

Abstract
Diseases such as cancer are often related to collaborative effects involving interactions of multiple genes within complex pathways, or to combinations of multiple SNPs. To understand the structure of such mechanisms, it is helpful to analyze genes in terms of the purely cooperative, as opposed to independent, nature of their contributions towards a phenotype. Here, we present an information-theoretic analysis that provides a quantitative measure of the multivariate synergy and decomposes sets of genes into submodules each of which contains synergistically interacting genes. When the resulting computational tools are used for the analysis of gene expression or SNP data, this systems-based methodology provides insight into the biological mechanisms responsible for disease.
Keywords: gene modules, microarray analysis, pathways, SNP data, synergy
Introduction
Systems biology is based on a holistic, rather than reductionist, view of biological systems. Therefore, the concept of synergy is fundamental and we would like to quantitatively analyze the ways by which the ‘whole' may be greater than the ‘sum of the parts' when focusing on specific systems of interacting components, such as genes, with respect to a phenotype, such as a particular cancer. With the help of high-throughput technologies, we now have vast amounts of data involving simultaneous values of biological variables, and we are presented with an opportunity to analyze such data in terms of the multivariate correlations among these variables.
For example, given a set of genes, we may wish to quantify the amount of information that the joint expression state of each of its subsets provides about a phenotype, such as a particular cancer, and then compare these amounts to each other. It is intuitively clear that such analysis can shed light into the structure of potential pathways that involve these genes and are responsible for the phenotype. Indeed, if we find, for example, that the amount of information provided by the joint expression of all genes in a set is higher than what could be attributed to additive independent contributions of its subsets, then this fact provides an indication that the additional information is due to some cooperative interaction involving all of the genes within a shared pathway.
Interestingly, although such problems have not yet been significantly addressed in molecular systems biology, related questions have been raised and investigated in the field of neuroscience under quite different contexts, such as asking how correlations among the joint activities of multiple neurons are related to a stimulus. In that case, measurements of the neurons' spiking patterns are made with the help of multi-electrode recordings. As a result of this research in neuroscience, an initial theoretical background has already been laid out (Brenner et al, 2000; Schneidman et al, 2003b) and can be directly applied in the field of molecular systems biology, where, rather than dealing with spike trains we have the advantage of access to much simpler variables, such as single expression values of genes or proteins, or SNP data. Furthermore, we are directly relating the data to a phenotype such as a particular disease, thus making the practical significance of this research immediate and potentially enormous.
We believe that the time is ripe for molecular systems biology to reap benefits from multivariate analysis. This research will be consistent with the paradigm shift at which systems biology is aimed, namely analyzing data on the level of gene modules, rather than of individual genes. In this review, we summarize and build on existing knowledge, and generalize based on recent definitions and methods of analyzing the multivariate synergy among interacting genes, which have already been applied on experimental biological data. Furthermore, because the various statements and symbols in the literature can be confusing and are occasionally contradictory or inaccurate, an additional purpose of this review is to clarify the fundamental concepts for the benefit of the research community by explaining the precise physical meaning and potential practical significance, or lack of it, of various related quantities.
Preliminaries from information theory
In information theory (Cover and Thomas, 2006), the uncertainty of a random variable X is measured by its entropy H(X); the uncertainty of a random variably X given knowledge of another random variable Y is measured by the conditional entropy H(X∣Y); and the uncertainty of a pair of random variables X, Y is measured by the entropy H(X,Y). These quantities are connected by the equation H(X,Y)=H(X)+H(Y∣X)=H(Y)+H(X∣Y). See Table I containing definitions of basic information theoretic quantities and their interrelationships.
Table 1.
