

Abstract
Age-related macular degeneration (ARMD) is the leading cause of irreversible visual loss in the Western world, affecting approximately 25 million people worldwide. The pathogenesis is complex and missense mutations in FBLN5 have been reported in association with ARMD. We have investigated the role of fibulin 5 in ARMD by completing the first European study of the gene FBLN5 in ARMD (using 2 European cohorts of 805 ARMD patients and 279 controls) and by determining the functional effects of the missense mutations on fibulin 5 expression. We also correlated the FBLN5 genotype with the ARMD phenotype. We found two novel sequence changes in ARMD patients that were absent in controls and expressed these and the other nine reported FBLN5 mutations associated with ARMD and two associated with the autosomal recessive disease cutis laxa. Fibulin 5 secretion was significantly reduced (P<0.001) for four ARMD (p.G412E, p.G267S, p.I169 T, and p.Q124P) and two cutis laxa (p.S227P, p.C217R) mutations. These results suggest that some missense mutations associated with ARMD lead to decreased fibulin 5 secretion with a possible corresponding reduction in elastinogenesis. This study confirms the previous work identifying an association between FBLN5 mutations and ARMD and for the first time suggests a functional mechanism by which these mutations can lead to ARMD. It further demonstrates that FBLN5 mutations can be associated with different phenotypes of ARMD (not limited to the previously described cuticular drusen type). Such knowledge may ultimately lead to the development of novel therapies for this common disease.
Keywords: age-related macular degeneration, ARMD, fibulin 5, FBLN5, cutis laxa, genotype–phenotype correlation, elastinogenesis
INTRODUCTION
Age-related macular degeneration (ARMD; MIM#s 603075, 153800, and 608895) is the commonest cause of irreversible visual loss in the Western world [Klaver et al., 1998; Tielsch et al., 1995], affecting approximately 25 million people worldwide (http://AMDalliance.org).
The progressive loss of central vision in ARMD occurs due to degenerative and neovascular changes within the retina at the macula. Specifically, extracellular deposits of protein and lipid develop beneath the retinal pigment epithelium (RPE) and within an elastin-containing structure known as Bruch's membrane and can be seen clinically as yellowish white spots in the retina [Ambati et al., 2003]. The RPE degenerates and choroidal neovascularization can spread through the RPE into the retina with disastrous consequences for central vision [Ambati et al., 2003].
Both environmental and genetic risk factors are known to be associated with ARMD [Christen et al., 1996; Seddon et al., 1996]. Sequence changes in the fibulin 5, complement factor H, human leukocyte antigen (HLA), and perhaps the toll-like receptor 4 genes have all been associated with ARMD [Hageman et al., 2005; Haines et al., 2005; Edwards et al., 2005; Klein et al., 2005; Zareparsi et al., 2005; Stone et al., 2004; Goverdhan et al., 2005].
Previously a comprehensive analysis of fibulin 5 (FBLN5; MIM#604580) in a cohort of 402 patients with ARMD and age-matched controls identified missense mutations in FBLN5 in 1.7% of ARMD patients recruited from the United States [Stone et al., 2004]. Due to the high prevalence of ARMD this equates to many hundreds of thousands of ARMD patients worldwide with FBLN5 mutations.
The fibulins are a family of extracellular matrix (ECM) proteins [Timpl et al., 2003] and are characterized by tandem arrays of epidermal growth factor–like (EGF-like) domains. Fibulins are widespread components of ECM and participate in diverse supramolecular structures with binding sites for several proteins including tropoelastin, fibrillin and proteoglycans. Phenotypes previously associated with mutations in fibulin genes include Malattia Leventinese and Doyne honeycomb retinal dystrophy (fibulin 3) [Stone et al., 1999], cutis laxa (fibulin 5) [Loeys et al., 2002; Markova et al., 2003], and ARMD in a single family (fibulin 6) [Schultz et al., 2003].
Fibulin 5 is a 66-kDa secreted protein of 448 amino acids [Nakamura et al., 1999]. It shares around 90% amino acid identity with other fibulin species. It is one of the shorter fibulins and has a central segment of five calcium binding EGF-like modules and a carboxy-terminal domain III of sequence identity with the other fibulins [Timpl et al., 2003]. It is widely expressed throughout the body including the RPE [Stone et al., 2004] and choroid (www.ncbi.nlm.nih.gov/UniGene). The missense FBLN5 variants identified in the ARMD patients [Stone et al., 2004] are spread along the protein with one lying very close to the arg-gly-asp (RGD) integrin binding domain (p.V60L), one affecting a calcium-binding EGF-like domain (p.I169 T), and three affecting the region thought to bind to LOXL1 (p.R351W, p.A363 T, and p.G412E) [Liu et al., 2004] .
Two groups have published a description of a fibulin 5 −/− mouse [Yanagisawa et al., 2002; Nakamura et al., 2002]. In both cases the mice exhibited a severe form of elastinopathy with loose skin, vascular abnormalities, and emphysematous lungs, demonstrating that FBLN5 is essential for elastinogenesis. Of note, Bruch's membrane, lying between the RPE and choroid, is a multilayered structure that contains a central layer of elastin, and abnormalities in this membrane play a key role in the pathogenesis of ARMD.
