

Abstract
Activation of sonic hedgehog (Shh) signaling occurs in the majority of pancreatic ductal adenocarcinomas. Here we investigate the mechanisms by which Shh contributes to pancreatic tumorigenesis. We find that Shh expression enhances proliferation of pancreatic duct epithelial cells, potentially through the transcriptional regulation of the cell cycle regulators cyclin D1 and p21. We further show that Shh protects pancreatic duct epithelial cells from apoptosis through the activation of phosphatidylinositol 3-kinase signaling and the stabilization of Bcl-2 and Bcl-XL. Significantly, Shh also cooperates with activated K-Ras to promote pancreatic tumor development. Finally, Shh signaling enhances K-Ras-induced pancreatic tumorigenesis by reducing the dependence of tumor cells on the sustained activation of the MAPK and phosphatidylinositol 3-kinase/Akt/mTOR signaling pathways. Thus, our data suggest that Shh signaling contributes to tumor initiation in the pancreas through at least two mechanisms and additionally enhances tumor cell resistance to therapeutic intervention. Collectively, our findings demonstrate crucial roles for Shh signaling in multiple stages of pancreatic carcinogenesis.
Keywords: K-ras, mouse model, pancreatic cancer, Shh
Pancreatic cancer is the fourth leading cause of cancer-related deaths in the United States, with ≈30,000 new cases annually (1, 2). Pancreatic ductal adenocarcinoma (PDAC) arises from precursor lesions called pancreatic intraepithelial neoplasia (PanIN), which are believed to be induced by the transformation of the epithelial cells of the small intercalated and intralobular ducts (3). PanIN lesions are characterized by specific histologic changes to the duct epithelium that correlate with the accumulation of alterations in the KRAS2 oncogene and loss of the INK4A (PanIN 1 and 2), TP53 (PanIN 2–3), and MADH4 (PanIN 3) tumor suppressor genes (3).
Hedgehog ligands bind to their receptors and activate a signaling cascade that culminates in the nuclear translocation of Gli transcription factors and the regulation of target genes (4–6). Interestingly, sonic hedgehog (Shh) is excluded from the developing pancreas, as well as the mature organ (7), yet is activated in early PanIN lesions, with increasing levels as lesions progress to invasive PDAC (8, 9). Further, ectopic expression of Shh under the control of the Pdx-1 promoter, active in pancreas progenitor cells, induces intestinal metaplasia in the pancreas accompanied by mutations in Kras (8). Significantly, inhibition of hedgehog signaling blocks proliferation and induces apoptosis, in culture and s.c. xenografts, in a subset of these cell lines (8, 9). Thus, the evidence suggests that activated Shh signaling is a critical early mediator of pancreatic cancer development.
Yet little is understood about the mechanisms by which Shh signaling contributes to this disease. We report here the consequences of Shh expression on the proliferation and survival of pancreatic duct epithelial cells (PDECs). We demonstrate that Shh expression enhances tumor initiation and growth and reduces tumor cell death after therapeutic intervention. These findings reveal critical roles for Shh signaling in pancreatic tumor initiation, growth, and survival, and greatly enhance our understanding of the role of Shh in pancreatic carcinoma.
Results
Targeting the Pancreatic Duct Epithelium.
To target the pancreatic duct epithelium, we generated transgenic mice in which expression of the receptor for avian leukosis virus subgroup A (ALV-A), TVA [encoded by the quail ALV-A susceptibility locus tumor virus A (tv-a)], is regulated by the keratin 19 promoter and enhancer elements (K19-tv-a mice) (10–13). Immunofluorescent staining of pancreas tissue sections demonstrated the presence of TVA specifically within the duct epithelium (B.C.L. and H. Varmus, unpublished data). We found that PDECs isolated from transgenic (but not nontransgenic) animals are susceptible to infection by ALV-A-based RCAS viruses (Fig. 1A, data not shown).
Fig. 1.

Shh expression and signaling in PDECs. (A) RCAS-GFP-infected PDECs in culture. (Left) Phase contrast image. (Right) Fluorescent image. (B) Formation of duct-like structures by GFP-expressing Trp53, Ink4a/Arf null PDECs embedded in matrigel. (C) Detection of Shh expression in RCAS-Shh- and RCAS-GFP-infected PDECs by immunoblotting with an anti-myc monoclonal antibody. Tumor suppressor genotypes are indicated. β-actin serves as a control. (D) RT-PCR analysis of Shh pathway components in RCAS-Shh- and RCAS-GFP-infected PDECs. β-actin serves as a control. w.t., wild type.
Our experience with the elastase-tv-a mice suggested that additional genetic alterations in tumor suppressor genes were required for tumor progression (14). We therefore crossed the K19-tv-a mice to animals bearing conditional mutant (floxed) alleles at the Trp53 and Ink4a/Arf tumor suppressor loci, as well as to mice expressing the Cre recombinase from the Ptf1a locus, which activates expression in pancreas progenitor cells (15–17). These crosses resulted in K19-tv-a, Trp53 flox, Ptf1a-Cre; K19-tv-a, Ink4a/Arf flox, Ptf1a-Cre; and K19-tv-a, Trp53 flox, Ink4a/Arf flox, Ptf1a-Cre animals. PDECs isolated from Cre-positive animals with floxed tumor suppressor alleles showed complete recombination at the tumor suppressor loci as assayed by PCR (data not shown). PDECs also formed duct-like structures when embedded in matrigel (18), a feature that is unaffected by tumor suppressor gene deletion (Fig. 1B).