Basic quantities of information theory and their interrelationships. X and Y are random variables with probability mass functions denoted, for simplicity, as p(x) and p(y)
| Symbol | Term | Formula | Physical meaning |
|---|---|---|---|
| H(X) | Entropy of X | Uncertainty of X | |
| H(X,Y) | Joint Entropy of X, Y | Uncertainty of pair X, Y | |
| H(X∣Y=y) | Conditional Entropy of X given Y=y | Uncertainty of X given Y = y | |
| H(X∣Y) | Conditional Entropy of X given Y | Uncertainty of X given Y | |
| I(X;Y) | Mutual Information common to X and Y | Reduction of uncertainty of X due to knowledge of Y (or vice versa). | |
| Relations confirmed in Venn diagram of Figure 1A: | |||
| I(X;Y)=H(X)−H(X∣Y)=H(Y)−H(Y∣X)=[H(X)+H(Y)]−H(X,Y) H(X,Y)=H(X)+H(Y∣X)=H(Y)+H(X∣Y). | |||
Given two random variables X and Y, the amount of information that each one of them provides about the other is the mutual information I(X;Y), which is equal to
Therefore, the physical meaning of I(X;Y) is ‘the reduction of the uncertainty of X due to knowledge of Y' (or vice versa) and this relationship can be depicted in a Venn diagram (Figure 1A) in which the single-variable entropies H(X), H(Y) are represented by two overlapping sets, whereas the two-variable entropy is represented by the union of these sets and the mutual information common to X and Y is represented by their intersection. Note that H(X)=I(X;X), so entropy is the ‘self-information.' Also note that conditioning of entropies in Venn diagrams is indicated by set subtraction, so that, for example, the set representing H(X∣Y) results from subtracting the set representing H(Y) from the set representing H(X).
Figure 1.

Venn diagrams indicating the mutual information common to multiple variables. (A) Mutual information common to two variables. Arrows stemming from the perimeter of a circle refer to the area inside the whole circle. Arrows stemming from the interior of a region refer to the area of that region. The mutual information I(X;Y) is defined by the intersection of the two sets, whereas the joint entropy H(X,Y)—not shown—is defined by the union of the two sets. (B) Mutual information common to three variables. The mutual information I(X;Y;Z) is defined by the intersection of the three sets. The bivariate synergy of any two of the variables with respect to the third is equal to the opposite of the mutual information common to the three variables and therefore positive synergy cannot be shown in a Venn diagram.
The mutual information common to two variables is always a non-negative quantity, consistent with the intuitively clear fact that uncertainty can only be reduced on the average as a result of additional knowledge. It is zero if and only if X and Y are independent of each other.
Like entropy, mutual information can be conditioned on the knowledge of another random variable by including this conditioning on all terms of the definition. For example, given a third random variable Z, the conditional mutual information I(X;Y∣Z) is equal to H(X∣Z)−H(X∣Y,Z), which means ‘the reduction of the uncertainty of X due to knowledge of Y (or vice versa), when Z is given'. It is zero if and only if X and Y are conditionally independent of each other (conditioned on knowledge of Z). In the Venn diagram of Figure 1B, we confirm that the set representing I(X;Y∣Z) results from subtracting the set representing H(Z) from the set representing I(X;Y).
The above definitions of mutual information and entropy remain valid when we substitute a random variable by a vector of random variables. However, the definition of the mutual information common to more than two variables is not a trivial generalization of the two-variable case. The mutual information common to three variables X, Y, Z can be defined as (McGill, 1954; Watanabe, 1960; Cover and Thomas, 2006)
The above equation can be visually confirmed by looking at the Venn diagram interpretation (Figure 1B): the single-variable entropies H(X), H(Y), H(Z) are represented by three overlapping sets whereas the multiple-variable entropies are represented by the unions of the corresponding sets. The mutual information I(X;Y;Z) common to the three variables is the intersection of the three sets. Indeed, any point that belongs in only one or two of these sets will be cancelled out in the equation. For example, the number of appearances of any point that belongs to the first and second set, but not in the third, will be (1+1+0)−(1+1+1)+1=0. Only the points that belong to all three sets will not be cancelled, because (1+1+1)−(1+1+1)+1=1.
It is easily proved that I(X;Y;Z) is equal to I(X;Y)−I(X;Y∣Z). Therefore, the physical meaning of I(X;Y;Z) is ‘the reduction of the mutual information common to two variables due to knowledge of a third variable.' Or, substituting the mutual information by its own physical meaning, it is ‘the reduction of ‘the reduction of the uncertainty of one variable due to knowledge of a second variable' due to knowledge of a third variable'. This quantity is symmetric in X, Y and Z and, contrary to the mutual information common to two variables, it is not necessarily non-negative (because contrary to uncertainty itself, ‘reduction of uncertainty' is not necessarily reduced due to additional knowledge), a fact that sometimes is considered ‘unfortunate' (Cover and Thomas, 2006) for quantifying information. However, as will be explained in this review, this is a fortunate fact for our purposes, because it allows for strictly positive synergy between two of these variables with respect to the third.
