

Abstract
Human nucleoside diphosphate (NDP) kinase A is a ‘house-keeping’ enzyme essential for the synthesis of nonadenine nucleoside (and deoxynucleoside) 5′-triphosphate. It is involved in complex cellular regulatory functions including the control of metastatic tumour dissemination. The mutation S120G has been identified in high-grade neuroblastomas. We have shown previously that this mutant has a folding defect: the urea-denatured protein could not refold in vitro. A molten globule folding intermediate accumulated, whereas the wild-type protein folded and associated into active hexamers. In the present study, we report that autophosphorylation of the protein corrected the folding defect. The phosphorylated S120G mutant NDP kinase, either autophosphorylated with ATP as donor, or chemically prosphorylated by phosphoramidate, refolded and associated quickly with high yield. Nucleotide binding had only a small effect. ADP and the non-hydrolysable ATP analogue 5′-adenyly-limido-diphosphate did not promote refolding. ATP-promoted refolding was strongly inhibited by ADP, indicating protein dephosphorylation. Our findings explain why the mutant enzyme is produced in mammalian cells and in Escherichia coli in a soluble form and is active, despite the folding defect of the S120G mutant observed in vitro. We generated an inactive mutant kinase by replacing the essential active-site histidine residue at position 118 with an asparagine residue, which abrogates the autophosphorylation. The double mutant H118N/S120G was expressed in inclusion bodies in E. coli. Its renaturation stops at a folding intermediate and cannot be reactivated by ATP in vitro. The transfection of cells with this double mutant might be a good model to study the cellular effects of folding intermediates.
Keywords: folding intermediate, histidine phosphorylation, molten globule, neuroblastoma, nucleoside diphosphate (NDP) kinase (nm23)
Abbreviations: p[NH]ppA, adenosine 5′-[β,γ-imido]triphosphate; ANS, 8-anilino-1-naphthalene sulfonate; BisANS, 4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid; DTT, dithiothreitol; I, folding intermediate; K-pn, Killer of prune; M, folded monomer; NDP, nucleoside diphosphate; Nm23, non-metastatic clone 23
INTRODUCTION
NDP (nucleoside diphosphate) kinases catalyse the reversible transfer of the γ-phosphoryl group of nucleoside triphosphates to NDPs [1,2]. The two-step reaction has a covalent intermediate that is transiently phosphorylated on His118 in human NDP kinase A [3,4]:
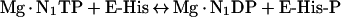 |
(1a) |
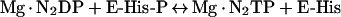 |
(1b) |
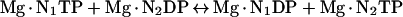 |
(1) |
Where E is the enzyme and P is the phosphorylated amino acid, in this case His118.
NDP kinases have a single nucleotide-binding site per subunit, where the NTP and the NDP bind sequentially during the catalytic cycle (reactions 1a and 1b).
In addition to their catalytic function, eukaryotic NDP kinases are involved in complex regulatory functions, some of which are not associated with kinase activity. Drosophila NDP kinase, the product of the awd gene, is essential for larval development [5]. Human NDP kinase B (also known as Nm23-H2, where Nm23 stands for non-metastatic clone 23) is a transcription factor for the proto-oncogene c-myc [6] and has nuclease activity [7,8]. Crosslinking experiments demonstrate that both NDP kinase A (also known as Nm23-H1) and B bind to DNA in vivo [9]. NDP kinase A is an anti-metastatic protein [10]. Low expression is associated with high metastatic potential in tumours and cells in culture. Overexpression of the protein in highly metastatic cell lines decreases their metastatic potential [11]. The mechanism of NDP kinase A action in metastasis progression is still not well understood. NDP kinase A was previously reported to act in a co-ordinated manner with the human equivalent of the Drosophila prune protein in the control of cancer metastasis [12].
