

Abstract
Lantibiotics such as gallidermin are lanthionine-containing polypeptide antibiotics produced by gram-positive bacteria that might become relevant for the treatment of various infectious diseases. So far, self-toxicity has prevented the isolation of efficient overproducing strains, thus hampering their thorough investigation and preventing their exploitation in fields other than the food area. We wanted to investigate the effect of lantibiotic precursor peptides on the producing strains in order to evaluate novel strategies for the overproduction of these promising peptides. In this study, gallidermin was chosen as a representative example of the type A lantibiotics. A Staphylococcus gallinarum Tü3928 mutant, whose gene for the extracellular pregallidermin protease GdmP was replaced by a kanamycin-resistance gene, was constructed. Mass spectrometry (MS) analysis indicated that this mutant produced fully posttranslationally modified gallidermin precursors with truncated versions of the leader peptide, but not the entire leader as predicted from the gdmA sequence. In filter-on-plate assays, these truncated pregallidermins showed no toxicity against Staphylococcus gallinarum Tü3928 up to a concentration of 8 g/liter (corresponding to approximately 2.35 mM), while gallidermin produced clear inhibitory zones at concentrations as low as 0.25 g/liter (0.12 mM). We showed that the lack of toxicity is due entirely to the presence of the truncated leader, since MS as well as bioassay analysis showed that the peptides resulting from tryptic cleavage of pregallidermins and gallidermin produced by S. gallinarum Tü3928 had identical masses and approximately the same specific activity. This demonstrates that even a shortened leader sequence is sufficient to prevent the toxicity of mature gallidermin. In nonoptimized fermentations, the gdmP mutant produced pregallidermin to a 50%-higher molar titer, suggesting that the absence of self-toxicity has a beneficial effect on gallidermin production and giving a first confirmation of the suitability of the overproduction strategy.
Antibiotic resistance has spread dramatically among pathogens throughout the last two decades. Of particular concern is the increasing emergence of multiresistant pathogens in health care settings. Nowadays, about 70% of nosocomial infections in the United States are resistant to at least one antibiotic (35), and in 2002, the first clinical isolate of Staphylococcus aureus with high-level resistance to vancomycin, a drug of last resort, was isolated (8). As the number of resistant pathogens continues to grow, the number of new antibiotics to fight them is declining steeply (37). Therefore, the development of new antimicrobial agents should be encouraged.
Type A lantibiotics are cationic, amphiphilic peptides that are produced by a wide range of gram-positive bacteria and show bactericidal activity against other gram-positive bacteria. They show a dual mode of action at nanomolar concentrations which involves pore formation and inhibition of peptidoglycan biosynthesis by specific interaction with cell wall precursor lipid II (5, 19, 60). Lantibiotics are ribosomally synthesized and undergo posttranslational modifications resulting in the formation of unusual residues, like the thioether amino acids lanthionine and methyllantionine which are crucial for a proper functional structure of the lantibiotic(24). So far, no lantibiotic has been developed into a clinical product, even though nisin has proven its potential to treat peptic ulcers caused by Clostridium difficile and Helicobacter pylori (13), gallidermin is effective against acne caused by Propionibacterium acnes and bovine mastitis caused by S. aureus (40, 62), and mersacidine can treat methicillin-resistant Staphylococcus aureus infections in mice (33). The major obstacle to the clinical development of type A lantibiotics is their generally very low production titer, which has triggered attempts to improve volumetric productivity (3, 54) which generally have failed to provide a significant increase in titer.
Gallidermin is a tetracyclic, 21-amino-acid, type A lantibiotic peptide which is produced by the nonpathogenic Staphylococcus gallinarum Tü3928 (25) and shows broad-spectrum activity against important gram-positive pathogens like S. aureus, Staphylococcus epidermidis, Streptococcus pyogenes, Streptococcus pneumoniae, Streptococcus faecalis, and P. acnes (20). Gallidermin (6L-epidermin) is closely related to epidermin, which is synthesized by the class II strain S. epidermidis Tü3298 (1, 51). The biosynthesis, modification, secretion, and activation of epidermin have been broadly studied (6, 11, 34, 41, 50-52). Since gallidermin and epidermin have a high degree of identity on the DNA and protein level, and several genes of the two gene clusters have been shown to be interchangeable (4, 38, 44), many assumptions on gallidermin biosynthesis and function have been made based on previous research on epidermin. The 11 genes responsible for epidermin synthesis are organized as a 13-kb gene cluster which is located on pTü32, a 54-kb plasmid of S. epidermidis Tü3298 (2, 50, 52). Epidermin formation starts with ribosomal synthesis of an extended prepropeptide encoded by the structural gene epiA, which is posttranslationally modified by gene products EpiB, EpiC, and EpiD (50). These are responsible for the dehydration of Thr and Ser residues to dehydrobutyrine and dehydroalanine (DhB and DhA), for subsequent lanthionine and methyllantionine formation, and for the formation of an S-aminovinyl-d-cysteine ring (4, 45). These posttranslational modifications lead to the prepeptide. EpiT and EpiH, which show high homology to ATP binding cassette (ABC) transporters, should be responsible for active translocation of the prepeptide out of the cell (17, 44, 45).
However, as the DNA sequence of the epiT locus does not encode a functional subunit (having an internal deletion that causes a frameshift and a second deletion at the 3′ end) the translocation of epidermin through the cytoplasmic membrane is presumably accomplished by a host-encoded system (44). In contrast, the homologous gene in the gallidermin synthesis cluster, gdmT, encodes a fully functional protein (44). Removal of the leader peptide from the prepeptide leads to the mature lantibiotic. This reaction takes place extracellularly and is catalyzed by EpiP, a subtilisin-like serine protease (1, 45). The regulator EpiQ activates the expression of every gene encoded in the epidermin gene cluster except epiP and the promoterless epiQ (42).
The mechanism of action of lantibiotics results in toxicity against the producing strains (45). S. epidermidis Tü3298 has evolved a protection mechanism consisting of a specialized ABC transporter, EpiEFG, that is thought to remove lantibiotic molecules from the cytoplasmic membrane (17, 39, 43, 47). However, this export system merely decreases the exposure to the lantibiotic and does not confer resistance to the producer (26). Due to this autotoxicity, epidermin as well as gallidermin titers in fermentations have generally remained low, on the order of 250 to 330 mg/liter (21, 27, 55, 56, 58).
Operating under the hypothesis that gallidermin toxicity is the major obstacle to its overproduction, we developed a novel two-step production strategy which is based on (i) the overproduction of a potentially nontoxic precursor of gallidermin, such as pregallidermin, followed by (ii) an enzymatic activation step leading to the active lantibiotic. Therefore, we investigated the production and the toxicity of pregallidermin, which was generated by a mutant strain impaired in the final proteolytic step of the synthesis pathway. As the biosynthesis of type A lantibiotics is highly conserved, such a strategy might harbor great potential for a whole series of potentially clinically relevant molecules.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains and plasmids are listed in Table 1. Escherichia coli strains were routinely grown in Luria-Bertani broth (46), whereas staphylococci were grown in B broth (29). All percentages refer to weight/volume unless otherwise specified. For production of gallidermin by S. gallinarum Tü3928, YE4 medium (27) or medium 21 (5% Ohly Kat yeast extract [Deutsche Hefewerke GmbH, Marl, Germany], 2% NaCl, 0.5% maltose, pH 7.2) was used. Media were supplemented when appropriate with ampicillin (100 μg/ml), chloramphenicol (10 μg/ml), kanamycin (50 μg/ml), or tetracycline (10 μg/ml), unless otherwise noted. All cultures were grown at 37°C and 225 rpm in a shaking incubator, unless otherwise noted.