Further generalizing (Fano, 1961), the mutual information common to n variables, I(X1; X2; …, Xn), can be defined as
 |
Again, the above formula is consistent with the Venn diagram (in higher-dimensional space) representation of information (Yeung, 1991). For example, note that
which shows that the area common to all sets is present in the result. It can similarly be shown that the area common to only some of, but not all, the sets is not present in the result.
It is easily proved that I(X1;X2;…;Xn) satisfies the following recursive rule:
where the conditional mutual information common to multiple variables is defined by simply including the conditioning in all terms of the original definition.
Therefore, the physical meaning of I(X1;X2;…;Xn) for large values of n is ‘the reduction of the mutual information common to n−1 variables due to knowledge of the nth variable,' which is equivalent to the mind boggling ‘the reduction of the reduction of the reduction … of the uncertainty of one variable due to knowledge of a second variable due to knowledge … of an nth variable.' This quantity often appears in related literature, and we include it in this review, because it will be useful for clarifying various alternative suggested definitions of synergy.
Random variables defined from biological measurements
How can we quantify the amount of information that, for example, the joint expression levels of n genes G1, G2, …, Gn provide about a phenotype C, such as a particular cancer? According to the previous section, information theory has a ready answer, I(G1,…,Gn;C), as long as we are referring to random variables. We can use input data from measurements to define all these quantities as random variables by creating probabilistic models from relative frequencies. In the case of SNPs, the data are automatically binary (or, more accurately, tri-level to differentiate between monoallelic and biallelic presence), whereas in the case of continuous gene expression values, we can use a binarization approach to distinguish whether each gene is ‘on' or ‘off,' so that the number of possible joint expression states becomes manageable (eight in our case). If, for example, we have a set of expression data for the n genes from many biological samples (tissues, henceforth referred to as just ‘samples') in the presence as well as the absence of a cancer C, then for each of the 2n binary expression states, we can count the number N0 of healthy (C=0) samples as well as the number N1 of cancerous (C=1) samples encountered in that state. We can then use the counts N0 and N1 to define a probabilistic model from the relative frequencies, which will reflect the statistics of the input expression data. The larger the input data set from the measurements in cancer as well as health, the more gene expression variability the model will be able to include, and the more features the results will be able to capture.
Once we have the model defined, then we can readily evaluate all the information-theoretic quantities referred to in the previous section directly from the counts N0 and N1. For example, let us assume that we have n genes and K samples. For each of the 2n binary expression states, we evaluate the probability P=(N0+N1)/K of occurrence of that state. For each state for which P is nonzero, we further evaluate the probability Q=N1/(N0+N1) of cancer in that state, defined by the corresponding binary expression values g1, …, gn. The conditional entropy H(C∣G1=g1,…,Gn=gn) for that particular state will be equal to h(Q), where (see Table I) the function h is defined by h(q)=−qlog2q−(1−q)log2(1−q). Therefore, the conditional entropy H(C∣G1,…,Gn) will be equal to the sum ∑Ph(Q) over all such states. The mutual information I(G1,…,Gn;C) is then simply evaluated as H(C)−H(C∣G1,…,Gn). See Figure 2A for simple example.
Figure 2.

Examples of numerical evaluation of mutual information and synergy. In both cases, they should be seen as illustrations of the concept and not as representative of actual biological examples, in which many samples are needed for meaningful modeling. Black squares indicate a gene being ‘on' and white squares indicate a gene being ‘off.' (A) Evaluation of mutual information between a set of five genes and cancer from four normal and four cancerous samples. (B) Evaluation of the synergy between two genes with respect to cancer, derived from four normal and four cancerous samples. Two extreme cases are shown, the first with maximum synergy +1, and the second with minimum synergy −1 (redundancy).
High-throughput molecular data are known to suffer from high levels of noise, which may adversely affect the accuracy of these markers. However, a more fundamental problem in many cases is the lack of sufficient data, even in noisy form. These data must be plentiful in the presence as well the absence of the phenotype to enrich the probabilistic model so that the resulting random variables are defined in a meaningful way. Once we have a sufficiently large number of measurements, then the problem of noise becomes alleviated because the approach is holistic and the deviations from the accurate values will be averaged out.
This methodology has been used (Varadan and Anastassiou, 2006; Varadan et al, 2006) in actual biological examples. In a subsequent section of this review, we also provide an illustrating example with numerical calculations.
In the remainder of this paper, we use the term ‘factors' and the symbols G1, G2,…,Gn to refer to random variables representing quantities such as the expression levels of several genes or the presence or absence of several SNPs that contribute towards an outcome. We will also use the term ‘phenotype' and the symbol C to refer to this outcome, which can be, for example, the presence or absence of a particular cancer.