The S120G mutation in NDP kinase A has been found in several high-grade neuroblastomas, but not in low-grade tumours or in control tissues [13]. This suggests that the presence of the mutant protein may be associated with tumour aggressiveness. Indeed the mutant appears to promote neuroblastoma more effectively than the wild-type protein when expressed in an NB69-derived human neuroblastoma cell line [14]. If the S120G mutation is transfected into human breast carcinoma cell lines, the resulting mutant NDP kinase no longer inhibits motility, and if the mutation is transfected into prostate cancer cells the mutant NDP kinase no longer inhibits colonization and invasion [15]. The S120G mutant has impaired affinity for the human prune protein [16]. Wild-type NDP kinase A reduces the acute desensitization of muscarinic K+ channels, whereas the S120G mutant kinase suppresses this effect and even accelerates desensitization [17].
Despite the proximity of Ser120 to the active site histidine residue and its conservation in all known NDP kinases, the S120G mutation only slightly decreases the kinase activity [18,19]. It decreases the stability of the protein to denaturation [18,20]. A folding defect is the most substantial effect of the mutation: the urea-denatured S120G mutant cannot refold in vitro [18]. Instead it accumulates as a ‘molten globule’ folding intermediate, as assessed by various biochemical and biophysical techniques. The wild-type enzyme refolds under the same conditions and associates to form active hexamers.
To understand the structural basis of the folding defect of the mutant S120G, its X-ray structure has been solved in our laboratory [21]. The mutation does not affect subunit conformation or subunit assembly. The mutant and wild-type hexamers are identical within the experimental error (root mean square deviation on Cα positions of 0.3 Å; 1 Å = 0.1 nm). Thus incorporation of mutant subunits into hexamers probably does not affect other subunits. Heterohexamer formation is a common property of hexameric NDP kinases [22]. The S120G mutation is probably not dominant negative in the sense that S120G mutant subunits will not change the function of other subunits in the hexamer. S120G mutant subunits and H118N mutant subunits have been shown to associate to form hexamers in vitro and are active [22a]. However, the S120G mutation may appear to be ‘dominant’ if a folding intermediate or lower order oligomers are present in cells with the S120G mutant NDP kinase, but not in cells with wild-type NDP kinase. A large amount of dimeric S120G mutant protein has been found in vitro by crosslinking [15,20]. Immunoprecipitation experiments showed that the S120G mutant protein interacts with a 28 kDa protein, however, this interaction did not occur with the wild-type protein [20].
A similar natural mutation is the K-pn (Killer of prune) mutation in the Drosophila NDP kinase. The replacement of the proline residue at position 97 by a serine residue results in a dominant, conditional and lethal phenotype [23]. The nature of the particular molecular species that confers the dominant character is still unknown, but biochemical evidence suggests that it is the folded monomer [24].
The S120G mutant NDP kinase A folds and associates into an active hexameric protein in mammalian cells and Escherichia coli, but cannot do this in vitro. We discovered that phosphorylation of the active site histidine residue, either enzymatically or chemically with phosphoramidate, corrects the folding defect of the S120G mutant. In agreement with the proposed mechanism, the folding defect persists if the active site histidine residue, which is phosphorylated during the catalytic cycle, is replaced by an asparagine residue in the S120G protein.
EXPERIMENTAL
Mutagenesis and protein purification
S120G and H118N mutant constructs were generated using the Transformer™ site-directed mutagenesis kit (Clonetech). The mutagenesis was performed on the pET21b (Novagen) construct containing NDP kinase A. The mutagenic primers designed to produce the point mutations were: 5′-CATTATACATGGC-GGTGATTCTGTGG-3′ and 5′-AGGAACATTATAAATGGCAGTGATTCT-3′ for S120G and H118N respectively (the altered nucleotides are in bold). The H118N/S120G double mutant in the pET21-nm23 plasmid was made with the QuikChange site-directed mutagenesis kit (Stratagene) following the manufacturer's instructions. Mutations were confirmed by restriction enzyme digestion analysis and nucleotide sequencing.