We chose to investigate FBLN5 variants in the European population, identified novel sequence variants, and determined their prevalence in two European cohorts of 514 patients (United Kingdom) and 288 patients (Netherlands). FBLN5 is a secreted protein which functions in the extracellular matrix [Timpl et al., 2003]. We thus evaluated whether these novel sequence variants and previously identified variants impaired secretion of mutant FBLN5 in cell culture experiments. We also determined phenotype–genotype correlations.
MATERIALS AND METHODS
At the University of Southampton a cohort of 514 ARMD patients and 188 control patients from the same clinic population were screened. All patients and control patients were over the age of 50 years and had undergone a dilated retinal examination. Mutation analysis of FBLN5 (GenBank accession number: NM_006329.2) was performed using a combination of single stranded conformation polymorphism and direct sequencing as reported previously [Lotery et al., 2000a,b, 2001]. Ethical permission for this study was obtained from the Southampton and Southwest Hants Local Research Ethics Committee (approval no. 347/02/t).
The Dutch cohort consisted of 288 unrelated Caucasian patients and 149 ethnically- and age-matched controls. Subjects were primarily recruited from the ophthalmic departments in Amsterdam and Rotterdam. All subjects underwent fundus photography. Transparencies were graded according to a modified version of the International Classification System [Bird et al., 1995]. Cases were subjects with soft distinct drusen and pigmentary irregularities, soft indistinct or reticular drusen, geographic atrophy, or neovascular macular degeneration (i.e., age-related maculopathy stages 2–4). Controls were subjects aged 65 years and older with no or only a few small hard drusen and no other macular pathology (age-related maculopathy stage 0). Control subjects were either unaffected spouses of cases, or subjects who attended the ophthalmology department for reasons other than retinal pathology. A combination of DHPLC analysis (Transgenomic, Omaha, NE; www.transgenomic.com) and direct sequencing was used for mutation analysis.
Computer-Based Predictions of Structure
The protein fold recognition program “Threader 3” [Jones et al., 1999] was used to predict the effects of missense mutations on protein folding. Threader assesses whether a test primary amino acid sequence (in this case a fibulin 5 sequence) can align with and fold like known protein structures from a library. For threading, the test sequence is divided dynamically into small segments. Threading of the test sequence upon each known structure is based on optimization of the pairwise (folding) energy sums and also the solvation energy sums. Threader generates Z-scores, which are quantitative measures of the quality of matches: Large, positive Z-scores point toward a successful prediction. Threading was not constrained by providing any predicted secondary structure for fibulin 5.
Expression of Fibulin 5 (FBLN5) in COS7 Cells
To investigate the expression of wild-type (WT) and mutant FBLN5 we generated expression constructs in pEF6-ssFLAG vector for WT FBLN5 and 13 different FBLN5 mutations. These included those mutations previously described as being associated with ARMD (p.V60L, p.R71Q, p.P87S, p.I169 T, p.R351W, p.A363 T, and p.G412E) [Stone et al., 2004], the mutations we have identified as being associated with ARMD (p.G267S, p.Q124P), one missense change we identified in a control patient without ARMD as a control (p.G202R), a mutation identified in both ARMD and control patients (p.V126 M), and two mutations previously described as causing cutis laxa (p.S227P [Loeys et al., 2002] and (p.C217R [Fischer et al., 2004]). Mutations were generated by site-directed mutagenesis using the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA; www.stratagene.com) as described [Wang et al., 2002] and the presence of each mutation was confirmed by DNA sequencing. The pEF6-ssFLAG constructs were used to transfect COS-7 cells and protein expression was induced. Nontransfected COS-7 cells do not express fibulin 5 (data not shown).
Cell Culture and Transfection
All experiments were carried out in triplicate. COS-7 cells were grown and transfected as described previously using 0.5 μg of DNA [Wang et al., 2002]. After 72 hr cells were harvested, and for Western blot analysis whole cell lysates were prepared as previously described and blotted [Wang et al., 2002]. Samples of the medium (nonconcentrated) in which the cells had grown were also blotted to detect secreted fibulin 5. The expressed tagged fibulin 5 was detected by Anti-FLAG antibody (Sigma, Gillingham, Dorset, England; www.sigmaaldrich.com) and the bands were quantified using Fluor-S Max MultiImager (Biorad, Hercules, CA; www.bio-rad.com).
For each sample in all experiments we compared the secreted fibulin 5 to fibulin 5 in the cell lysate, thus allowing us to determine a ratio for each sample. We did not compare absolute measurements between samples as we were using transient transfections but, instead, we compared the ratios of cell lysate and medium fibulin 5 for each mutant fibulin 5 construct with the ratio for the WT fibulin5 constructs.
RESULTS
Two novel heterozygous missense mutations were identified solely in three United Kingdom patients (p.Q124P in one patient and p.G267S in two apparently unrelated patients). No sequence variations were found in patients alone in the Netherlands cohort (Table 1). Two missense changes were identified in controls (p.V126 M found in 5 controls and 1 ARMD patient and p.G202R found in a control individual), suggesting these changes are polymorphic sequence variants.
Table 1.