Bivariate synergy
Given two factors G1, G2 and a phenotype C:
The information that G1 provides about C is equal to I(G1;C)
The information that G2 provides about C is equal to I(G2;C)
The information that G1 and G2 jointly provide about C is equal to I(G1,G2;C)
The synergy between G1 and G2 with respect to C is defined by
This definition is consistent with the intuitive concept that synergy is the additional contribution provided by the ‘whole' compared with the sum of the contributions of the ‘parts.' It has been used in neuroscience literature (Gawne and Richmond, 1993; Gat and Tishby, 1999; Schneidman et al, 2003a, 2003b) under different contexts. It involves averaging over all possible states that the pair of factors can assume; therefore, it is possible that the potential high synergy in one of these states will not be noticeable in the single-averaged quantity. If we wish, we can easily define the synergy of individual states and focus on these specific states (Brenner et al, 2000) rather than averaging them. This process may provide additional insights. However, gene expression often depends on complex Boolean functions of transcription factors involving many states responsible for the same outcome. Furthermore, focusing on individual states will further increase the sensitivity to the number of input measurements, as some of these states may appear so few times that any statistical measurement becomes meaningless. On the other hand, the averaging in the definition above addresses this concern in an optimum and balanced manner, as it is weighted according to the probabilities of the states.
It is easily proved that Syn(G1,G2;C)=−I(G1;G2;C), the opposite of the mutual information common to the three variables G1, G2, C, as was defined in the previous section, which implies that bivariate synergy is symmetric in G1, G2, C and also equal to Syn(G1,G2;C)=I(G1;G2∣C)−I(G1;G2), that is, the synergy of two of the variables with respect to the third is the ‘gain in the mutual information' of two of the variables due to knowledge of the third. A moment's thought will confirm that this is consistent with the definition of synergy. For example, this information gain can also be expressed as Syn(G1,C;G2)=I(G1;C∣G2)−I(G1;C), in other words if the information that G1 provides about C is higher if we know G2 than it is if we do not know G2, then this additional information is the synergy between the two genes with respect to C. Note that synergy is zero when the two quantities I(G1;G2∣C) and I(G1;G2) cancel by being equal to each other. The former is zero when G1 and G2 are ‘conditionally independent,' whereas the latter is zero when they are ‘activity independent' (Schneidman et al, 2003b). Therefore, neither kind of independence alone guarantees zero synergy (Han, 1980). We may also wish to normalize this quantity by dividing by H(C), in which case synergy will be bounded by +1 and –1, as in the following cases (Figure 2B).
An example of extreme bivariate positive synergy is the following: assume that each of the genes G1 and G2 is equally (50% of the time) expressed in both the presence and absence of cancer. At first glance, it would appear that the two genes are totally unrelated to C, because I(G1;C)=I(G2;C)=0. Indeed, these genes would never be found high up in any typical individual ‘gene ranking' computational method! However, upon scrutinizing the second-order statistics, we may find that, in all cancerous samples either both genes are expressed or both are not expressed, whereas in all healthy samples one of the two is expressed but not the other. In that case, C is determined with absolute certainty from the joint state of the two genes and I(G1,G2;C)=1, therefore the synergy is equal to +1.
An example of extreme bivariate negative synergy (redundancy) is the following: assume that half of the samples are cancerous with both G1 and G2 expressed and the other half of the samples are healthy and neither of the two genes is expressed. In that case, C is determined with absolute certainty by the single expression of either of the two genes, so I(G1;C)=I(G2;C)=I(G1,G2;C)=1, and the synergy is equal to –1.
Multivariate synergy
The extension of these concepts to systems of multiple interacting genes is important in molecular systems biology, one reason being that pathways often involve multiple genes, such as in the formation of multi-protein complexes serving as pathway components.
One way of generalizing the definition of bivariate synergy to include n contributing factors with respect to a phenotype (Chechik et al, 2002) is to compare the contribution of the full set with the additive contributions of the single individual factors according to the quantity
The advantage of this definition is its simplicity, but it clearly fails to consider the various ways by which ‘parts' may cooperatively define the ‘whole' if there is positive synergy within some subsets of the full set.
There have been two other different definitions suggested in the literature comparing the correlations among the n genes to the correlations observable among at most n−1 genes. They are both related to the mutual information I(G1;G2;…;Gn;C) common to multiple variables, defined earlier. However, the physical meaning of either of the resulting quantities is complicated and not useful for our purposes. These definitions are described in Supplementary text 1 together with examples demonstrating that they are inappropriate for our applications.