All proteins were expressed from a pET21 vector backbone. Wild-type protein and S120G and H118N mutants were purified at 4 °C and analysed as described previously [18], with the following modifications: the DNase-treated bacterial extract was applied to a QAE-Sepharose column equilibrated with 50 mM Tris/HCl buffer (pH 8.0) containing 2 mM DTT (dithiothreitol). The enzyme was eluted by a linear gradient of 0–1 M NaCl in the same buffer. Fractions containing the H118N protein were identified by electrophoresis and by measurement of activity of the Ser120 and wild-type proteins. The pH of the fractions containing the enzyme was corrected to 7.0, and the samples were diluted 3-fold with distilled water to decrease the ionic strength. The enzyme preparations were applied on to a Blue-Sepharose column. The column was washed with 20 mM sodium phosphate buffer (pH 7.0) containing 2 mM DTT. The enzymes, including the inactive H118N mutant, were eluted by 1 mM ATP in the same buffer. The enzymes were precipitated by dialysis against a saturated solution of ammonium sulfate, recovered by centrifugation at 6000 g for 10 mins at 4 °C and further purified by size-exclusion chromatography on a Sephacryl S-200 column equilibrated with 0.2 M sodium phosphate buffer (pH 7.0) containing 1 mM DTT. This step eliminated aggregated and dissociated proteins and ATP. The enzymes were essentially pure, as determined by SDS/PAGE. The final enzyme preparation was stored at −80 °C, or at −20 °C in 50% (v/v) glycerol, 20 mM sodium phosphate buffer (pH 7.0) and 2 mM DTT. Long-term storage results in the modification of physical properties, although the S120G mutant is still active. We systematically discarded preparations that were dissociated, bound BisANS (4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid) and did not display a co-operative unfolding curve.
The H118N/S120G mutant was produced in an insoluble form. Inclusion bodies were washed three times in Tris/HCl (pH 8.0) containing 2 mM DTT, then solubilized in 6 M GdmCl containing 50 mM Tris/HCl (pH 8.0) and 20 mM DTT, and purified by size-exclusion chromatography on a Sepharose CL-6B column equilibrated with 50 mM Tris/HCl (pH 8.0), 5 M urea, 20 mM glycine and 10 mM DTT. The protein, identified by SDS/PAGE, was first dialysed against 0.1% formic acid and then against distilled water and stored frozen in an unfolded state.
Enzymatic protein phosphorylation
NDP kinase was phosphorylated as described previously [25]. The equilibrium constant of phosphorylation reaction (1a) is low (see Introduction). The reaction was pushed to completion by coupling with the pyruvate kinase reaction. The progress of phosphorylation was continuously monitored with the lactate dehydrogenase reaction. The phosphorylated protein was denatured by a 20 min incubation in the presence of solid GdmCl at a final concentration of 6 M. The denaturing reagent and ATP were eliminated rapidly on a HiTrap desalting column equilibrated with 100 mM Tris/HCl (pH 8.0). The final protein concentration was 20 μg/ml. The absence of ATP in the final preparation was ascertained by UV spectroscopy.
Chemical protein phosphorylation
NDP kinase is phosphorylated on the active site histidine by phosphoramidate [1,4]. This reagent is not commercially available. The dipotassium salt of phosphoramidate was synthesized as described previously [26]. Chemical phosphorylation was achieved with 0.16 M phosphoramidate in 0.16 M Tris/HCl (pH 8.0) and 10 mM MgSO4 for 30 min. After denaturation, the NDP kinase was reactivated by gel filtration as described above.
Analytical techniques
Protein concentration was determined from the optical density at 280 nm using a molar absorption coefficient of 1.35 for 1 mg/ml, which was calculated from the amino acid composition [27]. Enzymatic activity was assayed by a spectrophotometric assay, with 1 mM ATP and 0.2 mM 8-bromoinosine-5′-diphosphate as substrates [25]. An LS50B spectrofluorimeter (Perkin Elmer) was used to measure intrinsic protein fluorescence intensity (excitation at 295 nm, emission at 340 nm) and BisANS fluorescence intensity (excitation at 390 nm, emission at 480 nm).