SequenceVariations in the FBLN5 Gene*
| Nucleotide change |
Effect or sequence change |
Degree of conservationa |
|||
|---|---|---|---|---|---|
| Amino acid changing variants | |||||
| United Kingdom data | ARMD (n = 514) | Controls (n = 188) | |||
| Exon 4 | c.371A>C | p.Gln124Pro | 1 | 0 | 8/8, chicken |
| Exon 6 | c.604G>A | p.Gly202Arg | 0 | 1 | 11/11, chicken |
| Exon 8 | c.799G>A | p.Gly267Ser | 2 | 0 | 10/10, chicken |
| Netherlands data | ARMD (n = 291) | Controls (n = 91) | |||
| Exon 4 | c.376G>A | p.Val126Met | 1 | 5 | 7/8, bovine |
| Synonymous coding variants | |||||
| United Kingdom data | ARMD affected (n = 514) | Controls (n = 188) | |||
| Exon 5 | c.484C>T | p.Leu162Leu | 1 | 0 | |
| Exon 9 | c.945T>C | p.Ile315Ile | 206 | 76 | |
| Exon 10 | c.1122C>T | p.Tyr374Tyr | 19 | 10 | |
| Exon 10 | c.1134T>C | p.Tyr378Tyr | 2 | 0 | |
| Netherlands data | ARMD (n = 291) | Controls (n = 91) | |||
| Exon 7 | c.735C>T | p.Cys245Cys | 2 | 0 | |
| Exon 9 | c.945T>C | p.Ile315lle | 99 | 37 | |
| Exon 10 | c.1122C>T | p.Tyr374Tyr | 18 | 7 | |
| Intronic variants | |||||
| United Kingdom data | ARMD (n = 514) | Controls (n = 188) | |||
| Intron 3 | c.125-53G>A | 1 | 0 | ||
| Intron 3 | c.125-45G>C | 5 | 1 | ||
| Netherlands data | ARMD (n = 291) | Controls (n = 91) | |||
| 5′UTR | c.1-33C>A | 1 | 0 | ||
| Intron 1 | c.18-28A>G | 21 | 8 | ||
| 3′UTR | c.1347+97G>A | 0 | 1 | ||
| 3′UTR | c.1347+121C>T | 1 | 0 | ||
| 3′UTR | c.1347+352G>C | 15 | 28 | ||
| 3′UTR | c.1347+423T>C | 7 | 6 | ||
| 3′UTR | c.1347+514A>G | 18 | 38 | ||
Degree of conservation values are the number of nonhuman species that share this residue with humans. The species with the greatest phylogenic distance from humans with a sequence that is homologous to that of humans is also given. cDNA numbering is taken from Ref Seq # NM_006329.2 where +1 corresponds to the A of the ATG initiation codon.
Number of conserved variations/total number of variations.
We used the protein fold recognition program Threader 3 to predict the effects on protein folding of these missense sequence changes together with those previously published in ARMD patients [Stone et al., 2004], the missense changes identified in control individuals (p.V126 M, p.G202R) and two mutations previously described in cutis laxa (p.S227P, p.C217R) [Loeys et al., 2002; Fischer et al., 2004]. The structure 1l0qa0 (391 residues) was the best match both 1) when the WT fibulin 5 sequence (448 residues) was threaded using default parameter values against each of the 6251 structures in the reference library (Z = 6.8, statistically very significant); and 2) when the top 15 matches from (1) were subjected to randomization tests (using the option r50), which identify true-positive matches and eliminate false-positives. A prediction of the effects on folding of each mutation was obtained indirectly by comparing the results from threading the mutant against 1l0qa0 with those from threading the WT against the same structure. Table 2 presents the threading energies, the Z-scores (which indicate the quality of matches), and the alignments for WT and each mutation. Of note, both the cutis laxa mutation p.C217R and the ARMD-associated change p.Q124P reduce the magnitude of the pairwise (folding) energy sum by >2.5%, and reduce the corresponding Z-score. For p.Q124P, compensation for the change in folding energy (Table 2; column 2) by an opposing change in solvation energy (column 4) leads to virtually no change in the total energy (column 6) and its Z-score (column 7). For p.C217R, the changes in pairwise and solvation energies are synergistic, leading to a reduction of the magnitude of total energy by >2.5%. The two mutations p.G267S and p.R351W improve the stability on folding by >2.5%, and with reduced percentage alignments.
Table 2.