TABLE 1.
Strains and plasmids
| Bacterial strain or plasmid | Relevant characteristic(s) | Reference and/or source |
|---|---|---|
| Bacterial strains | ||
| E. coli TOP10 | E. coli cloning host strain [F−mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(araleu) 7697 galU galK rpsL (Strr) endA1 nupG] | Invitrogen |
| S. aureus RN4220 | Intermediate strain for cloning | 32 |
| S. gallinarum Tü3928; DSMZ 17239 | Gallidermin producer, Tetr | 25; DSMZ, Braunschweig, Germany |
| S. gallinarum ΔgdmP::aphIII | Pregallidermin producer, Tetr Kanr | This study |
| S. epidermidis Tü3298 | Epidermin producer | 1; DSMZ |
| K. rhizophila | Gallidermin-sensitive indicator strain, previously known as Micrococcus luteus | 30; DSMZ |
| Plasmids | ||
| pDG782 | B. subtilis kanamycin resistance cassette vector, Ampr Kanr | 14 |
| pZErO-1 | E. coli cloning vector, Kanr | Invitrogen |
| pUC18NotI | E. coli cloning vector, Ampr | 15 |
| pGEM5 z(+) | E. coli cloning vector, Ampr | Stratagene |
| pPSM1058 | Staphylococcal knockout vector, pBT2 derivative, temperature sensitive, oriV Camr Ampr | 36 |
| pUC18NotI-P | pUC18NotI with a 651-bp PstI-HindIII fragment internal to gdmP | This study |
| pGV2 | pPSM1053 in which a 0.9-kb BamHI-SalI fragment was replaced by a NotI oligonucleotide | This study |
| pGV4 | pUC18NotI-P with a 1.5-kb ClaI-EcoRI fragment containing the Kanr gene from pDG782 fused to a part of gdmQ | This study |
| pGV5 | pGV2 with a 2.2-kb NotI fragment from pGV4 containing the fragment internal to gdmP and the Kanr gene fused to a part of gdmQ | This study |
Genetic procedures.
Standard genetic procedures were performed as reported elsewhere (46). Staphylococcal chromosomal DNA was isolated using QIAGEN genomic tips (QIAGEN, Hilden, Germany) according to the protocol supplied by the manufacturer, except that prior to extraction cells were incubated with 20 μg/ml of lysostaphin at 37°C for 1 h. Low-copy plasmid isolation from staphylococci was performed as described by Kies (28). In our hands, only staphylococcal plasmid DNA could be introduced into S. gallinarum; therefore, plasmid DNA prepared from E. coli had to be first electroporated into the restriction-negative S. aureus RN4220 as described elsewhere (49). Electrocompetent S. gallinarum cells were prepared, and electroporation was carried out as described previously (16), except that the Genepulser electroporator (Bio-Rad, Hercules, CA) was operated at 2.5 kV, 1,000 Ω, and 1 μF.
Enzymes for molecular cloning were purchased from New England Biolabs (Ipswich, MA). DNA purification steps were performed with JetQuick columns (Genomed GmbH, Löhne, Germany). PrimerSelect (Lasergene package; DNAStar, Inc.) software was utilized for primer design, and oligonucleotides were synthesized by Microsynth (Balgach, Switzerland). Routine PCR amplifications were performed in 25-μl reaction mixtures containing 0.2 mM deoxynucleoside triphosphates, 0.2 μM of each primer, 1× PCR buffer (Fermentas, Vilnius, Lithuania), 1.5 mM MgCl2, and 0.070 U Taq DNA polymerase (Fermentas, Vilnius, Lithuania). The standard cycling conditions were 94°C for 2 min, followed by 30 cycles of 94°C for 30 s, 52 to 55°C for 30 s, and 72°C for 1 min, and a final extension of 72°C for 10 min. The primers used in this study are listed in Table 2. Sequencing of cloned fragments was performed by GATC Biotech AG (Konstanz, Germany). Inverse PCR (iPCR) was performed as described elsewhere (53). PCR, with primers designed on the epidermin cluster sequence, was performed using the Expand long template PCR system (Roche, Basel, Switzerland). Southern blot analysis was performed as described elsewhere (46). Digoxygenin (DIG)-labeled probes were prepared with a PCR DIG probe synthesis kit (Roche, Basel, Switzerland) as described by the manufacturer. Detection was performed with anti-DIG alkaline phosphatase and the chemiluminiscent alkaline phosphatase substrate CSPD (Roche, Basel, Switzerland), following the manufacturer's instructions.
TABLE 2.
Primers used in this study
| Primer name | Primer sequence (5′-3′) |
|---|---|
| GALL3 F | AAGGGATCCTCAGGCGCAGAACCACGAA |
| GALL3 R | TGCGGTACCGCTCAAAGCAGTCCTAACCAAC |
| epiB F | TATGAGGATTTTGCCGTGATAAGTC |
| epiP R | TTGTTGTTGCAGCTGTGGGTAATGA |
| Gdmseq F2 | TAGGATCCCATTACCCACAGCTGCAACAAC |
| Episeq R2 | GGCGGGAATTCGTTAGAGGTGCTATATGAA |
| gdmP-PstI F | CATATCTGCAGGGTTTGTAGCGCATCATAATC |
| gdmP-HindIII R | CGGTCACAAGCTTAGTAAGTCCCAAGTAGAGTCC |
| gdmQ-EcoRI F2 | CGGAATTCGTCTATCAATTCATCATCAATG |
| gdmQ R | TGAAGGGAAATAATATGATTGCTATTAATATTGTAGGTG |
| kan-ClaI F | TTATCGATGCCGTATGTAAGGATTCAG |
| kan R | CCTACAATATTAATAGCAATCATATTATTTCCCTTCAAAACAATTCATCCAG |
| PROOF-K | ACGAACTCCAATTCACTGTTCCTTG |
| PROOF-P | GGTGAGGGGTGCTATATGAAGAAATT |
| PROOF-Q | CTTTGCACACCCTTAAATTATCTCTTAATC |
Sequencing of the complete gallidermin gene cluster.
The sequencing strategy relied on the already-available 4,373-bp-long sequence (GenBank no. U61158) that covers the central part of the gallidermin synthesis cluster (gdmF, gdmT, gdmH, gdmA, and a partial coding sequence of gdmE and gdmB) (44, 51). iPCR was performed using the GALL3 primer pair, which was designed on the available sequence. Amplification of a ClaI digest of S. gallinarum Tü3928 genomic DNA generated a 4-kb amplicon, including gdmG and gdmB, as verified by Southern blotting with epiB- and epiG-derived amplicons as probes. The amplicon was cloned and sequenced by primer walking. A 4.4-kb-long fragment resulted from amplification with the primer pair epiB F/epiP R, which was designed on the sequence of the epidermin gene cluster of S. epidermidis Tü3928 (GenBank no. X62386 and no. X99127) (9, 50). The primers are detailed in Table 2. After sequencing, primer gdmseq F2 was designed on the end of the partial gdmP sequence and was used together with the primer episeq R2, which was designed on the sequence downstream of epiP, in order to amplify a 1-kb fragment for sequencing. The sequences of the three novel fragments and the available part were assembled with SeqManII (Lasergene package; DNAStar, Inc.).
Development of an integration vector and construction and verification of an S. gallinarum ΔgdmP::aphIII mutant strain.