Shh Enhances Proliferation of PDECs.
To identify the effect of active Shh signaling on PDECs, we infected cultured TVA-positive PDECs with RCAS viruses encoding a myc epitope-tagged Shh molecule or GFP as a control (19). Expression of the introduced Shh was confirmed by immunoblotting protein lysates with an anti-c-myc antibody (Fig. 1C). RT-PCR analysis of the hedgehog pathway components Gli1 and ptch1, previously described hedgehog targets (20), demonstrated activation of hedgehog signaling (Fig. 1D). Interestingly, Smo mRNA levels were also elevated in Shh-expressing cells (Fig. 1D).
We next determined whether active Shh signaling stimulates the proliferation of PDECs. Shh- and GFP-expressing PDECs were plated at equal numbers, and cells were harvested and counted after 5, 10, and 15 days. We observed that Shh stimulates the proliferation of PDECs relative to their GFP-expressing counterparts, and that this effect is independent of tumor suppressor gene status (Fig. 2A).
Fig. 2.

Shh enhances the proliferation of PDECs. (A) Cell numbers for Shh- or GFP-expressing PDECs of the indicated tumor suppressor genotypes at the indicated times after plating. Dotted lines, Shh-expressing PDECs; solid lines, GFP-expressing PDECs. Results are representative of at least two experiments. (B) RT-PCR analysis of cell cycle-related transcripts in RCAS-Shh- and RCAS-GFP-infected PDECs. β-actin serves as a control. (C) Immunoblot analysis of cell cycle- and Ras-regulated signaling molecules in RCAS-Shh- and RCAS-GFP-infected PDECs. β-actin serves as a control. w.t., wild type.
Consistent with published data, RT-PCR analysis demonstrated a modest increase in cyclin D1 mRNA levels and a modest reduction in p21 mRNA levels in Shh-expressing cells relative to GFP-infected controls, although the effect on p21 was diminished in cells lacking p53 (Fig. 2B) (21, 22). This change is in contrast to mRNA levels of cyclin A, cyclin E, and p27, which are unchanged by Shh expression (Fig. 2B). Immunoblot experiments confirmed modestly elevated levels of cyclin D1 and modestly decreased levels of p21 protein in cells with active Shh signaling, although the effect on p21 protein was blunted in p53-deficient cells (Fig. 2C).
Activation of the MAPK and phosphatidylinositol 3-kinase (PI3-kinase) signaling pathways is commonly found in PDAC (23). Immunoblot analysis of lysates from serum-starved PDECs showed increased phosphorylation of Akt, indicative of pathway activation, in all Shh-expressing cells relative to GFP-expressing cells, and increased phosphorylation of Erk1/2, with a more pronounced effect in cells deficient at one or more tumor suppressor loci (Fig. 2C). Interestingly, expression of an activated Gli1 molecule (24) in PDECs did not stimulate these pathways (data not shown).
Flow-cytometry analysis showed that 73.4% of GFP-expressing PDECs are in G1 with 18.9% in S-phase, whereas Shh expression increases the S-phase fraction to 27.1% and reduces the G1 fraction to 59.1%. Inhibition of PI3-kinase with LY294002 or MAPK signaling with PD98059 reduces the S-phase fraction to 21.6% and 17.6%, respectively, and increases the G1 fraction to 68.7% and 68.2%, respectively, suggesting that the activation of these pathways is required for Shh-induced proliferation. Collectively, these data indicate that Shh may stimulate proliferation of PDECs through the regulation of multiple molecules.
Shh Enhances Survival of PDECs.
To determine whether Shh expression in PDECs confers resistance to apoptotic stimuli, we exposed PDECs to the cytotoxic agent cycloheximide (CHX) (25, 26), recently shown to induce apoptosis in pancreatic cancer cell lines (27). We found that Shh expression enhances survival after 24 or 48 h of exposure to 100 μM CHX independent of tumor suppressor status, as determined by trypan blue exclusion (Fig. 3A). We also found that Shh expression protects against death induced by UV irradiation (data not shown). Elevated levels of the cleaved, or active, form of caspase 3 were detected in GFP-expressing but not Shh-expressing PDECs, confirming the CHX-induced death as apoptosis (Fig. 3B). The CHX-induced apoptosis depends on Fas, caspase 8, and caspase 3 because inhibition of these molecules blocks cell death after CHX exposure (data not shown). Thus, Shh protects PDECs from Fas-dependent apoptosis, consistent with findings in other cell types (28).
Fig. 3.