“Threading” of the Fibulin 5 Mutants Against the Structure 1l0qa0*
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | −1026 | 6.80 | −7.81 | 2.41 | −1214 | 6.14 | 157.3 | 359 | 92.3 | 80.4 | → | |
| V60La | −1021 | 6.77 | −7.82 | 2.42 | −1210 | 6.14 | 155.9 | 359 | 92.3 | 80.4 | IE | → |
| R71Qa | −1026 | 6.80 | −7.77 | 2.41 | −1213 | 6.13 | 156.9 | 359 | 92.3 | 80.4 | IC | → |
| P87Sa | −1031 | 6.84 | −7.81 | 2.44 | −1222 | 6.21 | 158.5 | 359 | 92.3 | 80.4 | IE | → |
| Q124Pa | −970 | 6.39 | −9.75 | 2.78 | −1207 | 6.10 | 174.8 | 359 | 92.3 | 80.4 | IE | ↓ ↓ |
| V126Ma | −1010 | 6.74 | −7.77 | 2.41 | −1197 | 6.12 | 160.0 | 359 | 92.3 | 80.4 | IE | → |
| I169Ta | −1018 | 6.77 | −8.46 | 2.54 | −1224 | 6.16 | 157.2 | 359 | 92.3 | 80.4 | IE | ↓ |
| G202Rb | −1026 | 6.84 | −8.20 | 2.51 | −1225 | 6.27 | 158.6 | 359 | 92.3 | 80.4 | IC | → |
| C217Rc | −986 | 6.62 | −6.76 | 2.23 | −1148 | 5.88 | 155.2 | 359 | 92.3 | 80.4 | IE | ↓ ↓ |
| S227Pc | −1026 | 6.87 | −7.77 | 2.40 | −1211 | 6.13 | 159.0 | 359 | 92.3 | 80.4 | IC | ↓ ↓ |
| G267Sa | −1036 | 6.93 | −10.03 | 2.85 | −1279 | 6.56 | 190.4 | 354 | 90.8 | 79.0 | IC | ↓ |
| R351Wa | −1026 | 6.76 | −9.55 | 2.75 | −1258 | 6.34 | 175.8 | 357 | 91.8 | 79.9 | IC | → |
| A363Ta | −1026 | 6.79 | −8.06 | 2.44 | −1221 | 6.15 | 158.4 | 359 | 92.3 | 80.4 | IC | → |
| G412Ea | −1026 | 6.83 | −7.81 | 2.42 | −1215 | 6.18 | 157.3 | 359 | 92.3 | 80.4 | O | ↓ |
Threading of the test fibulin 5 sequences (column 1) upon 1l0qa 0 is based on optimization of the pairwise energy sums and the solvation energy sums. The raw pairwise energy sum (kcal mol−1, column 2) is adjacent to its corresponding Z-score (column 3). The raw solvation energy sum (kcal mol−1, column 4) is adjacent to its Z-score (column 5). The sum of pairwise and solvation energies (column 6), weighted as described in the Threader documentation, is given with its Z-score (column 7). The high sequence-similarity scores (column 8) might indicate common ancestry between 1l0qa 0 and fibulin 5. In addition, the number of residues (column 9) and the exceptionally high percentages of 1l0qa 0 (column 10) and fibulin 5 (column 11) that were aligned are listed. Column 12 lists whether a mutation is inside (I) or outside (O) the alignment, and the 1l0qa 0 secondary structure (E, extended; C, coil) at that point. Column 13 summarizes the secretion data (Fig. 1). Deviations >2.5% from figures obtained for wild-type fibulin 5 are shown in bold. Changes in the alignments are shown in italics. Z-scores are quantitative measures of the quality of matches. Large and positive Z-scores point toward a successful prediction: Z>4.0, very significant, indicating probably a correct prediction; 4.0>Z>3.5, significant with a good chance of being correct;3.5>Z>2.7, borderline;2.7>Z>2.0, poor;2.0>Z, very poor.
Mutations associated with ARMD.
Missense change identified in a control patient without ARMD.
Mutations associated with cutis laxa.
Changes in protein folding might interfere with secretion of the protein and to test this we expressed each of the sequence variants and WT fibulin 5 in COS7 cells together with those previously published in ARMD patients [Stone et al., 2004], the missense changes identified in control individuals (p.V126 M, p.G202R) and two mutations previously described in cutis laxa (p.S227P, p.C217R) [Loeys et al., 2002; Fischer et al., 2004]. Western blot analysis following harvesting of the transfected cells indicated WT fibulin 5 was present both in whole cell lysates and in the medium from the cell culture (Fig. 1A), confirming that fibulin 5 is secreted by these cells. The ratio of fibulin 5 detected in the medium compared to that in the whole cell lysate for 6 of the 11 disease-associated mutations was significantly reduced (P<0.001) (Fig. 1B) indicating a reduction in secretion for these mutant forms of fibulin 5 but no change in secretion was noted for the two sequence variants identified in controls. Four of these mutations are associated with ARMD (p.G412E, p.G267S, p.I169 T, and p.Q124P) and two have been found to cause cutis laxa (p.S227P, p.C217R) (Fig. 1B).
Figure 1.
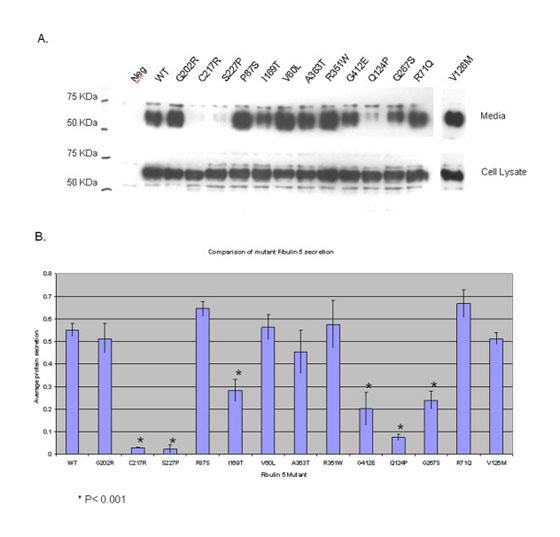
A: Secretion of WT and mutant forms of fibulin 5. WT and mutant fibulin 5 were expressed in transfected COS-7 cells. At 72 hr after transfection, cell lysate and medium samples were prepared and separated by 10% SDS–polyacrylamide gels followed by immunoblotting. These included WT, two mutations previously associated with cutis laxa (p.C217R and p.S227P), mutations previously associated with ARMD (p.R71Q, p.P87S, p.I169 T, p.V60L, p.A363 T, p.R351W, and p.G412E), novel mutations we identified in ARMD patients (p.Q124P, p.G267S), and two control missense mutations (p.V126 M, p.G202R). B: Quantification of secreted fibulin 5. For WT and each mutation the ratio of secreted fibulin 5 to fibulin 5 in the cell lysate was calculated.These were compared between WT and each mutations and results analyzed using Student's t-test. *P<0.001.