A fragment internal to gdmP was amplified by PCR using the primer pair gdmP-PstI F and gdmP-HindIII R. PCR amplification was performed with an annealing temperature of 58°C and generated a 651-bp-long amplicon, which was digested with PstI and HindIII and cloned into pUC18NotI to yield pUC18NotI-P. An aphIII-gdmQ fusion was generated by overlap extension PCR (7, 18, 22, 23) by fusing the aphIII gene without its terminator sequence to a fragment of gdmQ (maintaining the gdmQ Shine-Dalgarno sequence and start codon; Fig. 1). Amplification of the 364-bp-long sequence of gdmQ was performed with the primer pair gdmQ-EcoRI2 F and gdmQ R. The aphIII gene was amplified from pDG782 by PCR using primers kan-ClaI F and kan R. Overlap extension PCR was performed using a 1:1 molar mixture of the two amplicons as a template and outer primers gdmQ-EcoRI2 F and kan-ClaI F. The resulting 1,510-bp-long amplicon was digested with EcoRI and ClaI and inserted into pUC18NotI-P, generating plasmid pGV4. The E. coli-Staphylococcus shuttle vector pGV2 was constructed by excising a 1.4-kb SacI-NheI fragment containing a nuclease gene from pPSM1058 and replacing it with a SacI-NotI-SpeI linker fragment from the multiple-cloning site of pGEM5zf(+). The knockout vector pGV5 was assembled by cloning the approximately 2.2-kb NotI fragment from pGV4 containing the gdmP fragment and the aphIII-gdmQ fusion into pGV2. S. gallinarum Tü3928 was transformed with pGV5, and transformants were grown at 30°C on selective B agar plates containing 20 μg/ml chloramphenicol. Gene replacement was induced in S. gallinarum Tü3928(pGV5) as described previously (12), except that the first growth step at 30°C was repeated three times and cells were subsequently incubated at 42°C on B agar plates containing 7.5 μg/ml kanamycin (determined in previous experiments to be the MIC). Screening for strains with double homologous recombination was performed on B agar plates containing either 20 μg/ml chloramphenicol or 7.5 μg/ml kanamycin. The site of integration was verified by PCR, applying primers PROOF-K and PROOF-P and primers PROOF-Q and PROOF-P to chromosomal DNA from S. gallinarum Tü3928 and S. gallinarum ΔgdmP::aphIII.
FIG. 1.

Construction of an S. gallinarum Tü3928 mutant lacking gdmP. (Top) gdmP and gdmQ and the sequence between them in detail, including the stop codon of gdmP, the start codon of gdmQ, and the Shine-Dalgarno sequence (SD). (Bottom) gdmP, the kanamycin-resistance gene aphIII, and gdmQ in S. gallinarum ΔgdmP::aphIII. The small arrows represent primers for verification of homologous recombination, and the sequences that were chosen for homologous recombination are shaded in light and dark gray. The promoters of gdmP and aphIII are represented by large arrows. The expected lengths of the amplicons are indicated on the right (for details, see the text).
Gallidermin and pregallidermin analytics.
Detection of gallidermin and pregallidermin was performed with a protocol adapted from Fiedler et al. (12). Culture supernatants were centrifuged at 16,000 × g at 4°C for 10 min, filtered through a 0.2-μm cellulose-acetate membrane filter (Carl Roth GmbH, Karlsruhe, DE), and separated on a Prontosil Eurobond C18 5.0-μm column with a guard column (Bischoff, Leonberg, Germany) operated in a La Chrom high-performance liquid chromatography (HPLC) system (Merck, Germany) equipped with an L-7100 pump, L-7200 autosampler, L-7455 diode array detector, and L-7490 refractive index detector. Data collection was performed with Merck-Hitachi model D-7000 chromatography station software. The injection volume was 10 μl and column temperature was maintained at 25°C. The mobile phase was composed of 0.1% (vol/vol) aqueous H3PO4 (solvent A) and acetonitrile (solvent B) and followed a linear gradient from 90% A/10% B to 45% A/55% B in 14 min at a flow rate of 1 ml/min. Regeneration of the column and equilibrium to start conditions were achieved in 5 additional minutes.
The identity of gallidermin produced in our laboratory by S. gallinarum Tü3928 was confirmed by comparing its LC/electrospray ionization mass spectrometric (ESI-MS) spectra with those of commercially available gallidermin obtained from Alexis Biochemical Corp. (San Diego, CA). The MS analysis was adapted from the method of Kies et al. (29) and performed on a Finningan LCQ Deca LC/ESI-MS in cationic mode. A Bischoff Prontosil Eurobond C18 5.0-μm column was used for the stationary phase. The mobile phase consisted of 0.1% (vol/vol) aqueous trifluoroacetic acid (solvent C) and 100% acetonitrile (solvent B). Separation was performed with a gradient from 90% C/10% B to 45% C/55% B in 14 min at a flow rate of 1 ml/min. The relative abundance of ions in relation to the mass-to-charge (m/z) ratio of ions is indicated on the resulting spectra. The identity of the isolated pregallidermin and the composition of the tryptic digest of pregallidermin were verified by a different ESI-MS procedure. The samples were 20-fold diluted with 50% acetonitrile-0.2% aqueous formic acid and measured in the positive mode on a Q-Tof Ultima API mass spectrometer (Micromass, United Kingdom). The deconvoluted spectra were obtained using MaxEnt1 software and show the relative abundance of ions in relation to the mass of ions.
Bioassays were performed with the gallidermin-sensitive strain Kocuria rhizophila as previously described for epidermin (48) or with S. gallinarum Tü3928. For the latter, a culture was incubated overnight at 37°C, the optical density at 600 nm was adjusted to 0.1, and 100 μl was plated on LB agar plates. Twenty microliters of enriched pregallidermin solutions (0.5 to 8 g/liter in water) or commercial gallidermin solutions (0.13 to 8 g/liter) was pipetted onto sterile paper filters. Water was used as a control. The filters were air dried and then placed upside-down on S. gallinarum Tü3928 plates, which were incubated at 30°C for 24 h. For the tryptic cleavage, a control bioassay with trypsin solutions (from 10 mg/liter to 10 g/liter) in sterile water, pH 6, was performed.
Tricine-SDS-PAGE analysis.
Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed as described previously (31, 48). S. gallinarum ΔgdmP::aphIII and S. gallinarum Tü3928 cultures were grown in 20 ml of production medium 21 in 100-ml Erlenmeyer flasks. Samples were collected after 15 h at a concentration of 10.9 g (dry weight) of cells per liter for S. gallinarum ΔgdmP::aphIII and 9.8 g/liter for S. gallinarum Tü3928. Two microliters of the supernatant of the S. gallinarum ΔgdmP::aphIII culture (containing pregallidermin) as well as 2 μl of the supernatant of the S. gallinarum Tü3928 culture (containing gallidermin), 10 μl of solutions with different concentrations of commercial gallidermin (50 mg/liter and 100 mg/liter), and polypeptide SDS-PAGE molecular mass marker (Bio-Rad, Hercules, CA) were loaded onto acrylamide gels. These were composed of a 4% stacking gel and a 16.5% separating gel. As Coomassie staining is less sensitive than silver staining, and due to the small sizes of gallidermin and pregallidermin, the latter method was chosen. Blotting of the gels to 0.45-μm PROTRAN nitrocellulose membranes was performed in a mini Trans-Blot cell (Bio-Rad, Hercules, CA) at 4°C and 0.8 mA/cm2 for 30 min. The transfer buffer was composed of 25.5 mM Tris, 192 mM glycine, and 0.1% SDS in 20% (vol/vol) methanol. Silver staining of the membranes was performed as described elsewhere (12).
Tryptic cleavage and activation of pregallidermin.
Enriched pregallidermin was dissolved in water to a final concentration of 250 mg/liter, and the pH was adjusted to 6.0. Trypsin (20 U/mg; American Laboratories Inc., Omaha, NE) was added to a final concentration of 50 mg/liter, and proteolytic cleavage was carried out at 25°C for 20 h.