Shh promotes the survival of PDECs. (A) Viability of PDECs of the indicated tumor suppressor genotypes after treatment with a 100 μM concentration of CHX or vehicle as measured by trypan blue exclusion. Shaded bars, Shh-expressing PDECs; open bars, GFP-expressing PDECs. Results are representative of at least two experiments. (B) Immunoblot analysis of cleaved and uncleaved caspase 3 in protein lysates from RCAS-GFP- and RCAS-Shh-infected Ink4a/Arf, Trp53 null PDECs before and after a 24-hour treatment with 100 μM CHX. β-actin serves as a control. (C) Immunoblot analysis of Bcl-2 family members in protein lysates from RCAS-GFP- and RCAS-Shh-infected Ink4a/Arf, Trp53 PDECs treated with a 100 μM concentration of CHX or vehicle. β-actin serves as a control. (D) RT-PCR demonstrating Bcl-XL mRNA levels in untreated and CHX-treated Ink4a/Arf, Trp53 null PDECs. β-actin serves as a control. (E) Viability of Ink4a/Arf, Trp53 null PDECs after treatment with CHX and Bcl-2, MAPK, and PI3-kinase inhibitors. Shaded bars, Shh-expressing PDECs; open bars, GFP-expressing PDECs. Results are representative of two experiments. ∗, P < 0.01 for CHX + inhibitors compared with CHX alone.
The Bcl-2 family proteins are important regulators of apoptosis that control the integrity of the mitochondrial membrane and the release of cytochrome c (29). We found elevated levels of the antiapoptotic proteins Bcl-2 and Bcl-XL in Shh-expressing cells after CHX treatment, relative to GFP controls, whereas levels of the proapoptotic protein Bad were similar in both cell types (Fig. 3C). Interestingly, RT-PCR analysis of Bcl-2 and Bcl-XL mRNA levels suggested that this increase occurred posttranscriptionally (Fig. 3D). Interrogation with small molecules showed that Shh-induced protection from apoptosis was dependent on Bcl-2 and Bcl-XL and required active PI3-kinase signaling, but not active MAPK signaling (Fig. 3E). Importantly, Bcl-2 and Bcl-XL levels are reduced when PI3-kinase signaling is inhibited, suggesting that this signaling pathway is required for the stabilization of these proteins (Fig. 3C).
Shh Cooperates with K-Ras in Pancreatic Tumorigenesis.
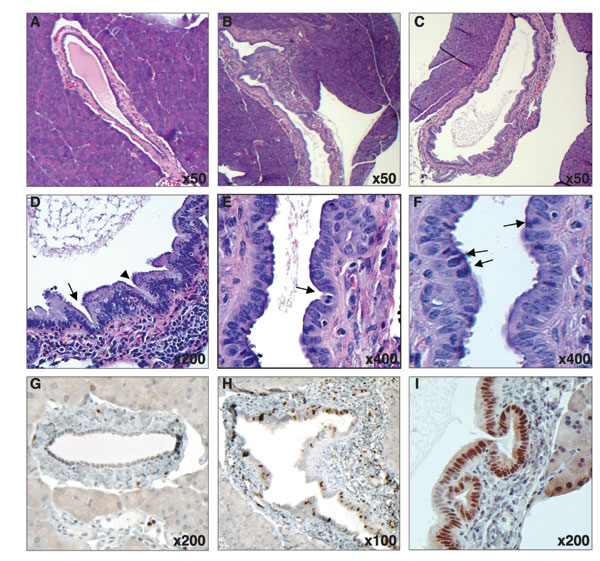
To determine whether Shh expression within the pancreas results in tumor formation, we used Shh-expressing PDECs in an orthotopic transplant model; 2 × 106 wild-type, Ink4a/Arf null, Trp53 null, and Ink4a/Arf, Trp53 null PDECs expressing either Shh or GFP were implanted into the pancreata of eight age-matched nude mice, and animals were monitored for 4 months. No grossly visible tumors developed; however, serial sectioning of harvested pancreata identified lesions with the features of PDAC precursor lesions in four of seven animals implanted with Trp53, Ink4a/Arf null Shh-expressing cells and two of three animals implanted with Trp53 null Shh-expressing cells [see supporting information (SI) Fig. 6]. All lesions displayed enhanced proliferation as denoted by Ki67 staining, and all lesions stained positive for Pdx1 (SI Fig. 6). No animals transplanted with Ink4a/Arf null or wild-type cells developed pancreatic lesions. These findings suggest that activation of the hedgehog pathway might initiate pancreatic tumorigenesis.
Activating KRAS2 mutations are present in ≈90% of all ductal adenocarcinomas. We found that Ink4a/Arf, Trp53 null PDECs infected with RCAS viruses encoding a FLAG epitope-tagged activated K-RasG12D molecule (hereafter simply denoted as K-Ras) proliferate faster than GFP controls (Fig. 4A). Significantly, when PDECs from Ink4a/Arf, Trp53 null pancreata are infected with both Shh- and K-Ras-bearing RCAS viruses, proliferation is enhanced when compared with PDECs expressing either Shh or K-Ras alone (Fig. 4A). We also found that activated Ras signaling protects PDECs from death when challenged with apoptotic stimuli (data not shown).
Fig. 4.