The following definition of the multivariate synergy for a set of n factors G1,G2,…,Gn, with respect to a phenotype C was recently (Varadan et al, 2006) proposed:
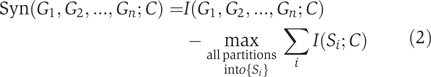 |
where partition is defined as a collection {Si} of disjoint subsets Si whose union is the full set, that is, 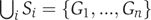 and
and 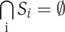 . For example, for n=3,
. For example, for n=3,
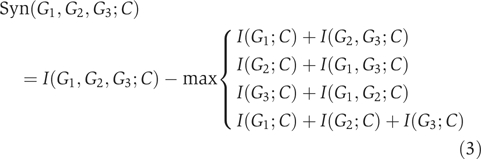 |
This is a natural generalization of the bivariate synergy, because it is also consistent with the intuitive concept that synergy is the additional amount of contribution for a particular task provided by an integrated ‘whole' compared with what can best be achieved after breaking the whole into ‘parts' by the sum of the contributions of these parts.
The partition of the set of factors that is chosen in this formula is the one that maximizes the sum of the amounts of mutual information connecting the subsets in that partition with the phenotype, and we will refer to it as the ‘maximum-information partition' of the set {G1,G2, …, Gn} with respect to the phenotype C.
As was the case with the bivariate synergy, we may wish to normalize by dividing this quantity by the entropy H(C), in which case the maximum possible synergy will be +1. In case of extreme redundancy, such as when G1=G2=⋯=Gn=C, the synergy can become as low as −(n−1). Examples of both cases appear in Supplementary text 1.
Note that the synergy, as defined above, refers to the combined cooperative effect of all n factors. If, for example, one of these factors is totally independent of all the other factors as well as the phenotype, then the synergy of the full set of n factors will be zero, even if the remaining n−1 factors form a synergistic set. In that case, that synergistic set will readily be identified by the maximum information partition, and can then be independently analyzed with the same methodology, as explained in the next section.
The tree of synergy
Assuming that the set of measurements is rich enough to generate a reasonably meaningful model of random variables, positive synergy indicates some form of direct or indirect interaction by participating in common pathways. Finding the maximum information partition is helpful towards deciphering such pathways, because the subsets in the maximum information partition are natural candidates of synergistic submodules, or pathway components. In turn, each of these subsets may undergo the same analysis, resulting in a hierarchical decomposition of the gene set into smaller modules. This decomposition is graphically depicted by a tree, referred to as ‘the tree of synergy,' defined as follows:
The tree of synergy of a set of factors {G1,G2,…,Gn} with respect to a phenotype C is a rooted and not necessarily binary tree with n leaves, each of which represents one of the factors Gi. Each intermediate node of the tree represents a subset of factors, those that are represented by the leaves of the clade formed by the node, and the root represents the whole set. The maximum-information partition, as defined above, of the whole set, is reflected by the branching of the root, so that the nodes that are directly stemming from the root represent the subsets defined by the maximum-information partition. Some of these nodes may be leaves, representing a single factor. If they are not leaves, then they represent a subset of factors, which has its own maximum-information partition, defined and evaluated as above, with respect to the phenotype. This methodology is repeated for all such subsets, until the full tree is formed. The root of the tree is labeled with the value of the synergy of the full set, and each intermediate node is also labeled with the value of the synergy of the corresponding subset. These values at the intermediate nodes are all non-negative numbers; otherwise, the definition of synergy would be contradicted. However, the root itself may be labeled with negative synergy (as in the example of the next section). In other words, the original full set of factors is not necessarily synergistic, but all its subsets that are present in the tree of synergy are synergistic.
The tree of synergy naturally reveals high-synergy subsets. To illustrate this fact, consider the case of n=3. For easier explanation, we omit the phenotype from the symbols, so that equation (3) is equivalently rewritten in simpler notation as
Using the same simplified notation, we can write the formulas for the three bivariate synergies of the corresponding subsets:
There are four possibilities: If the first term I1+I23 in Equation (4) defines the maximum-information partition, then this implies that I1+I23⩽I2+I13, I1+I23⩽I3+I12 and I1+I23⩽I1+I2+I3, from which it follows that Syn23⩾Syn13, Syn23⩾Syn12 and Syn23⩾0. In other words, the set {G2,G3} is the maximum-synergy subset of size 2, and its synergy is non-negative. We use identical symmetric reasoning for the next two possible cases. Finally, if the fourth term (I1+I2+I3) defines the maximum-information partition, then this implies that Syn12⩽0, Syn13⩽0 and Syn23⩽0.