Fermentations.
S. gallinarum Tü3928 and S. gallinarum ΔgdmP::aphIII from frozen stocks were reisolated on B agar plates and incubated for 15 h at 37°C. Three-milliliter precultures of medium 21 were inoculated from isolated colonies and grown for 12 h at 37°C and 190 rpm, from where they were inoculated into 100 ml of medium 21 in 500-ml Erlenmeyer flasks with one baffle under the same conditions. Finally, a 5-liter stirred-tank BioStat A bioreactor (Braun Biotech Int., Melsungen, Germany), equipped with automatic recording of dissolved oxygen tension (Pro 6000; Mettler-Toledo, Greifensee, Switzerland) and control of pH and temperature, containing 3 liter of medium 21, was inoculated with 50 ml of a preculture. The pH was controlled by the addition of 3 M NaOH or 3 M H2SO4. Biomass formation was determined off-line by the measurement of optical density at 600 nm and cellular dry weight determinations in triplicates. Foam was controlled by the addition of silicon antifoam DF 204 (BASF, Ludwigshafen, Germany). The dissolved oxygen tension was maintained above 50% first by adjusting stirrer speed and then by adjusting aeration. A feed of 50% maltose in water was initiated after the end of the batch phase at 6 h when the cells had reached a stationary phase.
Product isolation.
The isolation of gallidermin and pregallidermin was performed with a protocol adapted from Fiedler et al. (12). Gallidermin or pregallidermin was adsorbed from the cell-free fermentation broth onto XAD-7 (4%, vol/vol; Fluka, Buchs, Switzerland) over 90 min. The resin was filtered off, washed with water, and eluted with methanol. The eluate was reduced in vacuo until precipitation occurred. The precipitate was recovered by vacuum membrane filtration (pore size, 0.2 μm) and dissolved in sterile Millipore water before further analysis. HPLC analysis at this point showed a purity of 85% (by peak area at 230 nm) for the isolated gallidermin and pregallidermin. The pregallidermin preparation was further purified by preparative HPLC on a Prontosil PREP 2025 C18 10.0-μm column with a guard column (Bischoff, Leonberg, Germany). The injection volume was 0.8 ml and the column temperature was maintained at 25°C. The mobile phase was composed of 27.5% of 0.1% (vol/vol) aqueous trifluoroacetic acid and 72.5% acetonitrile. The pregallidermin fraction was sampled, and the eluate was reduced in vacuo until precipitation occurred. The 96%-pure (by peak area at 230 nm) pregallidermin precipitate was dried, weighed, and dissolved in sterile Millipore water to a concentration of 10 g/liter. A dilution series of this solution was used to generate a standard curve for quantification of pregallidermin in supernatants by a peak area at 230 nm.
Nucleotide sequence accession number.
The final 12,705-bp sequence of the gallidermin gene cluster was deposited in the GenBank database under accession no. DQ367437.
RESULTS
Sequencing of the gallidermin gene cluster.
The central part of the sequence of the gallidermin synthesis gene cluster (gdmF, gdmT, gdmH, gdmA, and partial sequences of gdmE and gdmB) was already available (16, 41). We determined the DNA sequences of the remaining gaps in the gallidermin gene cluster by iPCR with primers designed on the available gdm sequence or by PCR with primers based on the equivalent epidermin cluster sequences. The DNA sequence thus determined was in perfect agreement with the sequence determined previously by Hille (16), which became available to us only after the sequencing of the gallidermin synthesis gene cluster had been completed. We can confirm that the epidermin and gallidermin cluster is highly homologous on both the DNA and amino acid level, and for further information, we refer the reader to the original work (16).
Construction and verification of an S. gallinarum ΔgdmP::aphIII mutant strain.
The Shine-Dalgarno sequence of gdmQ overlaps with the gdmP stop codon (Fig. 1); thus, gdmQ is translationally coupled to gdmP, and the gdmP promoter may serve as a promoter for transcription of both genes (16). To ensure expression of gdmQ in a gdmP deletion mutant, the Bacillus subtilis aminoglycoside phosphotransferase gene (aphIII) (14), conferring kanamycin resistance, including its constitutive promoter in the suitable orientation, was introduced upstream of gdmQ into gdmP (Fig. 1). An internal gdmP fragment and an aphIII-gdmQ gene fusion were inserted into a derivative of the conditional shuttle vector pBT2, which is based on the temperature-sensitive replicon of pE194 (7). The resulting plasmid, pGV5, was transferred into S. gallinarum Tü3928, and selection for double homologous recombination was performed by incubation at the permissive temperature and selection at the nonpermissive temperature on kanamycin-containing plates. The resulting colonies were screened for double homologous recombination by replication onto chloramphenicol- and kanamycin-containing plates. Three colonies were found to be sensitive to chloramphenicol and resistant to kanamycin and assumed to be deletion mutants. These were further characterized by PCR analysis, and all displayed the expected amplicons. One of the three strains was named S. gallinarum ΔgdmP::aphIII and was further analyzed and characterized in this study.
Characterization of the S. gallinarum ΔgdmP::aphIII mutant strain.
Supernatants of S. gallinarum ΔgdmP::aphIII and S. gallinarum Tü3928 cultures grown in YE4 medium for 18 h were analyzed by HPLC for the presence of gallidermin or any precursor peptides. The supernatants from S. gallinarum Tü3928 cultures contained only gallidermin, whereas no peak corresponding to gallidermin could be identified in supernatants of S. gallinarum ΔgdmP::aphIII. Instead, a novel peak was detected at a slightly shorter retention time. By analyzing the supernatants of S. gallinarum ΔgdmP::aphIII cultures with silver-stained blots of Tricine-SDS-polyacrylamide gels, we detected the presence of a roughly 4-kDa peptide (Fig. 2). This indicated that indeed the mutant no longer produced gallidermin but produced a higher-molecular-mass precursor peptide. Interestingly, at comparable cellular dry weight concentrations, the supernatant of S. gallinarum Tü3928 contained remarkably more proteins than the supernatant of the mutant (Fig. 2); the proteins might accumulate in the supernatant as a result of interaction of gallidermin with the cytoplasmic membrane of the wild-type bacteria.
FIG. 2.
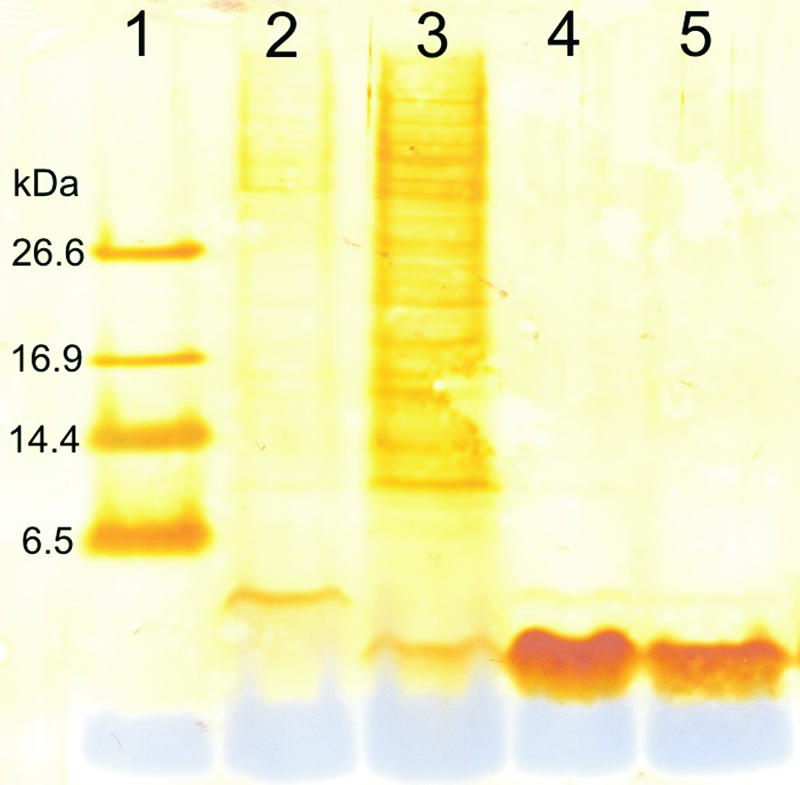
Tricine-SDS-PAGE of 2 μl of the supernatant of an S. gallinarum Tü3928 ΔgdmP::aphIII culture (containing pregallidermin) (lane 2) and 2 μl of the supernatant of an S. gallinarum culture (containing gallidermin) (lane 3). One microgram (lane 4) and 0.5 μg (lane 5) of commercial gallidermin as well as the polypeptide SDS-PAGE molecular mass marker (lane 1) were loaded. Molecular mass markers (kDa) are on the left. After electrophoresis, the separated proteins were blotted onto a nitrocellulose membrane and silver stained.