Cooperative interaction between Shh and activated K-Ras. (A) Proliferation curves for Ink4a/Arf, Trp53 null PDECs expressing GFP, Shh, K-Ras, or both Shh and K-Ras. (B) Cystic ductal lesion induced after orthotopic transplantation of K-Ras- and Shh-expressing Trp53 null PDECs. Note abundant reactive stroma. (C) H&E-stained tissue section illustrating the histology of tumors induced after orthotopic transplantation of Ink4a/Arf, Trp53 null PDECs expressing both Shh and activated K-Ras. Tumors are mostly undifferentiated (D), with regions of ductal differentiation (box in C, and E). (F) RT-PCR analysis of Shh pathway components in tumors induced after orthotopic transplantation of RCAS-k-ras-infected PDECs (R1–R6) or RCAS-Shh- and RCAS-k-ras-infected PDECs (RS1–RS7). Tumors R1–R3 and RS1–RS3 lack Ink4a/Arf. Tumors R4–R6 and RS4–RS7 lack Trp53 and Ink4a/Arf. β-actin serves as a control.
To identify whether K-Ras and Shh cooperate in pancreatic tumorigenesis, we infected wild-type, Ink4a/Arf null, Trp53 null, or Ink4a/Arf, Trp53 null PDECs with RCAS-Shh and/or RCAS-k-ras, implanted infected cells into the pancreata of four age-matched nude mice, and monitored animals for ≤2 months. RCAS-GFP-infected cells were used as a negative control.
Transplantation of tumor suppressor wild-type PDECs did not result in tumor formation. Cystic ductal tumors were identified in 11 of 15 mice transplanted with 0.5, 1, or 2 × 106 Trp53 null cells expressing Shh and K-Ras (Table 1 and Fig. 4B). By contrast, only 5 of 16 animals transplanted with Trp53 null cells expressing K-Ras had any pancreatic lesions; by histology, three of these lesions resembled precursor PanIN lesions (data not shown). Further, no microscopic lesions were identified in non-tumor-bearing pancreata, indicating that Shh enhances tumor initiation in Trp53 null cells. In addition, the tumors induced by the combination of Shh and K-Ras were of a significantly larger size than those induced by K-Ras alone, indicating an additional role for Shh in tumor growth (Table 1).
Table 1.
Tumor induction by RCAS-k-ras and RCAS-Shh plus RCAS-k-ras-infected PDECs after orthotopic transplant
| Genotype of cells implanted | No. of cells (million) | Tumor incidence | Average tumor volume‡ (mm3) |
|---|---|---|---|
| Trp53, Ink4a/Arf null Shh | 2 | 4/7§¶ | n/a |
| Trp53, Ink4a/Arf null GFP | 2 | 0/8§ | n/a |
| Ink4a/Arf null Shh | 2 | 0/7§ | n/a |
| Ink4a/Arf null GFP | 2 | 0/8§ | n/a |
| Trp53 null Shh | 2 | 2/3§¶ | n/a |
| Trp53 null GFP | 2 | 1/4§¶ | n/a |
| Trp53, Ink4a/Arf null Ras | 0.5 | 4/4 | 207.63 mm3 |
| Trp53, Ink4a/Arf null Ras | 1 | 4/4 | 397.25 mm3 |
| Trp53, Ink4a/Arf null Ras | 2 | 3/4 | 943.33 mm3 |
| Trp53, Ink4a/Arf null Shh/Ras | 0.5 | 4/4 | 713.75 mm3 |
| Trp53, Ink4a/Arf null Shh/Ras | 1 | 4/4 | 1087.63 mm3* |
| Trp53, Ink4a/Arf null Shh/Ras | 2 | 4/4 | 884.23 mm3 |
| Trp53, Ink4a/Arf null GFP | 2 | 0/8 | n/a |
| Ink4a/Arf null Ras | 0.5 | 2/4 | 14.5 mm3 |
| Ink4a/Arf null Ras | 1 | 2/4 | 29.75 mm3 |
| Ink4a/Arf null Ras | 2 | 4/4 | 80.63 mm3 |
| Ink4a/Arf null Shh/Ras | 0.5 | 3/4 | 36.4 mm3 |
| Ink4a/Arf null Shh/Ras | 1 | 4/4 | 48 mm3* |
| Ink4a/Arf null Shh/Ras | 2 | 4/4 | 359.56 mm3 |
| Ink4a/Arf null GFP | 2 | 0/7 | n/a |
| Trp53 null Ras | 0.5 | 2/4‖ | 2.03 mm3 |
| Trp53 null Ras | 1 | 1/4‖ | 1.0 mm3 |
| Trp53 null Ras | 2 | 2/8‖ | 2.38 mm3 |
| Trp53 null Shh/Ras | 0.5 | 2/3‖ | 4.0 mm3 |
| Trp53 null Shh/Ras | 1 | 3/4*‖ | 12.08 mm3† |
| Trp53 null Shh/Ras | 2 | 6/8‖ | 16.12 mm3 |
| Trp53 null GFP | 2 | 0/8 | n/a |
Experiment conducted for 60 days except where noted. ∗, P < 0.05. †, P < 0.01, by Student's t test for entire Shh/Ras cohort compared with Ras cohort.
‡Tumor volume calculated using the formula (W2 × L)/2, where W = shortest dimension and L = longest dimension.