Analytical size-exclusion chromatography was performed using a Superdex 75 column (Pharmacia Biotech) equilibrated with 100 mM Tris/HCl (pH 8.0), 150 mM NaCl, 5 mM MgCl2 and 1 mM DTT, in the absence or presence of 100 μM ATP or ADP, at a flow rate of 0.5 ml/min. The column was calibrated with Bio-Rad markers. Protein fluorescence was monitored using a flow cell on the LS50B spectrofluorimeter (excitation at 295 nm, bandwidth of 5 nm; emission at 340 nm, bandwidth of 15 nm). In other experiments, the effluent was mixed with BisANS at a final concentration of 1 μM just before the flow cell. BisANS fluorescence was measured (excitation at 370 nm, bandwidth of 5 nm; emission at 480 nm, bandwidth of 15 nm). Protein peaks containing the folding intermediates were indicated by BisANS binding.
All measurements were performed at 25 °C in 100 mM Tris/HCl (pH 8.0) containing 1 mM DTT. Figures show typical results of experiments performed at least twice with different enzyme preparations.
RESULTS AND DISCUSSION
Protein phosphorylation assists folding of the S120G mutant NDP kinase A
We coupled reaction (1a) with the pyruvate kinase reaction to obtain full protein autophosphorylation. Phosphorylation was continuously monitored spectrophotometrically with the lactate dehydrogenase reaction. A decrease in NADH reflected the generation of ADP by reaction (1a). We calculated a stoichiometry of 0.85 phosphoryl groups incorporated for each NDP kinase subunit from the data generated by this spectrophotometric analysis (Figure 1A). The H118N mutant could not, as expected, be phosphorylated. The final drift was caused by non-enzymatic hydrolysis of the phosphohistidine intermediate. Its half-life was 40 min, similar to values published previously [26]. Spontaneous dephosphorylation is a substantial limitation for protein stability studies. Equilibrium experiments could not be performed with the phosphorylated enzyme, because most of the phosphohistidine was hydrolysed during incubation. We denatured the phosphorylated NDP kinase and then passed the sample over a HiTrap desalting column to quickly (in about 1 min) eliminate the denaturant, thereby initiating kinase refolding. The phosphorylated NDP kinase recovered approx. 60% of its enzymatic activity (Figure 1B). Very little reactivation has been noted in similar experiments with unphosphorylated enzyme and with enzyme that was dephosphorylated with equilibration buffer containing 100 μM ADP in the in the desalting experiment (Figure 1B).
Figure 1. Effect of phosphorylation on reactivation of NDP kinase A S120G mutant.

(A) The S120G mutant was phosphorylated by 100 μM ATP using the regeneration system described in the text. Phosphorylation was monitored with the lactate dehydrogenase reaction. Final protein concentrations were 0.54 mg/ml (wild-type), 0.49 mg/ml (S120G) and 0.44 mg/ml (H118N). (B) Time-course for the reactivation of ATP-phosphorylated kinase. (●) ATP-phosphorylated enzyme; (○) ATP-phosphorylated enzyme dephosphorylated with 100 μM ADP; (■) phosphoramidate-phosphorylated enzyme; (□) phosphoramidate-phosphorylated enzyme, dephosphorylated with 100 μM ADP; and (▲) non-phosphorylated enzyme. Activity is expressed as the percentage of native NDP enzyme.
The NDP kinases may be chemically phosphorylated on the active site histidine residue using phosphoramidate [1,4]. The crystal structure of the Drosophila and Dictyostelium NDP kinases phosphorylated in this way, in their native states, showed that the other histidine residues found in the protein were not phosphorylated. The S120G mutant was phosphorylated, unfolded and then refolding started by gel filtration (Figure 1B). The reactivation gave quite a good yield, although lower than with the autophosphorylated enzyme. The effect was lost when the chemically phosphorylated enzyme was dephosphorylated by ADP. This result clearly indicates that the active site phosphorylation assisted the protein folding.