In order to produce larger amounts of gallidermin and the potential precursor peptide, 3-liter fermentations were carried out with the wild-type and the mutant strain. These were subsequently enriched, and their identity was confirmed by MS. Gallidermin that had accumulated in the supernatant of S. gallinarum Tü3928 resulted in a mass ion peak (M + H)+ at 2,167.2 ± 2 Da, which was identical to the one obtained with commercially available gallidermin and is in excellent agreement with the signal expected from the predicted 21-amino-acid peptide (2,165 Da) (Fig. 3A and B). The MS spectrum of the compound isolated from the supernatant of S. gallinarum ΔgdmP::aphIII revealed the presence of two peptides of 3,407.4 Da and 3,620.5 Da (Fig. 3C), corresponding well to a 33- and a 36-amino-acid-long pregallidermin fragment (Fig. 3C). The expected form, consisting of the entire GdmA peptide, would have resulted in a mass peak of 5,608 kDa, but was surprisingly not detected. Moreover, remarkably different proportions of the two peptides were present in the supernatant, with the larger one reaching only 10% of the MS spectrum intensity (Fig. 3C). Hereinafter, we will apply the name “pregallidermin” to the mixture of the two precursor peptides obtained.
FIG. 3.


Identity of the peptides produced by S. gallinarum Tü3928 and S. gallinarum ΔgdmP::aphIII. (A and B) LC/ESI-MS spectrum of commercial gallidermin (A) and LC/ESI-MS spectrum of gallidermin (B) produced in this study. The spectra show the relative abundance of ions in relation to the mass-to-charge (m/z) ratio of ions. The detection range was limited to below 2,000, and thus, the double-charged molecular ion is detected. (C) The graph shows the spectrum of nano-ESI-MS of pregallidermin variants produced by S. gallinarum ΔgdmP::aphIII. The two peaks correspond to 33-amino-acid and 36-amino-acid pregallidermin fragments. This and the spectrum in graph D show the relative abundance of ions in relation to the mass of ions. The sequences are the amino acid sequences of the 30-amino-acid-long pregallidermin leader peptide (as expected from the gdmA sequence) (C1), and the 14- and 12-amino-acid-long truncated peptides of pregallidermin variants from the culture supernatant of S. gallinarum ΔgdmP::aphIII (C2 and C3). (D) Nano-ESI-MS of gallidermin generated by tryptic cleavage of pregallidermin. The peak at 2,165 corresponds to that of commercial gallidermin. The spectrum shows the relative abundance of ions in relation to the mass of ions.
Characterization of the activity of pregallidermin.
In order to evaluate the antimicrobial activity of the enriched compounds, we investigated their effects on the indicator strain Kocuria rhizophila ATCC 9341 and on S. gallinarum Tü3928. While supernatants of S. gallinarum Tü3928 cultures displayed the expected activity against K. rhizophila, supernatants from cultures of S. gallinarum ΔgdmP::aphIII showed no antimicrobial activity against it (results not shown). By systematically increasing the concentration of the purified peptides, it was shown that solutions with as much as 8 g/liter pregallidermin did not lead to the formation of inhibitory zones (Fig. 4A). In contrast, inhibitory zones were observed from spotting with solutions of 0.25 g/liter of gallidermin (Fig. 4B).
FIG. 4.

Results of bioassays to establish the toxicities and specific activities of different peptide preparations. Twenty microliters of solutions with different concentrations of pregallidermin/gallidermin were spotted on filters and placed on agar plates. The results of toxicity tests of enriched pregallidermin (A) and commercial gallidermin (B) against S. gallinarum Tü3928 are shown. K, control (sterile water). (C) Bioactivities of commercial gallidermin (1) and gallidermin produced by proteolytic cleavage of pregallidermin with trypsin (2) against the indicator strain K. rhizophila. K, control (sterile water for gallidermin and trypsin solution for pregallidermin). (D) Bioactivities of trypsin at different concentrations against the indicator strain K. rhizophila.
Proteolytic cleavage with trypsin.
In order to confirm that pregallidermin enriched from S. gallinarum ΔgdmP::aphIII supernatants could be transformed into active gallidermin and that the absence of a self-toxicity was due only to the presence of the truncated leader peptide, we investigated proteolytic cleavage of pregallidermin. Analysis of the cleavage sites of known proteases and of the pregallidermin sequence indicated that trypsin should be a suitable candidate for the biotransformation. Reactions were performed in which 75 nmol of pregallidermin was treated with 1 U of trypsin. After 20 h, total conversion of pregallidermin had taken place, since the pregallidermin peak could no longer be detected by HPLC analysis. Instead, a novel peak was detected at a retention time identical to that of gallidermin. The reaction mixture was analyzed by ESI-MS, and a molecular ion peak corresponding exactly to the expected 2,165-Da mass of gallidermin was obtained (Fig. 3D). To further confirm these results, we used equivalent amounts of gallidermin treated with trypsin and of commercially available gallidermin in bioassays with K. rhizophila. Indeed, the gallidermin generated by trypsin treatment showed a specific antimicrobial activity similar to that of the commercially available gallidermin, while the control solution containing trypsin but no pregallidermin did not lead to the formation of an inhibition zone (Fig. 4C). Furthermore, trypsin solutions from 10 mg/liter to 10 g/liter were used in a control bioassay, to exclude bioactivity of trypsin (Fig. 4D).
Effect on production titer.
The results discussed above indicate that S. gallinarum ΔgdmP::aphIII indeed produces pregallidermin that, within the range of our analysis, is not toxic to S. gallinarum Tü3928 strains. A truncated form of the leader sequence confers inactivity to the peptides, even though their gallidermin moiety appears to be fully mature. In view of our goal of overproducing gallidermin, we investigated whether the elimination of gdmP and the resulting switch from the production of a toxic peptide to a nontoxic precursor would be sufficient to increase the concentration of the product. In order to determine final cultivation titers, we carried out 3-liter fed-batch fermentations with the wild-type and the mutant S. gallinarum strains. Gallidermin accumulated to a maximum of 280 mg/liter in S. gallinarum Tü3928 fermentations (Fig. 5A), whereas pregallidermin accumulated to 630 mg/liter in S. gallinarum ΔgdmP::aphIII supernatants (Fig. 5B), corresponding to a gallidermin concentration of 420 mg/liter. Therefore, an increase of 50% has already been achieved in these initial, nonoptimized fermentations.
