting such interactions thereby increasing the proportion of surface CFTR that can be endocytosed. In support of this model, actin disruption by latrunculin A was proposed to release AMPA receptors from their anchoring to the post-synaptic membrane, thus allowing them to enter the perisynaptic endocytic pathway [36]. Such a putative mechanism would also be consistent with the observation that cyclic-AMP is able to inhibit CFTR internalization even in the presence of wiskostatin; depolymerization of actin by wiskostatin may un-tether CFTR from the cell surface, whereas cyclic-AMP may inhibit a downstream step in the endocytic pathway. Alternatively, wiskostatin could enhance the kinetics of internalization for an untethered population of CFTR, although this model would not be consistent with the reported positive role of N-WASP in regulating the endocytosis of other proteins [20]. Regarding a possible effect of wiskostatin on recycling, it is interesting to note that the endocytic accumulation of CFTR reached steady-state rapidly (within two minutes) under control conditions, suggesting that the internalized CFTR was efficiently recycled back to the cell surface under these conditions. In contrast, there was slow accumulation of CFTR in the wiskostatin-treated cells that reached steady-state very gradually (Fig. 6). Thus, whether we assume all CFTR at the plasma membrane to be freely endocytosed or exist as two populations (tethered and untethered), one would have observed a rapid increase in the CFTR signal only in the initial few minutes if wiskostatin augmented the endocytic retrieval of CFTR alone. Therefore, the slower accumulation of CFTR in the wiskostatin-treated cells suggests that the recycling pathway had to be inhibited or saturated, which resulted in a net increase in the intracellular pool of CFTR. It is therefore likely that wiskostatin also compromises the recycling efficiency of CFTR. A more definitive conclusion would be possible if we could directly measure the recycling kinetics of CFTR in control and wiskostatin-treated cells. However, the rapid kinetics of CFTR endocytosis, especially in the wiskostatin-treated cells (Fig. 6), precluded accurate measurements of the initial (unidirectional) rates of CFTR recycling in BHK cells (unpublished observations).
Fig. 7.
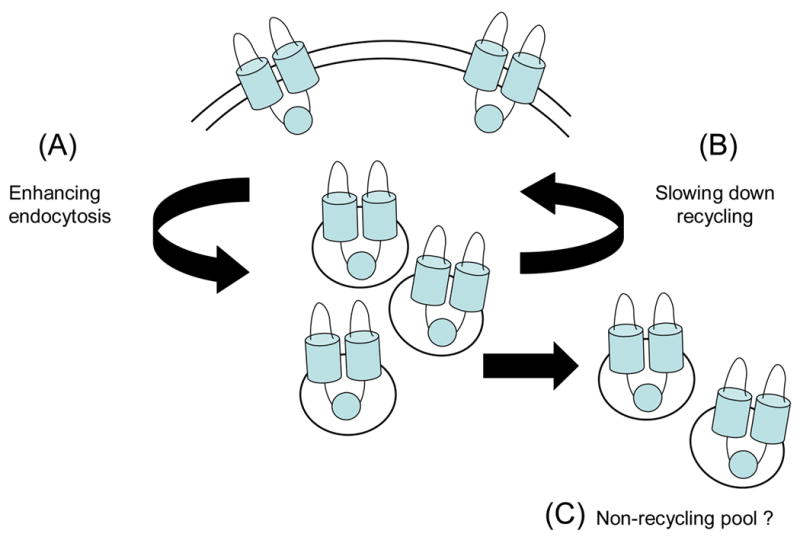
Model(s) to explain wiskostatin effects on CFTR trafficking. Wiskostatin could affect the distribution of CFTR between the cell surface and intracellular compartments by (A) enhancing endocytic retrieval of CFTR from the plasma membrane, (B) inhibiting the recycling of endocytosed CFTR back to the cell surface or (C) translocating CFTR to an off-pathway intracellular compartment.