In further renaturation experiments, we used unphosphorylated enzyme with MgATP present during the enzyme-refolding assay. It should be mentioned that the two procedures are not equivalent. In the experiment shown in Figure 1(B), most of the protein remained phosphorylated in the unfolded state. Thus the phosphoryl group may assist folding from the initial stages of this process. In contrast, if the unfolded and unphosphorylated kinase is diluted in MgATP-containing buffer, phosphorylation may occur after the formation of a catalytically competent form of the protein, which is presumably the correctly folded monomer. Refolding in the presence of MgATP is certainly more similar to the situation that occurs in the cellular environment. The active site histidine residue can be dephosphorylated by ADP. This demonstrates that the phosphorylated residue is the active site histidine.
Effect of nucleotides on the reactivation of the S120G mutant NDP kinase A
Full enzymatic activity of hexameric NDP kinases is associated with the correct quaternary structure. Folded NDP kinase A monomers are catalytically competent, but have a low specific activity that is about 1.5–2% of that of the hexamer [22a]. Similar results have been obtained with the Dictyostelium cytosolic NDP kinase [28]. Therefore enzymatic activity measurements indicated the correct folding and association. The urea-unfolded S120G mutant, diluted with buffer, recovered less than 3% of the control activity in the absence of ATP. The wild-type enzyme recovered 50% of the activity under the same conditions (Figure 2A). In the presence of ATP and 5 mM MgCl2, a large amount of mutant kinase was reactivated (Figure 2B). ATP and ADP form complexes with Mg2+ in the presence of 5 mM Mg2+ at pH 8.0, and are referred to below as MgATP and MgADP respectively. A low reactivation yield was noted when Mg2+ ions were absent (with 1 mM EDTA in solution). These different effects suggest that it is protein phosphorylation and not nucleotide binding that corrects the folding defect. We investigated the effect of ADP on NDP kinase reactivation to confirm this hypothesis. The effect of ADP should be very different depending on whether NDP kinase reactivation occurs through enzyme phosphorylation or nucleotide binding. If the activation mechanism involves protein phosphorylation, ADP should have no effect by itself, but it should drive the equilibrium of reaction (1a) to the left, dramatically decreasing the effect of MgATP. If reactivation is caused by nucleotide binding, ADP might have a weak activating effect as it binds to the enzyme. ADP should be a weak competitor against ATP because of their different affinities for NDP kinase; ATP binds NDP kinase with a much higher affinity (Kd of 0.25 μM) than ADP (Kd of 25 μM) [29]. Our study shows that ADP was not an activator and that it strongly inhibited the activating effect of MgATP. The non-hydrolysable ATP analogue p[NH]ppA {adenosine 5′-[β,γ-imido]triphosphate}, shown to bind to NDP kinases with an affinity similar to that of ADP [30], did not reactivate the NDP kinase (Figure 2A). It was a weaker inhibitor than ATP in the reactivation assay (Figure 2B). Thus NDP kinase is probably reactivated by phosphorylation (Figure 2B).
Figure 2. Effect of nucleotides on the reactivation of wild-type and S120G mutant NDP kinase A.

(A) Kinetics of reactivation. Urea-unfolded wild-type (squares) and S120G mutant (circles) NDP kinase A was diluted 20-fold in buffer in the absence (open symbols) or presence of 0.1 mM MgATP (closed symbols). The effects of 1 mM ADP (▲) and of 1 mM p[NH]ppA (♦) on the S120G mutant is also shown. The protein was used at a concentration of 5 μg/ml. (B) Extent of S120G mutant reactivation after 2 h in the presence of ATP (circles), ATP+100 μM ADP (squares), ATP+100 μM p[NH]ppA (▲) with 5 mM MgCl2 (closed symbols) and 1 mM EDTA (open symbols). The reaction was followed in the presence of the indicated concentration of ATP. (C) Reactivation of urea-unfolded S120G mutant diluted in buffer containing MgCl2. ATP, at a concentration of 100 μM, was present at time zero (●) or was added after 30 min (○). Triangles represent reactivation in the absence of ATP.