The conclusion is that the tree of synergy for three factors always automatically includes the pair of factors with maximum synergy in its intermediate node, as long as that synergy is positive, otherwise it directly branches into the three leaves.
If the number of factors under consideration is small, synergistic decomposition can be made using exhaustive search by enumerating all partitions of the set. As the number of factors increases, the total number of partitions, given by the Bell number (Kreher and Stinson, 1999), increases exponentially, but so is the number of required measurements for a meaningful model of random variables, which is currently limited, thus making computational complexity not a serious problem in the near future. Even considering only pairs, triplets and quadruplets of factors, in which case the complexity is manageable, provides a significant benefit in molecular systems biology, shifting away from the old paradigm of ‘one-gene-one-disease'.
Illustrating example
Examples of trees of synergy in a real application with experimental biological data have been already presented (Varadan et al, 2006). Here, we illustrate the capabilities with a clarifying example simulating a hypothetical ‘toy problem' scenario: assume that the products of two genes G1 and G2 interact by forming a dimer serving as a transcription factor required for the expression of a tumor suppressor protein and that, as a result of this mechanism, a particular cancer results from the lack of expression (perhaps due to mutations) of at least one of these two genes. Furthermore, assume that this cancer independently triggers the expression of another gene, G3, which is normally not expressed. As a result, G3 is highly correlated with the presence of cancer, although it does not participate in the pathways responsible for the disease.
The following simulation strategy is one simple way to generate hypothetical binary microarray expression data for these three genes compatible with the above scenario, assuming binary gene expression levels:
In healthy samples, the probability of G3=0 is 80% and of G3=1 is 20%.
In cancerous samples, the probability of G3=0 is 20% and of G3=1 is 80%.
And independently,
In healthy samples, the probability of both G1 and G2 to be 1 is 95%, whereas the remaining three joint states (00, 01 and 10) are equally likely.
In cancerous samples, the probability of both G1 and G2 to be 1 is 5%, whereas the remaining three joint states (00, 01 and 10) are equally likely.
A compatible state-count table (Figure 3A) was simulated under these assumptions, representing 100 healthy and 100 cancerous samples (precise values of counts are not important as slight variations will not change the qualitative outcome of this analysis). Note that the states 001, 011, 101, highlighted in red typeface, are those in which cancer is predominant (N1≫N0) and correspond to the Boolean logic ((NOT G1) OR (NOT G2)), AND G3, as expected from the assumptions. A methodology for deriving Boolean functions from state-count tables has previously been presented (Varadan and Anastassiou, 2006).
Figure 3.

From the ‘state-count table' to the ‘tree of synergy.' (A) An example of a state-count resulting from hypothetical microarray measurements of three genes G1, G2, G3 in both the presence and absence of a particular cancer C. N0 and N1 are the counts of each state in the absence and presence of cancer, respectively. (B) The amounts of mutual information between each subset of the set of three genes and the presence of cancer, with simplified notation (see text). (C) The tree of synergy resulting from these sets making repeated use of the formula defining multivariate synergy. This decomposition separates the full set into two redundant subsets, one of which is the synergistic pair of genes G1, G2, consistent with the assumptions under which the state-count table was simulated.
Using the resulting relative frequencies to define the random variables for the expression levels of the genes and the phenotype, we find, using simplified notation as in equation (4), the values for mutual information between gene subsets and cancer (Figure 3B). A simple MATLAB program deriving these values appears as Supplementary text 2. We can apply equation (4) to these values to identify the maximum-information partition and the value of the synergy. The resulting tree of synergy (Figure 3C) accurately decomposes the set of three genes into, on the one hand, the subset of the first two interacting genes G1 and G2 and, on the other hand, the third independent gene G3. The synergy between these two sets, indicated at the root of the tree, is negative and equal to I123−(I12+I3)=−0.1971; in other words, their redundancy was detected and thus they were properly isolated from each other. This is desirable, because, according to our assumptions, only the first of these sets plays a causative role in the phenotype. However, as indicated in the tree, the set of the two genes G1 and G2 has positive synergy, equal to I12−(I1+I2)=+0.0582, consistent with the assumption that they interact with each other with respect to cancer. Although mechanistic analysis of synergy detects correlations without differentiating between cause and effect, when coupled with additional biological knowledge it is clearly helpful towards inferring responsible pathways.