It is noteworthy that CFTR remained undegraded over a period of two hours in the wiskostatin-treated cells, which suggests that the protein was possibly trapped in the recycling compartment (or an off-pathway compartment). Picciano, et. al., have reported that CFTR is trapped in an intracellular compartment when a regulator of membrane recycling (Rme-1) is inhibited [6]. Conceivably, wiskostatin could have similar effects. Preliminary results indicate that there is partial recovery of surface CFTR levels when cells are transferred to regular medium following wiskostatin treatment (data not shown), suggesting that CFTR is not permanently trapped within the cells.
In conclusion, the results reported in this paper implicate a role of N-WASP and actin in regulating CFTR expression and activity at the cell surface. Wiskostatin, an allosteric inhibitor of N-WASP activity, has potent and complex effects on CFTR endocytosis and recycling. The net effect was a marked redistribution of CFTR from the cell surface into intracellular compartments without CFTR degradation. Physiologic or pathologic factors (e.g., Rho-GTPase inhibitors, bacterial toxins, etc) that modulate N-WASP activity and influence actin assembly could influence CFTR surface expression by this mechanism.
Acknowledgments
The authors thank Lois Musgrove and Ge Li for technical support. This work was supported by an NIH grant (HL 58341).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Prince LS, Workman RB, Jr, Marchase RB. Rapid endocytosis of the cystic fibrosis transmembrane conductance regulator chloride channel. Proc Natl Acad Sci U S A. 1994;91:5192–6. doi: 10.1073/pnas.91.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lukacs GL, Segal G, Kartner N, Grinstein S, Zhang F. Constitutive internalization of cystic fibrosis transmembrane conductance regulator occurs via clathrin-dependent endocytosis and is regulated by protein phosphorylation. Biochem J. 1997;328:353–61. doi: 10.1042/bj3280353. Pt 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bradbury NA, Cohn JA, Venglarik CJ, Bridges RJ. Biochemical and biophysical identification of cystic fibrosis transmembrane conductance regulator chloride channels as components of endocytic clathrin-coated vesicles. J Biol Chem. 1994;269:8296–302. [PubMed] [Google Scholar]
- 4.Weixel KM, Bradbury NA. Mu 2 binding directs the cystic fibrosis transmembrane conductance regulator to the clathrin-mediated endocytic pathway. J Biol Chem. 2001;276:46251–9. doi: 10.1074/jbc.M104545200. [DOI] [PubMed] [Google Scholar]
- 5.Hu W, Howard M, Lukacs GL. Multiple endocytic signals in the C-terminal tail of the cystic fibrosis transmembrane conductance regulator. Biochem J. 2001;354:561–72. doi: 10.1042/0264-6021:3540561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Picciano JA, Ameen N, Grant BD, Bradbury NA. Rme-1 regulates the recycling of the cystic fibrosis transmembrane conductance regulator. Am J Physiol Cell Physiol. 2003;285:C1009–18. doi: 10.1152/ajpcell.00140.2003. [DOI] [PubMed] [Google Scholar]
- 7.Howard M, Jiang X, Stolz DB, Hill WG, Johnson JA, Watkins SC, Frizzell RA, Bruton CM, Robbins PD, Weisz OA. Forskolin-induced apical membrane insertion of virally expressed, epitope-tagged CFTR in polarized MDCK cells. Am J Physiol Cell Physiol. 2000;279:C375–82. doi: 10.1152/ajpcell.2000.279.2.C375. [DOI] [PubMed] [Google Scholar]