§Euthanized at 4 months.
¶Microscopic lesions.
‖Cystic lesions.
Transplantation of Ink4a/Arf and Trp53, Inka/Arf double-null cells expressing K-Ras, or both K-Ras and Shh, led to the efficient formation of tumors (Table 1). However, the tumors induced by the combination of Shh and K-Ras were significantly larger than those induced by K-Ras alone, again indicating a role for Shh in pancreatic tumor growth (Table 1).
Histologically, the tumors induced by K-Ras, or Shh and K-Ras, in the Ink4a/Arf or Ink4a/Arf, Trp53 null backgrounds were undifferentiated carcinomas. The majority of the tumor mass had a sarcomatoid appearance, with regions of glandular differentiation (Fig. 4 C–E). The expression of Shh did not impact the differentiation status of the tumor. Immunoblotting of protein lysates from tumors induced by K-Ras, or K-Ras and Shh, identified the presence of the inducing oncoproteins in all tumors (SI Fig. 7A). Further, RT-PCR for the hedgehog pathway components ptch, smo, and Gli1 demonstrated elevated expression of each of these components in Shh-expressing tumors relative to those induced by K-Ras alone (Fig. 4F). Thus, collectively, our findings suggest that Shh cooperates with activated K-Ras in the development and growth of pancreatic tumors.
To further characterize these tumor cells, we established cell lines from four tumors expressing K-Ras only and lacking both Trp53 and Ink4a/Arf and from seven tumors expressing Shh and K-Ras and lacking Ink4a/Arf or Trp53 and Ink4a/Arf. Immunocytochemistry revealed that all cell lines were positive for K-Ras and Shh where expected and contained cells positive for keratin 8 and Pdx1, confirming their origin as PDECs (SI Fig. 7B).
We next analyzed the response of the tumor cell lines to the inhibition of downstream Ras signaling pathways by treating them with either 25 μM PD98059 to inhibit the MAPK pathway or 20 μM LY294002 to inhibit PI3-kinase signaling. Increased cell death did not occur in any cell line after treatment, as measured by trypan blue exclusion (data not shown). Pathway inhibition at 72 h was equivalent in all cell lines as shown by immunoblotting for phosphorylated Erk or phosphorylated Akt (Fig. 5 A and B). We observed that, although cell lines expressing K-Ras alone failed to proliferate in the presence of these inhibitors, cells expressing both Shh and K-Ras continued to proliferate in the presence of these compounds (Fig. 5 A and B). Similar findings were observed after treatment with rapamycin, which inhibits mTOR, and simultaneous treatment with rapamycin and PD98059 failed to completely inhibit the growth of cells expressing K-Ras and Shh, although there was an additive effect on cell proliferation (data not shown). Thus, these data indicate that Shh signaling in pancreatic cancer cells can reduce the requirement for sustained Ras signaling.
Fig. 5.

Shh and K-Ras in therapeutic intervention. Proliferation curves of untreated cell lines (dashed lines) or cells treated with specific pathway inhibitors (solid lines) after treatment with the MAPK inhibitor PD98059 (PD) (A), the PI3-kinase inhibitor LY294002 (LY) (B), the smoothened antagonist cyclopamine (CYC) (C), or LY294002 and cyclopamine (D). Immunoblotting of lysates from treated cells for phospho-ERK and phospho-Akt, and RT-PCR for Gli1, confirms comparable pathway inhibition in all cell lines.
To ascertain whether sustained Shh signaling is required for the proliferation of tumor cells, we treated the cell lines with the smoothened antagonist cyclopamine. Inhibition of the pathway was confirmed by RT-PCR for Gli1 (Fig. 5C). We found that cell lines expressing both Shh and K-Ras were less sensitive to hedgehog pathway inhibition than those cell lines that expressed K-Ras alone (Fig. 5C). Similar data were obtained by using shRNA targeting smoothened (data not shown). These data also indicate that K-Ras-expressing tumor cell lines activate hedgehog signaling, albeit weakly, consistent with published findings (30), and are dependent on the activation of this pathway for proliferation. To determine whether the simultaneous inhibition of Shh- and Ras-mediated signaling would block the proliferation of our tumor cell lines, we treated Shh plus K-Ras cell lines simultaneously with cyclopamine and LY294002. We observed that this strategy prevented the proliferation of cell lines expressing Shh and activated K-Ras (Fig. 5D). Thus, our data suggest a cooperative interaction between K-Ras and Shh in the proliferation of pancreatic cancer cells.
Discussion
Previous studies have suggested a role for Shh signaling in pancreatic carcinogenesis (8, 9, 31). Yet, before our study, the mechanisms by which hedgehog signaling contributes to pancreatic tumor initiation and progression were unknown. We have now demonstrated that ectopic expression of Shh in cultured PDECs enhances their proliferation in a MAPK- and PI3-kinase-dependent manner. We additionally showed that Shh protects PDECs from apoptotic stimuli partly through the posttranscriptional regulation of Bcl-2 and Bcl-XL. Significantly, activation of the PI3-kinase pathway is required for Shh-induced stabilization of Bcl-2 and Bcl-XL, although the mechanisms through which this occurs are unknown. Thus, activated hedgehog signaling induces two of the “hallmarks of cancer” in PDECs, the presumptive target cells for PDAC, and in this way may contribute to the initiation of pancreatic tumorigenesis (32).