Genotype–Phenotype Correlation
Two of the three Southampton patients had a clinical phenotype of cuticular drusen. Patient 1 (p.Q124P) had small round uniform drusen but no choroidal neovascularization and no evidence of pigment epithelial detachments. Patient 2 (p.G267S) had pure choroidal neovascularization in one eye but no drusen. Patient 3 (p.G267S) had cuticular drusen and central localized pigment epithelial detachments. (Fig. 2).
Figure 2.
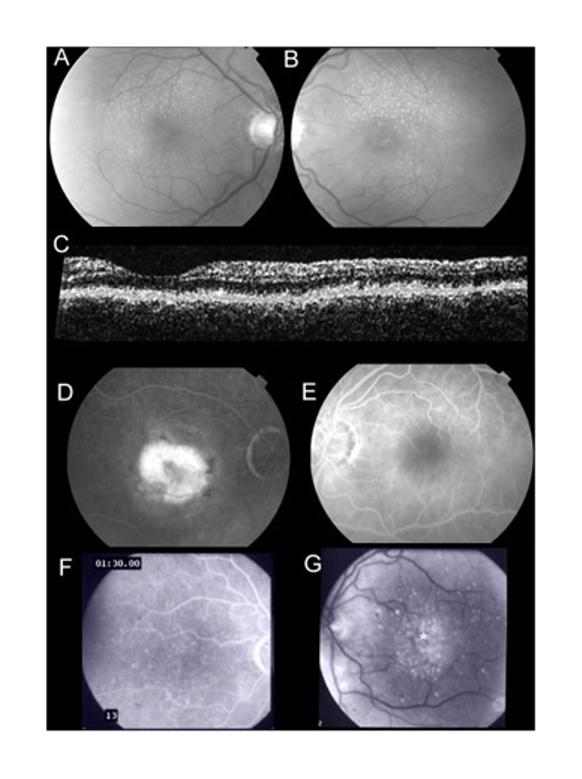
A,B: Retinal photographs of Patient 1 (p.Q124P). Small round (cuticular) drusen are symmetrically present. C:Optical coherence tomogram of Patient 1 demonstrates nodular thickening of the retinal pigment epithelium (*). D,E: Fluorescein angiograms of Patient 2 (p.G267S) demonstrate choroidal neovascularization in one eye only. F,G: Fluorescein angiogram photographs of Patient 3 (p.G267S) demonstrate cuticular drusen with central areas of focal retinal pigment epithelial detachment (*).
DISCUSSION
This study identified novel heterozygous fibulin 5 mutations in 3 out of 514 UK patients with ARMD, a prevalence of 0.6%. In the Netherlands cohort, however, we found no fibulin 5 sequence variants specific to patients. Previously, missense FBLN5 mutations were identified in 7 out of 402 U.S. patients with ARMD (1.7%) [Stone et al., 2004] and these mutations were proposed to result in an increased susceptibility to ARMD. The work we describe here is the first similar study in a different population of ARMD patients. The results from the UK cohort support the hypothesis that a number of rare FBLN5 variants are unique for ARMD patients, which suggests these mutations may affect fibulin 5 function. The results from the Netherlands cohort might reflect the lower number of patients studied or genetic founder effects specific for the Dutch population. It is well recognized that the first report [Stone et al., 2004] describing an association between a gene and a disease often overestimates the fraction of disease caused by mutation in the gene in question [Ioannidis et al., 2001] and these results would suggest this may be a factor here. Again this emphasizes the need for subsequent studies such as ours, both to assess the effect in further populations and also to further investigate the biological consequences of the genetic mutations.
The statistical association of sequence variants with a multifactorial condition such as ARMD is only the first step in determining any causative relationship. Fibulin 5 is a secreted extracellular matrix protein that has a role in elastinogenesis through interactions with integrins [Nakamura et al., 2002], tropoelastin [Tsuruga et al., 2004], and LOXL1 [Liu et al., 2004]. Missense mutations affecting protein folding might interfere with fibulin 5 moving through the secretory pathway or with subsequent protein interactions. Interference with protein trafficking and secretion is an increasingly recognized mechanism by which missense mutations cause genetic disease [Aridor and Hannan, 2000]. It is thought to occur when the missense mutation leads to abnormal protein folding, thus inducing the “unfolded protein response” in the endoplasmic reticulum, leading to intracellular degradation of the mutant protein; and it can occur with missense changes at different points in a protein [Aridor and Hannan, 2000].