The absence of an effect of ADP may seem surprising, because binding to NDP kinases is known to increase their thermodynamic stability. The probable explanation is the following: the binding of ADP and other nucleotides stabilizes the hexamer, whereas phosphorylation probably corrects the folding defect of the S120G mutant by stabilizing the native monomer (see below). Secondly, in vivo, ATP concentration is high in all cells, with an [ATP]/[ADP] ratio between 5 and 10. The folding in vivo yield may depend on the actual [ATP]/[ADP] ratio. The folding intermediate may persist in cells that have a low [ATP]/[ADP] ratio, and it may be trapped by chaperone proteins and have deleterious cellular effects.
The folding intermediate accumulating in the absence of MgATP slowly changes so that it cannot be reactivated. We found that reactivation was fast if the urea-denatured protein was diluted in MgATP-containing buffer. However, if the denatured protein was incubated in the absence of nucleotide before adding MgATP, the reactivation rate was lower (Figure 2C).
Effect of MgATP on folding/association followed by size-exclusion chromatography
Size-exclusion chromatography is useful since the various species present in a protein sample may be separated and identified. In contrast, enzymatic activity measurements detect the enzymatically competent species only, whereas optical spectroscopic techniques yield mean values of the measured parameter. The native hexamer, folding intermediate and native monomer are well separated by size-exclusion chromatography. The native hexamer was eluted first at an elution volume of 11.0 ml, then the folding intermediate was eluted at 12.8 ml and finally the native monomer was eluted at 13.8 ml. The elution volumes were reproducible within 0.1 ml. We followed the folding/association pattern in the following manner: we injected the unfolded protein into the column equilibrated with buffer in the absence or presence of 100 μM MgATP. The native protein served as a control. We identified the folding intermediate by mixing the column effluent with BisANS and measuring its fluorescence in a flow cuvette. BisANS, like ANS (8-anilino-1-napthalene sulfonate), binds specifically to folding intermediates and does not bind native or unfolded proteins [31]. The binding of BisANS distinguished the folding intermediate from other forms that have a similar Stokes radius, for example the dimers. However, quantitative estimation was not possible because the increase in the hexamer peak was not accompanied by a proportional decrease in BisANS binding.
The native wild-type NDP kinase A eluted as a hexamer at 11.0 ml (Figure 3A). When unfolded protein was injected, most of the sample recovered the hexameric structure. The shoulder observed at 12.2 ml corresponds to a folding intermediate which binds BisANS. We described previously the peculiar folding/association pattern of human NDP kinase A [22,32,33]. Typical hexameric NDP kinases, including the Dictyostelium cytosolic and Drosophila enzymes, unfold without the accumulation of dissociated species. In the refolding experiment, they fold as native monomers, which subsequently associate into hexamers. However, during the folding of the human NDP kinase A it is an equilibrium folding intermediate that accumulates and not folded monomers. The native S120G mutant and the native wild-type enzyme eluted with the same profile, indicating that the S120G mutant is a hexamer in solution, as in the crystal. We did not detect dissociated or BisANS-binding forms. This was simply a diagnostic test for the quality of the protein preparation. Following injection of the unfolded S120G mutant protein in the absence of MgATP we only detected the folding intermediate, characterized by BisANS binding (results not shown). MgATP promotes correct folding and association (Figure 3B). Control experiments showed that ATP in the absence of Mg2+, and ADP in the presence or absence of Mg2+ have no effect.
Figure 3. Effect of MgATP on the renaturation/assembly of wild-type and mutant NDP kinase studied by size-exclusion chromatography.