It remains unclear how Shh signaling activates the MAPK and PI3-kinase pathways. Consistent with our recent findings, analysis of PDECs expressing an activated Gli1 molecule demonstrated that the stimulation of these pathways occurs, at least in part, through Gli-independent mechanisms (31). Given the functional redundancy of Gli transcription factors, we were unable to ascertain whether they are required for Shh-mediated MAPK and PI3-kinase stimulation. Analysis of the expression levels of the Her2 and EGFR tyrosine kinases, which are potent activators of these pathways and are frequently overexpressed in early PanIN lesions, failed to identify elevated expression in Shh-expressing cells (33). Neither does it involve activating mutations in k-ras, as suggested by previous work (8), a result consistent with our recent findings with an activated Gli2 allele (31).
Orthotopic transplantation of PDECs with constitutive activation of Shh signaling led to the formation of early ductal pancreatic lesions. These lesions displayed many features of precursor PanIN lesions (3, 23). However, the extent of these features was considered insufficient by an experienced pathologist (D.S.K.) for definitive classification as mouse PanIN based upon the criteria established by the Penn Workshop (34). Interestingly, while these lesions formed, they failed to advance to frank tumors during the timeframe of our experiments. However, we did find that Shh enhanced tumor initiation by an activated K-Ras molecule and accelerated tumor growth, consistent with cooperation between Shh and IGF signaling in other model systems (35). Thus, our results collectively demonstrate a role for the Shh pathway in PDAC initiation and progression and are consistent with the activation of the pathway in early PanIN lesions and the sustained activation of the pathway during tumor progression (8, 9).
Our findings with Shh-expressing PDECs also provide an interesting contrast with activated K-Ras signaling in these cells. Transplantation of RCAS-k-ras-infected PDECs lacking either Ink4a/Arf or Ink4a/Arf and Trp53 led to the development of undifferentiated carcinomas within 60 days of transplant. Yet our cell culture studies demonstrated that in Ink4a/Arf, Trp53 null PDECs, Shh stimulated proliferation to a similar extent as an activated K-Ras molecule. We did find, however, that K-Ras provided a slightly more robust survival advantage to CHX-treated PDECs. The enhanced ability of K-Ras to transform PDECs may reflect this enhanced survival advantage or, alternatively, the activation of the Ral signaling pathway, which has been shown in other experimental systems to mediate, in large part, the transforming properties of activated Ras proteins (36, 37). Our results further underscored the functional significance of activating KRAS gene mutations as critical initiating events in pancreatic tumorigenesis; indeed, previously generated mouse models demonstrated that activation of K-Ras is sufficient to induce pancreatic tumors in vivo (38, 39). Detailed comparisons between PDECs expressing either Shh or K-Ras may therefore provide critical clues to the mechanisms underlying pancreatic cancer development.
Interestingly, we also found that Shh signaling reduced tumor cell dependence on the initiating oncogenic lesion. All four cell lines expressing K-Ras alone, and lacking Trp53 and Ink4a/Arf, remained sensitive to inhibition of the MAPK and PI3-kinase signaling pathways, demonstrating their continued requirement for tumor cell proliferation. This result contradicts previously published findings suggesting that activation of the PI3-kinase pathway was sufficient for tumor maintenance and may reflect either cell type or species differences, or the methods used to induce cell transformation (40). Yet all cell lines that express both K-Ras and Shh, including those that retain wild-type Trp53 alleles, continued to proliferate despite inhibition of these signaling pathways. Thus, constitutive Shh pathway activation reduced the dependence of tumor cells on activated K-Ras. This finding is significant because it predicts that a subset of PDAC, those with high levels of constitutively active Shh signaling, will be resistant to treatment with therapeutics that target Ras-regulated signaling pathways, several of which are currently in development and clinical trials.
We also observed that tumor cells expressing both Shh and K-Ras continued to proliferate when Shh signaling was inhibited. However, simultaneous inhibition of the Shh and PI3-kinase pathways blocked cell proliferation. Thus, Shh and K-Ras appeared to cooperate by providing independent signals that regulated tumor cell proliferation, although the precise mechanisms of this cooperation remain unclear. Analysis of the genomic, epigenetic, and gene expression profiles of the cell lines generated during our studies may shed light on potential collaborative changes involved in these processes.
Thus, our studies indicated a role for Shh in pancreatic tumor initiation and growth and also highlighted the potential impact of this pathway on therapeutic interventions in this malignancy.
Materials and Methods
Transgenic Mice and Animal Care.
The generation of keratin-19-tv-a transgenic mice is described elsewhere. The Ink4a/Arflox/lox, Trp53lox/lox, and Ptf1a-Cre strains have been previously described (15–17). Athymic nude mice were purchased from Charles River Laboratories (Wilmington, MA). Animals were kept in specific pathogen-free housing with abundant food and water under guidelines approved by the University of Massachusetts Medical School Institutional Animal Care and Use Committee.