- 8.Qualmann B, Kessels MM, Kelly RB. Molecular links between endocytosis and the actin cytoskeleton. J Cell Biol. 2000;150:F111–6. doi: 10.1083/jcb.150.5.f111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merrifield CJ, Qualmann B, Kessels MM, Almers W. Neural Wiskott Aldrich Syndrome Protein (N-WASP) and the Arp2/3 complex are recruited to sites of clathrin-mediated endocytosis in cultured fibroblasts. Eur J Cell Biol. 2004;83:13–8. doi: 10.1078/0171-9335-00356. [DOI] [PubMed] [Google Scholar]
- 10.Lamaze C, Fujimoto LM, Yin HL, Schmid SL. The actin cytoskeleton is required for receptor-mediated endocytosis in mammalian cells. J Biol Chem. 1997;272:20332–5. doi: 10.1074/jbc.272.33.20332. [DOI] [PubMed] [Google Scholar]
- 11.Forte JG, Ly B, Rong Q, Ogihara S, Ramilo M, Agnew B, Yao X. State of actin in gastric parietal cells. Am J Physiol. 1998;274:C97–104. doi: 10.1152/ajpcell.1998.274.1.C97. [DOI] [PubMed] [Google Scholar]
- 12.Sandvig K, van Deurs B. Selective modulation of the endocytic uptake of ricin and fluid phase markers without alteration in transferrin endocytosis. J Biol Chem. 1990;265:6382–8. [PubMed] [Google Scholar]
- 13.Fujimoto LM, Roth R, Heuser JE, Schmid SL. Actin assembly plays a variable, but not obligatory role in receptor-mediated endocytosis in mammalian cells. Traffic. 2000;1:161–71. doi: 10.1034/j.1600-0854.2000.010208.x. [DOI] [PubMed] [Google Scholar]
- 14.Swiatecka-Urban A, Boyd C, Coutermarsh B, Karlson KH, Barnaby R, Aschenbrenner L, Langford GM, Hasson T, Stanton BA. Myosin VI regulates endocytosis of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2004;279:38025–31. doi: 10.1074/jbc.M403141200. [DOI] [PubMed] [Google Scholar]
- 15.Prat AG, Xiao YF, Ausiello DA, Cantiello HF. cAMP-independent regulation of CFTR by the actin cytoskeleton. Am J Physiol. 1995;268:C1552–61. doi: 10.1152/ajpcell.1995.268.6.C1552. [DOI] [PubMed] [Google Scholar]
- 16.Prat AG, Cunningham CC, Jackson GR, Jr, Borkan SC, Wang Y, Ausiello DA, Cantiello HF. Actin filament organization is required for proper cAMP-dependent activation of CFTR. Am J Physiol. 1999;277:C1160–9. doi: 10.1152/ajpcell.1999.277.6.C1160. [DOI] [PubMed] [Google Scholar]
- 17.Peterson JR, Bickford LC, Morgan D, Kim AS, Ouerfelli O, Kirschner MW, Rosen MK. Chemical inhibition of N-WASP by stabilization of a native autoinhibited conformation. Nat Struct Mol Biol. 2004;11:747–55. doi: 10.1038/nsmb796. [DOI] [PubMed] [Google Scholar]
- 18.Ivanov AI, Hunt D, Utech M, Nusrat A, Parkos CA. Differential roles for actin polymerization and a myosin II motor in assembly of the epithelial apical junctional complex. Mol Biol Cell. 2005;16:2636–50. doi: 10.1091/mbc.E05-01-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leung DW, Morgan DM, Rosen MK. Biochemical properties and inhibitors of (N-)WASP. Methods Enzymol. 2006;406:281–96. doi: 10.1016/S0076-6879(06)06021-6. [DOI] [PubMed] [Google Scholar]
- 20.Benesch S, Polo S, Lai FP, Anderson KI, Stradal TE, Wehland J, Rottner K. N-WASP deficiency impairs EGF internalization and actin assembly at clathrin-coated pits. J Cell Sci. 2005;118:3103–15. doi: 10.1242/jcs.02444. [DOI] [PubMed] [Google Scholar]
- 21.Kanzaki M, Watson RT, Khan AH, Pessin JE. Insulin stimulates actin comet tails on intracellular GLUT4-containing compartments in differentiated 3T3L1 adipocytes. J Biol Chem. 2001;276:49331–6. doi: 10.1074/jbc.M109657200. [DOI] [PubMed] [Google Scholar]
- 22.Jiang ZY, Chawla A, Bose A, Way M, Czech MP. A phosphatidylinositol 3-kinase-independent insulin signaling pathway to N-WASP/Arp2/3/F-actin required for GLUT4 glucose transporter recycling. J Biol Chem. 2002;277:509–15. doi: 10.1074/jbc.M108280200. [DOI] [PubMed] [Google Scholar]