The urea-unfolded proteins (50 μl of 70 μg/ml solutions) were injected on to a Superdex 75 column. Shown are the wild-type NDP kinase A (A), S120G mutant (B), H118N mutant (C) and the H118N/S120G double mutant (D). ATP was either absent (○) or present at 100 μM (●). The native enzymes were injected for control experiments (no symbols). (E) The elution profile of the phosphorylated enzyme, in the absence (●) and presence (○) of 100 μM ADP in the elution buffer. The ordinate axis represents the intrinsic protein fluorescence intensity in arbitrary units.
The H118N mutant (Figure 3C) eluted as a mixture of hexamer and folding intermediate in the absence or presence of MgATP. The amount of hexamer slightly increases in the presence of MgATP. This indicates that MgATP binding may facilitate protein folding to some extent, because the H118N mutant cannot be phosphorylated. The H118N/S120G double mutant eluted as a single peak that corresponded to the position of the folding intermediate. The presence of ATP did not affect the elution pattern (Figure 3D).
Phosphorylation of the S120G mutant led to enzyme reactivation by facilitating correct folding and subunit assembly (Figure 1B). When the unfolded phosphorylated S120G mutant was injected into the column, most of it refolded/assembled on the column and eluted as the hexamer. Some of the protein eluted later at a position characteristic of the folding intermediate. In contrast, if the protein was dephosphorylated by including ADP in the equilibration buffer, all the protein eluted as a folding intermediate (Figure 3E). Dephosphorylation does occur in this experiment, but only after the protein has folded into a catalytically competent form, which is probably the folded monomer.
Effect of MgATP on the refolding of urea-denatured wild-type and mutant NDP kinases, followed by fluorescence spectroscopy
We next studied the effect of MgATP on the refolding of NDP kinase A, and of its mutants, to distinguish between an effect of MgATP on the folding process and subunit association. This is difficult because the folding intermediate aggregated at the high concentrations that are necessary for NMR spectroscopy (C. T. Craescu, unpublished work) or for near-UV CD spectroscopy (results not shown). The far-UV CD spectroscopy is not informative since the folding intermediate accumulated during the refolding of the S120G mutant contains a high percentage of secondary structure [18]. Intrinsic protein fluorescence was also not informative: the folding intermediate and the native protein have emission maximums at 340 nm, but the intermediate has a lower fluorescence intensity than the native protein. The differential binding of BisANS to different molecular forms was the most useful technique in this case. Unfolded wild-type and mutant NDP kinases were diluted in buffer in the presence or absence of MgATP, then BisANS was added immediately (Figure 4). The renaturing wild-type protein bound less BisANS than the mutants. Only the wild-type and the S120G mutant displayed a decrease in BisANS binding in the presence of MgATP compared with in its absence. The H118N mutant and the H118N/S120G double mutant were insensitive to the presence of MgATP. Mutation of the active site histidine residue abolished phosphorylation, but not ATP binding [29]. In all cases, spectra were insensitive to the presence of ADP, or ATP in the absence of MgCl2. Even though the H118N mutation is a conservative mutation, it affected protein refolding.
Figure 4. Effect of MgATP on urea-denatured NDP kinase A folding intermediate formation after dilution with buffer.

Wild-type protein (circles), S120G mutant (squares), H118N mutant (diamonds) and H118N/S120G double mutant (triangles). The protein concentration was 10 μg/ml and the residual urea concentration was 0.4 M. MgATP was absent (closed symbols) or present at 100 μM (open symbols). a.u., arbitrary units.
Finally, we studied the renaturation of NDP kinases in various concentrations of urea, in the absence and presence of MgATP, monitored by BisANS binding (Figure 5). MgATP did not affect the H118N/S120G double mutant. This is expected if phosphorylation is crucial for folding. In addition, the S120G mutant bound less BisANS in the presence of MgATP at low concentrations of urea, but not in the presence of 2–3 M urea. This supports the idea that MgATP promotes the reactivation of the folding-deficient S120G mutant through phosphorylation of the native monomer. A ‘global’ effect would decrease the folding intermediate concentration at all urea concentrations. This has been noted indeed with the natural osmolyte trimethylamine-N-oxide (F. Georgescauld, I. Mocan, M.-L. Lacombe and I. Lascu, unpublished work). Phosphoryl transfer may occur only after the formation of the active site. MgATP assisted, to some degree, the refolding of the H118N mutant, which cannot be phosphorylated (Figure 3).