Discussion
Analyzing the correlations among multiple contributing factors, such as gene expression levels and SNPs, can provide much needed insight into the structure of the causative factors, including biological mechanisms responsible for disease. This review summarized recent results on such techniques aiming to quantify the degree of cooperative interactions among such multiple factors, and to properly decompose them into smaller synergistic sets reflecting the structure of these interactions. This methodology works best in conjunction with approaches that first use optimization methods for searching over subsets to identify potential modules of multiple factors that jointly provide maximum information about the phenotype (Varadan and Anastassiou, 2006). After identifying such a module, analysis of the synergy among its members provides further valuable input into the problem of inferring related pathways that include these factors.
The concepts and the methodology presented in this review are complementary to those of existing techniques of analyzing expression data, such as variations of clustering (Eisen et al, 1998) and support vector machine-based (Boser et al, 1992) methods. For example, the aim of most existing methods of selecting sets of genes associated with disease is the ability to correctly classify between health and disease, or between different disease types. This ability can be quantified as the amount of information that the expression state of the gene set provides about the presence of a disease, that is, only the first term of the definition of synergy in equation (2). This is, of course, an important task. An additional important task, however, is to extract, out of this information, the part that is due to the purely cooperative nature among the genes in the set as a whole. This part results after subtracting the maximum possible information about the presence of disease attributed to independent contributions of subsets under all possible partitions of the gene set.
This distinction can be illustrated by a simple example. If we identify a pair of genes G1 and G2 with high value of I(G1,G2;C), where C designates the presence of a cancer, then this pair can be an appropriate choice for a classifier for cancer. However, the good classification performance may be due to the independent individual contributions of the two genes and does not necessarily imply that there is joint cooperative contribution. Indeed, if the synergy I(G1,G2;C)−[I(G1;C)+I(G2;C)] is not a positive number, then the genes' contributions are not cooperative, and we would have probably found these genes anyway using any individual ‘gene ranking' method, because the values of I(G1;C) and I(G2;C) would be high. If, on the other hand, the synergy of a gene pair is a large positive number, then we will have good reason to believe that the two genes ‘interact,' directly or indirectly, with respect to the presence of cancer. Gene selection based on search of high-synergy subsets can be a powerful tool for identifying protein–protein interactions with respect to a phenotype, and goes further than traditional approaches, because it can also identify high-synergy subsets of larger sizes.
Other existing techniques for the analysis of gene interactions use graphical models such as artificial neural networks, Boolean networks or Bayesian networks. In these models, the representative graphs are constructed from input data using specialized learning algorithms attempting to capture the structure of multiple relationships among genes and phenotypes. Again, the concepts and methods presented in this review play complementary roles. To take a particular example, consider a Bayesian network approach (Pearl, 1988) in which multivariate probability distributions are represented graphically so that each node in the graph represents a gene and one of the nodes represents the class label (such as the presence of cancer). Defining a graph structure that is optimally consistent with input data, such as gene expression levels, is a hard problem, and it is often necessary to use pre-existing knowledge about pathways to generate such a structure. Assuming that the Bayesian network has been defined, the ‘Markov blanket' (defined as the minimal set of variables that shield the node from the rest of the variables) of the class label node can be a set of genes likely to be associated with the presence of cancer. Analyzing this set in terms of its synergistic decomposition will provide valuable complementary information that can help refine the structure and properties of the Bayesian network. More generally, synergy provides a novel numerical measure to evaluate the cooperativity of interactions among multiple genes found using other computational methods, as well as suggesting possible ways to ‘fine-tune' structures and memberships of sets to improve the biological accuracy of the resulting models.
The main strength of the analysis of synergy in gene sets is the potential of deciphering the structures of pathways associated with a phenotype. It identifies sets of interacting genes ab initio, that is, without using pre-existing biological knowledge. However, once these sets are identified, additional biological knowledge is needed not only because it can identify known pathways that are compatible with the found genes and their synergistic decomposition, but also to address certain limitations of the methodology.
The main such limitation is the inability to identify the causal relationship between the phenotype and high-synergy modules of genes: It is not clear which is the cause and which is the effect, and it is possible that the synergistic decomposition will reveal a mixed set of modules, some of which are causative and some not, as was the case in the hypothetical illustrating example in the previous section.