- 23.Shumay E, Gavi S, Wang HY, Malbon CC. Trafficking of beta2-adrenergic receptors: insulin and beta-agonists regulate internalization by distinct cytoskeletal pathways. J Cell Sci. 2004;117:593–600. doi: 10.1242/jcs.00890. [DOI] [PubMed] [Google Scholar]
- 24.Zhu T, Dahan D, Evagelidis A, Zheng S, Luo J, Hanrahan JW. Association of cystic fibrosis transmembrane conductance regulator and protein phosphatase 2C. J Biol Chem. 1999;274:29102–7. doi: 10.1074/jbc.274.41.29102. [DOI] [PubMed] [Google Scholar]
- 25.Naren AP, Nelson DJ, Xie W, Jovov B, Pevsner J, Bennett MK, Benos DJ, Quick MW, Kirk KL. Regulation of CFTR chloride channels by syntaxin and Munc18 isoforms. Nature. 1997;390:302–5. doi: 10.1038/36882. [DOI] [PubMed] [Google Scholar]
- 26.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 27.Weixel K, Bradbury NA. Analysis of CFTR endocytosis by cell surface biotinylation. Methods Mol Med. 2002;70:323–40. doi: 10.1385/1-59259-187-6:323. [DOI] [PubMed] [Google Scholar]
- 28.Wang W, Oliva C, Li G, Holmgren A, Lillig CH, Kirk KL. Reversible silencing of CFTR chloride channels by glutathionylation. J Gen Physiol. 2005;125:127–41. doi: 10.1085/jgp.200409115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun J, Barbieri JT. ExoS Rho GTPase-activating protein activity stimulates reorganization of the actin cytoskeleton through Rho GTPase guanine nucleotide disassociation inhibitor. J Biol Chem. 2004;279:42936–44. doi: 10.1074/jbc.M406493200. [DOI] [PubMed] [Google Scholar]
- 30.Swiatecka-Urban A, Moreau-Marquis S, Maceachran DP, Connolly JP, Stanton CR, Su JR, Barnaby R, O'Toole G A, Stanton BA. Pseudomonas aeruginosa inhibits endocytic recycling of CFTR in polarized human airway epithelial cells. Am J Physiol Cell Physiol. 2006;290:C862–72. doi: 10.1152/ajpcell.00108.2005. [DOI] [PubMed] [Google Scholar]
- 31.Chasan B, Geisse NA, Pedatella K, Wooster DG, Teintze M, Carattino MD, Goldmann WH, Cantiello HF. Evidence for direct interaction between actin and the cystic fibrosis transmembrane conductance regulator. Eur Biophys J. 2002;30:617–24. doi: 10.1007/s00249-001-0188-9. [DOI] [PubMed] [Google Scholar]
- 32.Fischer H, Illek B, Machen TE. The actin filament disrupter cytochalasin D activates the recombinant cystic fibrosis transmembrane conductance regulator Cl- channel in mouse 3T3 fibroblasts. J Physiol. 1995;489:745–54. doi: 10.1113/jphysiol.1995.sp021088. Pt 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Short DB, Trotter KW, Reczek D, Kreda SM, Bretscher A, Boucher RC, Stutts MJ, Milgram SL. An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton. J Biol Chem. 1998;273:19797–801. doi: 10.1074/jbc.273.31.19797. [DOI] [PubMed] [Google Scholar]
- 34.Sun F, Hug MJ, Lewarchik CM, Yun CH, Bradbury NA, Frizzell RA. E3KARP mediates the association of ezrin and protein kinase A with the cystic fibrosis transmembrane conductance regulator in airway cells. J Biol Chem. 2000;275:29539–46. doi: 10.1074/jbc.M004961200. [DOI] [PubMed] [Google Scholar]
- 35.Haggie PM, Stanton BA, Verkman AS. Increased diffusional mobility of CFTR at the plasma membrane after deletion of its C-terminal PDZ binding motif. J Biol Chem. 2004;279:5494–500. doi: 10.1074/jbc.M312445200. [DOI] [PubMed] [Google Scholar]
- 36.Zhou Q, Xiao M, Nicoll RA. Contribution of cytoskeleton to the internalization of AMPA receptors. Proc Natl Acad Sci U S A. 2001;98:1261–6. doi: 10.1073/pnas.031573798. [DOI] [PMC free article] [PubMed] [Google Scholar]