We hypothesized that the missense mutations in fibulin 5 may lead to susceptibility to ARMD due to reduced fibulin 5 secretion; as an initial approach to investigating this we modeled the effects of mutations on protein folding using Threader 3. These results suggested some sequence variants might affect folding and thus potentially secretion of mutant protein. We therefore expressed WT and mutant forms of fibulin 5 and compared secretion. Six sequence changes did indeed interfere with secretion and four of these were predicted to interfere with folding. In contrast, other sequence changes were also predicted to change protein folding but did not change secretion. This may indicate protein folding changes that did not activate the unfolded protein response or, alternatively, reflect the limitations of computer modeling.
Mutations leading to autosomal recessive cutis laxa caused a dramatic reduction in secretion of fibulin 5 when compared with WT (P<0.001) and a similar reduction in secretion was observed for four of the nine mutations associated with ARMD (p.G412E, p.G267S,p.I169 T, and p.Q124P). In vitro expression studies such as these are often the first indication of missense mutations reducing protein secretion and thus causing a phenotype. Fibulin 5 is moderately expressed by the human RPE and not by the photoreceptors [Simone van Soest et al., personal communication] and therefore fibulin 5 through its role in elastinogenesis may contribute to the maintenance of the integrity of Bruch's membrane in the eye. Correspondingly, a reduction in secretion might interfere with this.
Several members of the fibulin gene family have been associated with macular degeneration. In previous studies by Stone et al. [2004] and other groups [Schultz et al., 2003], variations in other fibulins (fibulin-1, 2, 4, and 6) were also reported specifically associated with ARMD patients. In addition, a mutation in fibulin-3 causes a more severe form of macular degeneration (earlier onset), Malattia Leventinese or Doyne honeycomb retinal dystrophy with high similarities to ARMD [Stone et al., 1999]. Although no fibulin-3 mutation has been found in ARMD, the fibulin 3 protein accumulates in Bruch's membrane in ARMD [Marmorstein et al., 2002]. Secretion of mutant fibulin-3 is also significantly reduced [Marmorstein et al., 2002]. The FBLN5 gene is not highly polymorphic and the identification of new mutations in different ARMD cohorts but not in control populations (this study and the previous one by Stone et al., 2004) suggests that the missense sequence changes have a functional effect on fibulin 5 function and that there is a causal relationship between FBLN5 and the pathogenesis of ARMD rather then this being a chance association. This study further demonstrates that FBLN5 mutations can be associated with different phenotypes of ARMD (not limited to cuticular druse type).
Our results suggest that some missense mutations associated with ARMD lead to decreased fibulin 5 secretion. Since fibulin 5 is essential for elastinogenesis, our hypothesis is that this reduced secretion in turn results in a corresponding reduction in elastinogenesis. However, this is as yet unconfirmed for the ARMD related mutations and requires further investigation.
It is particularly noteworthy that one of the ARMD associated mutations (p.Q124P) leads to a similar reduction in secretion to the two mutations known to cause cutis laxa in which elastinogenesis is disrupted [Loeys et al., 2002; Fischer et al., 2004]. This raises the possibility that patients with this form of cutis laxa may have early onset ARMD, and their parents (heterozygous for these mutations) may themselves be at a higher risk of ARMD than the general population. Furthermore, these ARMD FBLN5 sequence variants may lead to cutis laxa if inherited as homozygous mutations and the ARMD patients in whom we have identified a sequence variant in fibulin 5 may be “carriers” for cutis laxa. We have found reduced secretion of FBLN5 for both cutis laxa mutations and for some ARMD mutations. This suggests there may be a relationship between dose of fibulin 5 and disease severity. Homozygous mutations may interfere with elastinogenesis and result in the early onset cutis laxa phenotype, whereas heterozygous mutations may result in a later onset phenotype of ARMD. Environmental stresses such as smoking may also influence the development of the ARMD late onset phenotype. Further studies investigating the effects of these variants and gene–environment interactions will be important. Not only to clarify how they may affect fibulin 5 function, but also to determine whether we should give lifestyle advice for the parents of patients with cutis laxa and consider additional ophthalmologic supervision and possible early treatment for these individuals with vitamin supplements [AREDS Research Group, 2001].
Previously, FBLN 5 associated ARMD has been associated with a specific retinal phenotype of cuticular drusen [Stone et al., 2004]. We have observed this same phenotype in two of our three patients with novel FBLN 5 mutations. However, interestingly, two patients with the same p.G267S sequence change demonstrated markedly different phenotypes. Such variation in retinal phenotype has been well described before in monogenetic retinal diseases [Weleber et al., 1993]. FBLN5 ARMD can therefore also be a cause of choroidal neovascularization in the absence of drusen.
This study identifies a mechanism by which FBLN5 mutations can lead to ARMD. Such knowledge should ultimately lead to the development of novel therapies for this devastating disease. It also expands the retinal phenotypes associated with FBLN5 mutations and identifies the prevalence of ARMD in two European cohorts. Such information will be valuable for future gene directed therapies.
ACKNOWLEDGMENTS
Research nurse support was provided by the Southampton Wellcome Trust Clinical Research Facility. Kay Kielty and Adam Huffman are acknowledged for providing access to, and assistance with, computing facilities in the Wellcome Trust Centre for Cell-Matrix Research, The University of Manchester. Angela Cree, University of Southampton, is acknowledged for technical support.