FIG. 5.

Results from 3-liter fed-batch fermentations performed to overproduce gallidermin (gdm) and pregallidermin (pregdm). Wild-type S. gallinarum Tü3928 fermentations led to a maximal yield of gallidermin of 280 mg/liter (A) and 3-liter fed-batch fermentations with the mutant S. gallinarum ΔgdmP::aphIII strain led to a maximal yield of pregallidermin of 630 mg/liter (B), which corresponds to a gallidermin concentration of 420 mg/liter. After 17 h, 15 g (NH4)2SO4 was added (X). CDW, cellular dry weight.
DISCUSSION
Overproduction of lantibiotics has so far met with only very limited success, which might be attributed to the toxicity of the molecules for its own producer. Fundamentally, there are three possible strategies to overcome this bottleneck and increase the amount of active molecule produced per reactor volume: (i) rapid removal of the active molecule from the cells (by selective in situ product removal) (20, 57, 58) or removal of the supernatant from the reactor (3, 54) (ii) the isolation of producer strains that are insensitive to elevated concentrations of the active molecule (57), and (iii) the production of a nontoxic variant of the molecule that can be easily transformed into the active form after its production. Although the first method has led in some cases to increases in volumetric productivities (3, 58), it did not lead to increases in product concentration, which would be advantageous from the perspective of product purification. No indications of success for the second method are available from the literature. This might be traced back to the multiple modes of action of type A lantibiotics on their gram-positive producers. Consequently, we investigated the feasibility of the production of a nontoxic variant.
An easy strategy towards this aim might consist of the elimination of the final posttranslational modification step, i.e., extracellular proteolytic cleavage of the leader sequence from the type A lantibiotic precursor peptide. This approach is supported by previous studies on nisin showing that N-terminal extensions are detrimental to the activity of the lantibiotic. The modified but uncleaved nisin Z precursor peptide has been found to be completely inactive against Micrococcus flavus (59), and a precursor in which the leader peptide of nisin was exchanged for the subtilin leader segment did not show any activity against lipid II-containing liposomes (61).
Consequently, we investigated this rationale for gallidermin, which has been shown to exhibit a particularly promising antimicrobial profile (10, 40, 62). Specifically, S. gallinarum ΔgdmP::aphIII was generated by insertional disruption of gdmP, which encodes a subtilisin-like serine protease responsible for gallidermin activation. Gallidermin could not be detected by HPLC analysis of culture supernatants of the engineered strain, while a new peak was observed, implying the production of pregallidermin. Two pregallidermin variants were identified, which each had a higher molecular mass than gallidermin (Fig. 2) and were shown to be truncated versions of the mature pregallidermin (Fig. 3). Their molecular masses corresponded to those of prepeptides having, respectively, a 12- and a 14-amino-acid-long leader peptide. As no alternative start codons are present in the gdmA sequence, these truncated prepeptides are likely to be generated by the action of other proteases. The two variants were present in different proportions in supernatants, with the larger one reaching only 10% of the spectrum intensity. These results were in agreement with those of a previous study reporting that two preepidermin fragments were detected in supernatants of an S. epidermidis ΔepiP mutant strain (29). Furthermore, the strain producing truncated pregallidermins did not show self-toxicity even at higher concentrations than the ones expected in overexpression experiments (Fig. 4).
Still, the possibility remained that the produced prepeptides did not carry a mature gallidermin moiety, but a nonfunctional variant of identical mass. However, hydrolysis of pregallidermin by tryptic cleavage resulted in fully active gallidermin of exactly the expected mass and, within the accuracy of a bioassay, identical specific activity to commercially available gallidermin (Fig. 3 and 4). This indicates that the truncated leader sequence is sufficient to prevent gallidermin toxicity. Furthermore, this suggests a simple processing strategy to efficiently obtain gallidermin from pregallidermin by biotransformation.
Finally, we looked for a first indication of the beneficial effects that elimination of product toxicity might have on gallidermin production. For this, we applied the same protocol as for gallidermin production from S. gallinarum Tü3928 to the fermentation of the mutant strain. Even in these nonoptimized settings, the mutant strain produced gallidermin to a 50% higher concentration (in mol/liter) than the wild-type strain. In the case of the wild-type strain, gallidermin concentrations increased over the first 10 h of the fermentation and then leveled off. Biomass concentration increased afterwards only slightly and at significantly reduced specific growth rates, suggesting adverse effects of the accumulating gallidermin. In contrast, production of pregallidermin by the mutant continued for another 10 h and leveled out only as the biomass stopped increasing, indicating a medium limitation rather than self-toxicity, as pregallidermin has been shown not to be toxic at these concentrations. This resulted in a higher final pregallidermin concentration, which was also—in molar terms—higher than the gallidermin concentration achieved in the first fermentation.
In summary, the fundamentals of a novel overproduction strategy for type A lantibiotics based on the elimination of their self-toxicity were established in this study. Exemplified by the overproduction of pregallidermin as a nontoxic gallidermin variant, this strategy might be of much broader usefulness, as type A lantibiotics are synthesized through almost identical production pathways. In fact, they are all ribosomally synthesized as inactive prepeptides that are posttranslationally modified, then exported and proteolytically activated extracellularly. Current efforts in our laboratory are directed to the optimization of the fermentation process and of the production strain to further enhance the production titer. We reason that the production strategy illustrated in this study can be the foundation of a process suitable for an industrial-scale production of pharmaceutical-grade type A lantibiotics, including the widely used nisin, which could play a significant role in the battle against antibiotic-resistant pathogens such as methicillin- and vancomycin-resistant S. aureus.
Acknowledgments
This work was supported by Anbics Management GmbH and a grant from the Swiss Commission for Technology and Innovation to G. Valsesia and G. Medaglia.
We are indebted to A. Dumoulin, M. Bischoff, B. Berger-Bächi, D. Hilvert, and P. Hunziker for their invaluable help with Staphylococcus molecular biology and MS analyses.
Footnotes
Published ahead of print on 28 December 2006.