Isolation, Culture, and Infection of Mouse PDECs.
Isolation and culture of mouse PDECs were performed as previously described (18). The RCAS-GFP and RCAS-Shh vectors have been described previously (41–43). Details on the construction of the RCAS-k-ras vector are provided in SI Methods. For proliferation assays, 105 cells were plated per well and counted at 5-day intervals. All experiments were performed in duplicate. For apoptosis assays, 106 cells were plated per well and treated with 100 μM CHX for 24 or 48 h. For pathway interrogation, cells were incubated with indicated inhibitors for 1 h before CHX treatment.
Immunoblotting.
Protein lysates were transferred to PVDF membranes (Hybond), and immunoblotting was performed as described (14). The complete list of primary antibodies and dilutions is provided in SI Table 2.
RT-PCR.
A total of 0.5 μg of purified total RNA was used for RT-PCR by using the SuperScript III One-Step RT-PCR kit (Invitrogen, Carlsbad, CA). Amplification parameters and primer pair sequences are provided in SI Table 4.
Orthotopic Implantation of PDECs.
Mice were anesthetized by i.p. injection of a mixture of 100 mg/kg ketamine and 10 mg/kg xylazine (Henry Schein Inc., Melville, NY) and 10 μl of the appropriate PDECs resuspended in matrigel injected into the pancreata of nude mice as described (44).
Immunostaining.
Immunostaining was performed as previously described (14). Primary antibody dilutions are listed in SI Table 3.
Culture and Treatment of Tumor Cell Lines.
Cell lines were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS and antibiotics in a humidified incubator at 37°C (5% CO2). Cell lines were plated at a density of 1 × 105 cells per well; allowed to adhere; treated with 25 μM PD98059, 20 μM LY294002, 10 μM cyclopamine, or vehicle; and counted at 24, 48, and 72 h after treatment.
Supplementary Material
Acknowledgments
The authors thank Paul Krimpenfort and Anton Berns (Netherlands Cancer Institute, Amsterdam, The Netherlands) for Trp53 and Ink4a/Arf conditional mutant mice; Dan Fults (University of Utah, Salt Lake City, UT) for RCAS-Shh DF1 producer cells; Anil Rustgi and Therese Deramaudt (University of Pennsylvania, Philadelphia, PA) for assistance with PDEC isolation; Eric Sandgren (University of Wisconsin, Madison, WI) for keratin 19 promoter and enhancer constructs; Michael Green (University of Massachusetts, Worcester, MA) for apoptosis reagents; and Lucio Castilla and members of the Lewis and Hebrok laboratories for critical reading of the manuscript and helpful discussions. This work was supported by the Pancreatic Cancer Alliance, a Burroughs Wellcome Fund Career Award in the Biomedical Sciences, American Cancer Society Grant IRG93–033, the Worcester Foundation for Biomedical Research (B.C.L), and National Institutes of Health Grant RO1 CA112537 (to M.H.).
Abbreviations
- CHX
cycloheximide
- PanIN
pancreatic intraepithelial neoplasia
- PDAC
pancreatic ductal adenocarcinoma
- PDECs
pancreatic duct epithelial cells.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0701158104/DC1.
References
- 1.Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ. CA Cancer J Clin. 2003;53:5–26. doi: 10.3322/canjclin.53.1.5. [DOI] [PubMed] [Google Scholar]
- 2.Warshaw AL, Fernandez-del Castillo C. N Engl J Med. 1992;326:455–465. doi: 10.1056/NEJM199202133260706. [DOI] [PubMed] [Google Scholar]
- 3.Hruban RH, Goggins M, Parsons J, Kern SE. Clin Cancer Res. 2000;6:2969–2972. [PubMed] [Google Scholar]
- 4.Freeman M. Nature. 2000;408:313–319. doi: 10.1038/35042500. [DOI] [PubMed] [Google Scholar]
- 5.Pasca di Magliano M, Hebrok M. Nat Rev Cancer. 2003;3:903–911. doi: 10.1038/nrc1229. [DOI] [PubMed] [Google Scholar]
- 6.Cohen MM., Jr Am J Med Genet A. 2003;123:5–28. doi: 10.1002/ajmg.a.20495. [DOI] [PubMed] [Google Scholar]
- 7.Kim SK, Hebrok M. Genes Dev. 2001;15:111–127. doi: 10.1101/gad.859401. [DOI] [PubMed] [Google Scholar]
- 8.Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernandez-del Castillo C, Yajnik V, et al. Nature. 2003;425:851–856. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshleman JR, Watkins DN, Beachy PA. Nature. 2003;425:846–851. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 10.Bader BL, Franke WW. Differentiation. 1990;45:109–118. doi: 10.1111/j.1432-0436.1990.tb00464.x. [DOI] [PubMed] [Google Scholar]
- 11.Hu L, Gudas LJ. J Biol Chem. 1994;269:183–191. [PubMed] [Google Scholar]