Footnotes
Grant sponsor: Wellcome Trust; T.F.C Frost and Delaslo Charitable Trusts; Grant sponsor: British Council Prevention Blindness; Grant sponsor: HobartTrust.
REFERENCES
- Ambati J, Ambati BK, Yoo SH, Ianchulev S, Adamis AP. Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol. 2003;48:257–293. doi: 10.1016/s0039-6257(03)00030-4. [DOI] [PubMed] [Google Scholar]
- AREDS Research Group A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age- related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119:1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor M, Hannan LA. Traffic jam: a compendium of human diseases that affect intracellular transport processes. Traffic. 2000;1:836–851. doi: 10.1034/j.1600-0854.2000.011104.x. [DOI] [PubMed] [Google Scholar]
- Bird AC, Bressler NM, Bressler SB, Chisholm IH, Coscas G, Davis MD, de Jong PT, Klaver CC, Klein BE, Klein R, Mitchell P, Sarks JP, Sarks SH, Soubrane G, Taylor H, Vingerling JR. An international classification and grading system for age-related maculopathy and age-related macular degeneration. The International ARM Epidemiological Study Group. Surv Ophthalmol. 1995;39:367–374. doi: 10.1016/s0039-6257(05)80092-x. [DOI] [PubMed] [Google Scholar]
- Christen WG, Glynn RJ, Manson JE, Ajani UA, Buring JE. A prospective study of cigarette smoking and risk of age-related macular degeneration in men. JAMA. 1996;276:1147–1151. [PubMed] [Google Scholar]
- Edwards AO, Ritter R, III, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- Fischer J, Jobard F, Oudot T, Geneviève D, Mégarbané A, Pauchard J, Saker S, Godeau G, Damour O, Peyrol S, Blanchet-Bardon C, de Prost Y, Hadj-Rabia S, Devillers M, Sommer P. Identification of novel mutations in elastin and fibulin 5 in three patients affected with cutis laxa. Third European Symposium Elastin 2004; Manchester, UK. 30th June–3rd July 2004.2004. [Google Scholar]
- Goverdhan SV, Howell MW, Mullins RF, Osmond C, Hodgkins PR, Self J, Avery K, Lotery AJ. Association of HLA class I and class II polymorphisms with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2005;46:1726–1734. doi: 10.1167/iovs.04-0928. [DOI] [PubMed] [Google Scholar]
- Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJH, Silvestri G, Russell SR, Klaver CCW, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R. From the cover: a common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, SchnetzBoutaud N, Agarwal A, Postel EA, Pericak-Vance MA. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. Replication validity of genetic association studies. Nat Genet. 2001;29:306–309. doi: 10.1038/ng749. [DOI] [PubMed] [Google Scholar]
- Jones DT, Tress M, Bryson K, Hadley C. Successful recognition of protein folds using threading methods biased by sequence similarity and predicted secondary structure. Proteins Suppl. 1999;3:104–111. doi: 10.1002/(sici)1097-0134(1999)37:3+<104::aid-prot14>3.3.co;2-g. [DOI] [PubMed] [Google Scholar]
- Klaver CC, Wolfs RC, Vingerling JR, Hofman A, de Jong PT. Age-specific prevalence and causes of blindness and visual impairment in an older population: the Rotterdam Study. Arch Ophthalmol. 1998;116:653–658. doi: 10.1001/archopht.116.5.653. [DOI] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, Sangiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhao Y, Gao J, Pawlyk B, Starcher B, Spencer JA, Yanagisawa H, Zuo J, Li T. Elastic fiber homeostasis requires lysyl oxidase-like 1 protein. Nat Genet. 2004;36:178–182. doi: 10.1038/ng1297. [DOI] [PubMed] [Google Scholar]
- Loeys B, Van Maldergem L, Mortier G, Coucke P, Gerniers S, Naeyaert JM, De Paepe A. Homozygosity for a missense mutation in fibulin-5 (FBLN5) results in a severe form of cutis laxa. Hum Mol Genet. 2002;11:2113–2118. doi: 10.1093/hmg/11.18.2113. [DOI] [PubMed] [Google Scholar]
- Lotery AJ, Munier FL, Fishman GA, Weleber RG, Jacobson SG, Affatigato LM, Nichols BE, Schorderet DF, Sheffield VC, Stone EM. Allelic variation in the VMD2 gene in best disease and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2000a;41:1291–1296. [PubMed] [Google Scholar]
- Lotery AJ, Namperumalsamy P, Jacobson SG, Weleber RG, Fishman GA, Musarella MA, Hoyt CS, Heon E, Levin A, Jan J, Lam B, Carr RE, Franklin A, Radha S, Andorf JL, Sheffield VC, Stone EM. Mutation analysis of 3 genes in patients with Leber congenital amaurosis. Arch Ophthalmol. 2000b;118:538–543. doi: 10.1001/archopht.118.4.538. [DOI] [PubMed] [Google Scholar]