REFERENCES
- 1.Allgaier, H., G. Jung, R. G. Werner, U. Schneider, and H. Zähner. 1985. Elucidation of the structure of epidermin, a ribosomally synthesized, tetracyclic heterodetic polypeptide antibiotic. Angew. Chem. Int. Ed. 24:1051-1053. [Google Scholar]
- 2.Augustin, J., R. Rosenstein, B. Wieland, U. Schneider, N. Schnell, G. Engelke, K. D. Entian, and F. Götz. 1992. Genetic analysis of epidermin biosynthetic genes and epidermin-negative mutants of Staphylococcus epidermidis. Eur. J. Biochem. 204:1149-1154. [DOI] [PubMed] [Google Scholar]
- 3.Bhugaloo-Vial, P., W. Grajek, X. Dousset, and P. Boyaval. 1997. Continuous bacteriocin production with high cell density bioreactors. Enzyme Microb. Technol. 21:450-457. [Google Scholar]
- 4.Bierbaum, G., F. Götz, A. Peschel, T. Kupke, M. van der Kamp, and H. G. Sahl. 1996. The biosynthesis of the lantibiotics epidermin, gallidermin, Pep5 and epilancin K7. Antonie Leeuwenhoek 69:119-127. [DOI] [PubMed] [Google Scholar]
- 5.Bonelli, R. R., T. Schneider, H. G. Sahl, and I. Wiedemann. 2006. Insights into in vivo activities of lantibiotics from gallidermin and epidermin mode-of-action studies. Antimicrob. Agents Chemother. 50:1449-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brotz, H., M. Josten, I. Wiedemann, U. Schneider, F. Götz, G. Bierbaum, and H. G. Sahl. 1998. Role of lipid-bound peptidoglycan precursors in the formation of pores by nisin, epidermin and other lantibiotics. Mol. Microbiol. 30:317-327. [DOI] [PubMed] [Google Scholar]
- 7.Brückner, R. 1997. Gene replacement in Staphylococcus carnosus and Staphylococcus xylosus. FEMS Microbiol. Lett. 151:1-8. [DOI] [PubMed] [Google Scholar]
- 8.Chang, S., D. M. Sievert, J. C. Hageman, M. L. Boulton, F. C. Tenover, F. P. Downes, S. Shah, J. T. Rudrik, G. R. Pupp, W. J. Brown, D. Cardo, and S. K. Fridkin. 2003. Brief report: infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N. Engl. J. Med. 348:1342-1347. [DOI] [PubMed] [Google Scholar]
- 9.Cockayne, A., P. J. Hill, N. B. Powell, K. Bishop, C. Sims, and P. Williams. 1998. Molecular cloning of a 32-kilodalton lipoprotein component of a novel iron-regulated Staphylococcus epidermidis ABC transporter. Infect. Immun. 66:3767-3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dannesen, P. J. W., M. J. M. Bonten, and R. A. Weinstein. 1998. Multiresistant bacteria as a hospital epidemic problem. Ann. Med. 30:176-185. [DOI] [PubMed] [Google Scholar]
- 11.Devos, W. M., O. P. Kuipers, J. R. van der Meer, and R. J. Siezen. 1995. Maturation pathway of nisin and other lantibiotics: posttranslationally modified antimicrobial peptides exported by gram-positive bacteria. Mol. Microbiol. 17:427-437. [DOI] [PubMed] [Google Scholar]
- 12.Fiedler, H. P., T. Hörner, and H. Decker. 1988. Purification of the hydrophilic antibiotics epidermin, gallidermin and nikkomycin Z by preparative reversed-phase HPLC. Chromatographia 26:215-220. [Google Scholar]
- 13.Goldstein, B. P. December1998. A method using nisin or other lanthocin for the treatment of diarrheal disease and for eliminating particular bacterial populations from the colon. U.S. patent WO9,856,398.
- 14.Guerout-Fleury, A. M., K. Shazand, N. Frandsen, and P. Stragier. 1995. Antibiotic-resistance cassettes for Bacillus subtilis. Gene 167:335-336. [DOI] [PubMed] [Google Scholar]
- 15.Herrero, M. V., V. de Lorenzo, and K. N. Timmis. 1990. Transposon vectors containing nonantibiotic selection markers for cloning and stable chromosomal insertion of foreign DNA in gram-negative bacteria. J. Bacteriol. 172:6557-6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hille, M. 2002. Untersuchungen zur Biosynthese der Lantibiotika Gallidermin and Epidermin. Ph.D. thesis. Eberhard-Karls-Universität, Tübingen, Germany.
- 17.Hille, M., S. Kies, F. Götz, and A. Peschel. 2001. Dual role of GdmH in producer immunity and secretion of the staphylococcal lantibiotics gallidermin and epidermin. Appl. Environ. Microbiol. 67:1380-1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho, S. N., H. D. Hunt, R. M. Horton, J. K. Pullen, and L. R. Pease. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51-59. [DOI] [PubMed] [Google Scholar]
- 19.Hoffmann, A., U. Pag, I. Wiedemann, and H. G. Sahl. 2002. Combination of antibiotic mechanisms in lantibiotics. Farmaco 57:685-691. [DOI] [PubMed] [Google Scholar]
- 20.Hörner, T., V. Ungermann, H. Zähner, H. P. Fiedler, R. Utz, R. Kellner, and G. Jung. 1990. Comparative studies on the fermentative production of lantibiotics by staphylococci. Appl. Microbiol. Biotechnol. 32:511-517. [DOI] [PubMed] [Google Scholar]
- 21.Hörner, T., H. Zähner, R. Kellner, and G. Jung. 1989. Fermentation and isolation of epidermin, a lanthionine containing polypeptide antibiotic from Staphylococcus epidermidis. Appl. Microbiol. Biotechnol. 30:219-225. [Google Scholar]
- 22.Horton, R. M., Z. L. Cai, S. N. Ho, and L. R. Pease. 1990. Gene-splicing by overlap extension: tailor-made genes using the polymerase chain reaction. BioTechniques 8:528-536. [PubMed] [Google Scholar]
- 23.Horton, R. M., H. D. Hunt, S. N. Ho, J. K. Pullen, and L. R. Pease. 1989. Engineering hybrid genes without the use of restriction enzymes: gene-splicing by overlap extension. Gene 77:61-68. [DOI] [PubMed] [Google Scholar]
- 24.Jack, R., F. Götz, and G. Jung. 1997. Lantibiotics, p. 323-368. In H. Kleinkauf and H. von Dören (ed.), Products of secondary metabolism, 2nd ed., vol. 7. Wiley-VCH, Weinheim, Germany. [Google Scholar]
- 25.Kellner, R., G. Jung, T. Hörner, H. Zähner, N. Schnell, K. D. Entian, and F. Götz. 1988. Gallidermin: a new lanthionine-containing polypeptide antibiotic. Eur. J. Biochem. 177:53-59. [DOI] [PubMed] [Google Scholar]
- 26.Kempf, M., U. Theobald, and H. P. Fiedler. 1999. Correlation between the consumption of amino acids and the production of the antibiotic gallidermin by Staphylococcus gallinarum. Biotechnol. Lett. 21:959-963. [Google Scholar]
- 27.Kempf, M., U. Theobald, and H. P. Fiedler. 1999. Economic improvement of the fermentative production of gallidermin by Staphylococcus gallinarum. Biotechnol. Lett. 21:663-667. [Google Scholar]
- 28.Kies, S. 2001. Regulation von Virulenzfaktoren und der Biosynthese des Lantibiotikums Epidermin in Staphylococcus epidermidis. Ph.D. thesis. Eberhard-Karls-Universität, Tübingen, Germany.