Another limitation is the fact that the decomposition of gene sets from the ‘tree of synergy' cannot account for overlapping biological pathways that may all contribute towards the phenotype. This inability is a direct consequence of the requirement that partitions of the full set include disjoint subsets. In such cases, synergistic decomposition is likely to still reveal the dominant pathway, whereas additional biological knowledge can be of help to compensate for this limitation.
Finally, when analyzing microarray data, it is preferable to avoid binarizing the gene expression levels to preserve full information. We are currently working on extending our computational methodology to measure synergy directly from the set of continuous expression levels.
What is missing is the large amount of publicly available measurements required for any meaningful definition of multivariate correlations. For example, it would be desirable and relatively cost-effective to analyze numerous standardized microarray measurements from biopsies of not only cancerous samples, but also those determined to be free of cancer serving as controls—ideally thousands of such experiments in each case. We hope that such efforts will materialize, for example merged within existing complementary cancer initiatives involving sequencing. In that case, we believe that employing multivariate synergy methodologies, as those described in this review, will provide novel tools valuable to medical research.
Supplementary Material
Supplementary text 1
Supplementary text 2
Acknowledgments
I thank Vinay Varadan for helpful discussions and the section editor, Thomas Lemberger, for the clarifying examples and figures that he suggested, which made this paper more accessible and easier to understand.
References
- Boser BE, Guyon IM, Vapnik VN (1992) A training algorithm for optimal margin classifiers. In 5th Annual ACM Workshop on COLT, Haussler D (ed), pp 144–152. Pittsburgh, PA, USA: ACM Press [Google Scholar]
- Brenner N, Strong S, Koberle R, Bialek W, de Ruyter van Steveninck R (2000) Synergy in a neural code. Neural Comput 12: 1531–1552 [DOI] [PubMed] [Google Scholar]
- Chechik G, Globerson A, Anderson M, Young E, Nelken I, Tishby N (2002) Group redundancy measures reveal redundancy reduction in the auditory pathway. In Advances in Neural Information Processing Systems, Dietterich TG, Becker S, Ghahramani Z (eds), pp 173–180. Cambridge, MA: MIT Press [Google Scholar]
- Cover T, Thomas J (2006) Elements of Information Theory. New York, NY, USA: Wiley Interscience [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 95: 14863–14868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fano RM (1961) Transmission of Information. New York, NY, USA: MIT Press [Google Scholar]
- Gat I, Tishby N (1999) Synergy and redundancy among brain cells of behaving monkeys. In Advances in Neural Processing Systems 11, Kearns M, Solla S, Cohn D (eds), pp 465–471. Cambridge, MA: MIT Press [Google Scholar]
- Gawne T, Richmond B (1993) How independent are the messages carried by adjacent inferior temporal cortical neurons? J Neurosci 13: 2758–2771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han TS (1980) Multiple mutual informations and multiple interactions in frequency data. Inform Control 46: 26–45 [Google Scholar]
- Kreher DL, Stinson DR (1999) Combinatorial Algorithms: Generation, Enumeration and Search. Boca Raton, FL, USA: CRC Press [Google Scholar]
- McGill WJ (1954) Multivariate information transmission. IRE Trans Info Theory 4: 93–111 [Google Scholar]
- Pearl J (1988) Probabilistic Reasoning in Intelligent Systems: Networks of Plausible Inference. San Francisco, CA, USA: Morgan Kaufmann Publishers [Google Scholar]
- Schneidman E, Bialek W, Berry MJ II (2003b) Synergy, redundancy, and independence in population codes. J Neurosci 23: 11539–11553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman E, Still S, Berry M, Bialek W (2003a) Network information and connected correlations. Phys Rev Lett 91: 238701. [DOI] [PubMed] [Google Scholar]
- Varadan V, Anastassiou D (2006) Inference of disease-related molecular logic from systems-based microarray analysis. PLoS Comp Biol 2: 585–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadan V, Miller D III, Anastassiou D (2006) Computational inference of the molecular logic for synaptic connectivity in C elegans. Bioinformatics 22: e497–e506 [DOI] [PubMed] [Google Scholar]
- Watanabe S (1960) Information theoretical analysis of multivariate correlation. IBM J Res Dev 4: 66–82 [Google Scholar]
- Yeung RW (1991) A new outlook on Shannon's information measures. IEEE Trans Info Theory 37: 466–474 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary text 1
Supplementary text 2