- Lotery AJ, Malik A, Shami SA, Sindhi M, Chohan B, Maqbool C, Moore PA, Denton MJ, Stone EM. CRB1 mutations may result in retinitis pigmentosa without para-arteriolar RPE preservation. Ophthalmic Genet. 2001;22:163–169. doi: 10.1076/opge.22.3.163.2222. [DOI] [PubMed] [Google Scholar]
- Markova D, Zou Y, Ringpfeil F, Sasaki T, Kostka G, Timpl R, Uitto J, Chu ML. Genetic heterogeneity of cutis laxa: a heterozygous tandem duplication within the fibulin-5 (FBLN5) gene. Am J Hum Genet. 2003;72:998–1004. doi: 10.1086/373940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein LY, Munier FL, Arsenijevic Y, Schorderet DF, McLaughlin PJ, Chung D, Traboulsi E, Marmorstein AD. Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99:13067–13072. doi: 10.1073/pnas.202491599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Ruiz-Lozano P, Lindner V, Yabe D, Taniwaki M, Furukawa Y, Kobuke K, Tashiro K, Lu Z, Andon NL, Schaub R, Matsumori A, Sasayama S, Chien KR, Honjo T. DANCE, a novel secreted RGD protein expressed in developing, atherosclerotic, and balloon-injured arteries. J Biol Chem. 1999;274:22476–22483. doi: 10.1074/jbc.274.32.22476. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Lozano PR, Ikeda Y, Iwanaga Y, Hinek A, Minamisawa S, Cheng CF, Kobuke K, Dalton N, Takada Y, Tashiro K, Ross J, Jr, Honjo T, Chien KR. Fibulin-5/DANCE is essential for elastogenesis in vivo. Nature. 2002;415:171–175. doi: 10.1038/415171a. [DOI] [PubMed] [Google Scholar]
- Schultz DW, Klein ML, Humpert AJ, Luzier CW, Persun V, Schain M, Mahan A, Runckel C, Cassera M, Vittal V, Doyle TM, Martin TM, Weleber RG, Francis PJ, Acott TS. Analysis of the ARMD1 locus: evidence that a mutation in HEMICENTIN-1 is associated with age-related macular degeneration in a large family. Human Molecular Genetics. 2003;12:3315–3323. doi: 10.1093/hmg/ddg348. [DOI] [PubMed] [Google Scholar]
- Seddon JM, Willett WC, Speizer FE, Hankinson SE. A prospective study of cigarette smoking and age-related macular degeneration in women. JAMA. 1996;276:1141–1146. [PubMed] [Google Scholar]
- Stone EM, Lotery AJ, Munier FL, Heon E, Piguet B, Guymer RH, Vandenburgh K, Cousin P, Nishimura D, Swiderski RE, Silvestri G, Mackey DA, Hageman GS, Bird AC, Sheffield VC, Schorderet DF. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet. 1999;22:199–202. doi: 10.1038/9722. [DOI] [PubMed] [Google Scholar]
- Stone EM, Braun TA, Russell SR, Kuehn MH, Lotery AJ, Moore PA, Eastman CG, Casavant TL, Sheffield VC. Missense variations in the fibulin 5 gene and age-related macular degeneration. N Engl J Med. 2004;351:346–353. doi: 10.1056/NEJMoa040833. [DOI] [PubMed] [Google Scholar]
- Tielsch JM, Javitt JC, Coleman A, Katz J, Sommer A. The prevalence of blindness and visual impairment among nursing home residents in Baltimore. N Engl J Med. 1995;332:1205–1209. doi: 10.1056/NEJM199505043321806. [DOI] [PubMed] [Google Scholar]
- Timpl R, Sasaki T, Kostka G, Chu ML. Fibulins: a versatile family of extracellular matrix proteins. Nat Rev Mol Cell Biol. 2003;4:479–489. doi: 10.1038/nrm1130. [DOI] [PubMed] [Google Scholar]
- Tsuruga E, Yajima T, Irie K. Induction of fibulin-5 gene is regulated by tropoelastin gene, and correlated with tropoelastin accumulation in vitro. Int J Biochem Cell Biol. 2004;36:395–400. doi: 10.1016/s1357-2725(03)00238-3. [DOI] [PubMed] [Google Scholar]
- Wang T, Waters CT, Rothman AM, Jakins TJ, Romisch K, Trump D. Intracellular retention of mutant retinoschisin is the pathological mechanism underlying X-linked retinoschisis. Hum Mol Genet. 2002;11:3097–3105. doi: 10.1093/hmg/11.24.3097. [DOI] [PubMed] [Google Scholar]
- Weleber R, Carr R, Murphey W, Sheffield V, Stone E. Phenotypic variation including retinitis pigmentosa, pattern dystrophy, and fundus flavimaculatus in a single family with a deletion of the codon 153 or 154 of the peripherin/RDS gene. Arch Ophthalmol. 1993;111:1531–1542. doi: 10.1001/archopht.1993.01090110097033. [DOI] [PubMed] [Google Scholar]
- Yanagisawa H, Davis EC, Starcher BC, Ouchi T, Yanagisawa M, Richardson JA, Olson EN. Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature. 2002;415:168–171. doi: 10.1038/415168a. [DOI] [PubMed] [Google Scholar]
- Zareparsi S, Buraczynska M, Branham KE, Shah S, Eng D, Li M, Pawar H, Yashar BM, Moroi SE, Lichter PR, Petty HR, Richards JE, Abecasis GR, Elner VM, Swaroop A. Toll-like receptor 4 variant D299G is associated with susceptibility to age-related macular degeneration. Hum Mol Genet. 2005;14:1449–1455. doi: 10.1093/hmg/ddi154. [DOI] [PubMed] [Google Scholar]