- 12.Grippo PJ, Sandgren EP. Am J Pathol. 2000;157:805–813. doi: 10.1016/S0002-9440(10)64594-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orsulic S. Mamm Genome. 2002;13:543–547. doi: 10.1007/s00335-002-4003-4. [DOI] [PubMed] [Google Scholar]
- 14.Lewis BC, Klimstra DS, Varmus HE. Genes Dev. 2003;17:3127–3138. doi: 10.1101/gad.1140403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Nat Genet. 2001;29:418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 16.Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. Nat Genet. 2002;32:128–134. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- 17.Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A. Nature. 2001;413:83–86. doi: 10.1038/35092584. [DOI] [PubMed] [Google Scholar]
- 18.Schreiber FS, Deramaudt TB, Brunner TB, Boretti MI, Gooch KJ, Stoffers DA, Bernhard EJ, Rustgi AK. Gastroenterology. 2004;127:250–260. doi: 10.1053/j.gastro.2004.03.058. [DOI] [PubMed] [Google Scholar]
- 19.Rao G, Pedone CA, Coffin CM, Holland EC, Fults DW. Neoplasia. 2003;5:198–204. doi: 10.1016/S1476-5586(03)80052-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalderon D. Cell. 2000;103:371–374. doi: 10.1016/s0092-8674(00)00129-x. [DOI] [PubMed] [Google Scholar]
- 21.Kenney AM, Rowitch DH. Mol Cell Biol. 2000;20:9055–9067. doi: 10.1128/mcb.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohta M, Tateishi K, Kanai F, Watabe H, Kondo S, Guleng B, Tanaka Y, Asaoka Y, Jazag A, Imamura J, et al. Cancer Res. 2005;65:10822–10829. doi: 10.1158/0008-5472.CAN-05-0777. [DOI] [PubMed] [Google Scholar]
- 23.Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genes Dev. 2006;20:1218–1249. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- 24.Browd SR, Kenney AM, Gottfried ON, Yoon JW, Walterhouse D, Pedone CA, Fults DW. Cancer Res. 2006;66:2666–2672. doi: 10.1158/0008-5472.CAN-05-2198. [DOI] [PubMed] [Google Scholar]
- 25.Gong J, Li X, Darzynkiewicz Z. J Cell Physiol. 1993;157:263–270. doi: 10.1002/jcp.1041570208. [DOI] [PubMed] [Google Scholar]
- 26.Collins RJ, Harmon BV, Souvlis T, Pope JH, Kerr JF. Br J Cancer. 1991;64:518–522. doi: 10.1038/bjc.1991.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koehler JA, Drucker DJ. Diabetes. 2006;55:1369–1379. doi: 10.2337/db05-1145. [DOI] [PubMed] [Google Scholar]
- 28.Sacedon R, Diez B, Nunez V, Hernandez-Lopez C, Gutierrez-Frias C, Cejalvo T, Outram SV, Crompton T, Zapata AG, Vicente A, Varas A. J Immunol. 2005;174:1456–1461. doi: 10.4049/jimmunol.174.3.1456. [DOI] [PubMed] [Google Scholar]
- 29.Kuwana T, Newmeyer DD. Curr Opin Cell Biol. 2003;15:691–699. doi: 10.1016/j.ceb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 30.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 31.Pasca di Magliano M, Sekine S, Ermilov A, Ferris J, Dlugosz AA, Hebrok M. Genes Dev. 2006;20:3161–3173. doi: 10.1101/gad.1470806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanahan D, Weinberg RA. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 33.Bardeesy N, DePinho RA. Nat Rev Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 34.Hruban RH, Rustgi AK, Brentnall TA, Tempero MA, Wright CV, Tuveson DA. Cancer Res. 2006;66:14–17. doi: 10.1158/0008-5472.CAN-05-3914. [DOI] [PubMed] [Google Scholar]
- 35.Rao G, Pedone CA, Valle LD, Reiss K, Holland EC, Fults DW. Oncogene. 2004;23:6156–6162. doi: 10.1038/sj.onc.1207818. [DOI] [PubMed] [Google Scholar]
- 36.Hamad NM, Elconin JH, Karnoub AE, Bai W, Rich JN, Abraham RT, Der CJ, Counter CM. Genes Dev. 2002;16:2045–2057. doi: 10.1101/gad.993902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lim KH, Baines AT, Fiordalisi JJ, Shipitsin M, Feig LA, Cox AD, Der CJ, Counter CM. Cancer Cell. 2005;7:533–545. doi: 10.1016/j.ccr.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 38.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 40.Lim KH, Counter CM. Cancer Cell. 2005;8:381–392. doi: 10.1016/j.ccr.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 41.Holland EC. Toxicol Pathol. 2000;28:171–177. doi: 10.1177/019262330002800122. [DOI] [PubMed] [Google Scholar]
- 42.Fults D, Pedone C, Dai C, Holland EC. Neoplasia. 2002;4:32–39. doi: 10.1038/sj.neo.7900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orsulic S, Li Y, Soslow RA, Vitale-Cross LA, Gutkind JS, Varmus HE. Cancer Cell. 2002;1:53–62. doi: 10.1016/s1535-6108(01)00002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mohammad RM, Al-Katib A, Pettit GR, Vaitkevicius VK, Joshi U, Adsay V, Majumdar AP, Sarkar FH. Clin Cancer Res. 1998;4:887–894. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}