- 29.Kies, S., C. Vuong, M. Hille, A. Peschel, C. Meyer, F. Götz, and M. Otto. 2003. Control of antimicrobial peptide synthesis by the agr quorum sensing system in Staphylococcus epidermidis: activity of the lantibiotic epidermin is regulated at the level of precursor peptide processing. Peptides 24:329-338. [DOI] [PubMed] [Google Scholar]
- 30.Kovacs, G., J. Burghardt, S. Pradella, P. Schumann, E. Stackebrandt, and K. Marialgeti. 1999. Kocuria palustris sp. nov. and Kocuria rhizophila sp. nov., isolated from rhizoplane of the narrow-leaved cattail (Typha angustifolia). Int. J. Syst. Bacteriol. 49:167-173. [DOI] [PubMed] [Google Scholar]
- 31.Kovarik, A., K. Hlubinova, A. Vrebenska, and J. Prachar. 1986. An improved colloidal silver staining method of protein blots on nitrocellulose membranes. Folia Biol. 33:253-257. [PubMed] [Google Scholar]
- 32.Kreiswirth, B. N., S. Löfdahl, M. J. Betley, M. O'Reilly, P. M. Schlievert, M. S. Bergdoll, and R. P. Novick. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709-712. [DOI] [PubMed] [Google Scholar]
- 33.Kruszewska, D., H. G. Sahl, G. Bierbaum, U. Pag, S. O. Hynes, and A. Ljungh. 2004. Mersacidin eradicates methicillin-resistant Staphylococcus aureus (MRSA) in a mouse rhinitis model. J. Antimicrob. Chemother. 54:648-653. [DOI] [PubMed] [Google Scholar]
- 34.Kuipers, O. P., G. Bierbaum, B. Ottenwalder, H. M. Dodd, N. Horn, J. Metzger, T. Kupke, V. Gnau, R. Bongers, P. van den Bogaard, H. Kosters, H. S. Rollema, W. M. de Vos, R. J. Siezen, G. Jung, F. Götz, H. G. Sahl, and M. J. Gasson. 1996. Protein engineering of lantibiotics. Antonie Leeuwenhoek 69:161-169. [DOI] [PubMed] [Google Scholar]
- 35.Leeb, M. 2004. A shot in the arm. Nature 431:892-893. [DOI] [PubMed] [Google Scholar]
- 36.Madsen, S. M., H. C. Beck, P. Ravn, A. Vrang, A. M. Hansen, and H. Israelsen. 2002. Cloning and inactivation of a branched-chain amino acid aminotransferase gene from Staphylococcus carnosus and characterization of the enzyme. Appl. Environ. Microbiol. 68:4007-4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nathan, C. 2004. Antibiotics at the crossroads. Nature 431:899-902. [DOI] [PubMed] [Google Scholar]
- 38.Ottenwalder, B., T. Kupke, S. Brecht, V. Gnau, J. Metzger, G. Jung, and F. Götz. 1995. Isolation and characterization of genetically engineered gallidermin and epidermin analogs. Appl. Environ. Microbiol. 61:3894-3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Otto, M., A. Peschel, and F. Götz. 1998. Producer self-protection against the lantibiotic epidermin by the ABC transporter EpiFEG of Staphylococcus epidermidis Tu3298. FEMS Microbiol. Lett. 166:203-211. [DOI] [PubMed] [Google Scholar]
- 40.Peel, J. E., and B. Suri. January1998. Use of gallidermin and epidermin for preventing or treating mastitis and for reducing withholding time of milk. U.S. patent 5,710,124-A.
- 41.Peschel, A. 1995. Biosynthese des Lantibiotikums Epidermin: Untersuchungen zur Regulation, Immunität und Sekretion. Ph.D. thesis. Eberhard-Karls-Universität, Tübingen, Germany.
- 42.Peschel, A., J. Augustin, T. Kupke, S. Stevanovic, and F. Götz. 1993. Regulation of epidermin biosynthetic genes by EpiQ. Mol. Microbiol. 9:31-39. [DOI] [PubMed] [Google Scholar]
- 43.Peschel, A., and F. Götz. 1996. Analysis of the Staphylococcus epidermidis genes epiF, -E, and -G involved in epidermin immunity. J. Bacteriol. 178:531-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peschel, A., N. Schnell, M. Hille, K. D. Entian, and F. Götz. 1997. Secretion of the lantibiotics epidermin and gallidermin: sequence analysis of the genes gdmT and gdmH, their influence on epidermin production and their regulation by EpiQ. Mol. Gen. Genet. 254:312-318. [DOI] [PubMed] [Google Scholar]
- 45.Sahl, H. G., and G. Bierbaum. 1998. Lantibiotics: biosynthesis and biological activities of uniquely modified peptides from gram-positive bacteria. Annu. Rev. Microbiol. 52:41-79. [DOI] [PubMed] [Google Scholar]
- 46.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 47.Saris, P. E. J., T. Immonen, M. Reis, and H. S. Sahl. 1996. Immunity to lantibiotics. Antonie Leeuwenhoek 69:151-159. [DOI] [PubMed] [Google Scholar]
- 48.Schägger, H., and G. von Jagow. 1987. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166:368-379. [DOI] [PubMed] [Google Scholar]
- 49.Schenk, S., and R. A. Laddaga. 1992. Improved method for electroporation of Staphylococcus aureus. FEMS Microbiol. Lett. 94:133-138. [DOI] [PubMed] [Google Scholar]
- 50.Schnell, N., G. Engelke, J. Augustin, R. Rosenstein, V. Ungermann, F. Götz, and K. D. Entian. 1992. Analysis of genes involved in the biosynthesis of the lantibiotic epidermin. Eur. J. Biochem. 204:57-68. [DOI] [PubMed] [Google Scholar]
- 51.Schnell, N., K. D. Entian, F. Götz, T. Horner, R. Kellner, and G. Jung. 1989. Structural gene isolation and prepeptide sequence of gallidermin, a new lanthionine containing antibiotic. FEMS Microbiol. Lett. 58:263-267. [DOI] [PubMed] [Google Scholar]
- 52.Schnell, N., K. D. Entian, U. Schneider, F. Götz, H. Zähner, R. Kellner, and G. Jung. 1988. Prepeptide sequence of epidermin, a ribosomally synthesized antibiotic with 4 sulfide-rings. Nature 333:276-278. [DOI] [PubMed] [Google Scholar]
- 53.Shams-Eldin, H., F. Debierre-Grockiego, and R. T. Schwarz. 2003. Chased PCR: a modified inverse PCR technique to characterize flanking regions of AT-rich DNA fragments. J. Mol. Microbiol. Biotechnol. 6:1-5. [DOI] [PubMed] [Google Scholar]
- 54.Taniguchi, M., K. Hoshino, H. Urasaki, and M. Fujii. 1994. Continuous production of an antibiotic polypeptide (nisin) by Lactococcus lactis using a bioreactor coupled to a microfiltration module. J. Ferment. Bioeng. 77:704-708. [Google Scholar]
- 55.Theobald, U. 2001. Fermentation of lantibiotics epidermin and gallidemin, p. 93-101. In V. Braun and F. Götz (ed.), Microbial fundamentals of biotechnology. Wiley-VCH, Bonn, Germany.
- 56.Theobald, U., and M. Kempf. 1998. A novel tool for medium optimization and characterization in the early stages of a metabolite production process. Biotechnol. Tech. 12:893-897. [Google Scholar]
- 57.Ungermann, V. 1992. Untersuchungen zur Produktbildung und Scale-Up der Produktion von Gallidermin einem lanthioninhaltigen Peptidantibiotikum aus Staphylococcus gallinarum Tü3928. Ph.D. thesis. Eberhard-Karls-Universität, Tübingen, Germany.
- 58.Ungermann, V., K. Goeke, H.-P. Fiedler, and H. Zähner. 1991. Optimization of fermentation and purification of gallidermin and epidermin, p. 410-420. In G. Jung and H.-G. Sahl (ed.), Nisin and novel lantibiotics. ESCOM Science Publishers, Leiden, The Netherlands.
- 59.van der Meer, J. R., J. Polman, M. M. Beerthuyzen, R. J. Siezen, O. P. Kuipers, and W. M. Devos. 1993. Characterization of the Lactococcus lactis nisin A operon genes nisP, encoding a subtilisin-like serine protease involved in precursor processing, and nisR, encoding a regulatory protein involved in nisin biosynthesis. J. Bacteriol. 175:2578-2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Heusden, H. E., B. de Kruijff, and E. Breukink. 2002. Lipid II induces a transmembrane orientation of the pore-forming peptide lantibiotic nisin. Biochemistry 41:12171-12178. [DOI] [PubMed] [Google Scholar]
- 61.Wiedemann, I., E. Breukink, C. van Kraij, O. P. Kuipers, G. Bierbaum, B. de Kruijff, and H. G. Sahl. 2001. Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J. Biol. Chem. 276:1772-1779. [DOI] [PubMed] [Google Scholar]
- 62.Zähner, H., F. Götz, T. Hörner, R. G. Werner, H. Allgaier, G. Jung, and R. Kellner. 1998. New peptide antibiotic gallidermin is produced by Staphylococcus gallinarum and used esp. for treating eczema, impetigo, cellulitis and acne. U.S. patent 5,843,709-A.