Figure 5. Effect of MgATP on folding intermediate accumulation during the renaturation of NDP kinase A.

Urea-unfolded protein was diluted into buffer and incubated for 16 h in the indicated concentration of urea in the absence (closed symbols) or presence (open symbols) of 100 μM MgATP. BisANS was added at a final concentration of 1 μM. (A) Wild-type (circles) and S120G mutant (squares) NDP kinase. (B) H118N mutant (diamonds) and H118N/S120G double mutant (triangles) NDP kinase.
Conclusions: biological significance
Our findings suggest the following scheme for the folding/association of NDP kinase A and its mutants (Scheme 1). A scheme with I (folding intermediate) off-pathway and a ‘triangular’ scheme cannot be excluded until kinetic studies are undertaken. The conclusions of the present study would not change significantly with these other schemes.
Scheme 1. Proposed folding/association pathway of the S120G NDP kinase A mutant.

The folding intermediate (I) and the hexamer (H) could be identified directly (as shown in Figure 3), whereas the native monomer and the dimer are hypothetical species. Phosphorylation shifts to right the I↔M equilibrium.
The key difference of the human NDP kinase A from other hexameric NDP kinases is that I rather than M (folded monomer), accumulates during protein refolding. This is reminiscent to the refolding of Dictyostelium cytosolic NDP kinase at an acidic pH [34]. The S120G mutation decreases the equilibrium constant of the I↔M reaction. How may phosphorylation assist subunit folding and assembly? The simplest hypothesis is that MgATP binds to the folded monomers and that protein autophosphorylation shifts the equilibrium towards M according to the mass-action law. Subunit assembly into hexamers makes the entire reaction irreversible. ADP cancels, in part, the ATP effect by dephosphorylating M. The K-pn point mutation, P97S, in Drosophila NDP kinase does not affect folding, but does affect subunit association. The effect of the mutation cannot be corrected by phosphorylation in vitro (I. Lascu, unpublished work).
When the protein is synthesized in vivo by ribosomes, it may follow the simple scheme described above. The in vivo [ATP]/[ADP] ratio is high, typically 5–10. Under these conditions the unfavourable effect of ADP is small. Once the native M is formed, it may be trapped in hexamers with NDP kinase A or B subunits. However, the situation may be more complex. The partially folded mutant protein may be sequestered by chaperone proteins or it may aggregate. Cellular crowding has been shown to favour associations [35]. It could increase the rate of normal hexamer formation, and also those of chaperone binding and aggregation. These two side-effects may be associated with the observed cellular effects of the S120G mutant NDP kinase A [15–17].
An interesting question is whether the molten globule folding intermediate can bind ATP and be autophosphorylated. Our data suggest that the catalytically-competent form is the folded monomer, which agrees with the pre-equilibrium hypothesis [36]. For example, MgATP did not shift the equilibrium between the folding intermediate and the native monomer in the presence of 2–3 M urea (Figure 5). Various proteins (wild-type and mutant) are misfolded in the absence of substrates, but adopt the native form in their presence [37,38]. In at least one case, the cofactor flavin mononucleotide (FMN) could not interact with intermediate states [39]. In contrast, Andersson et al. [40] demonstrated that Zn2+ and Co2+ induced refolding of carbonic anhydrase by their interactions with the molten globule state. It has been shown recently that metal ions interact with the unfolded form of the protein and promote folding into the native form, rather than the folding intermediate [41]. One might think that small ligands, including metal ions, can interact with partially or completely unfolded proteins. However, binding of ligands, such as ATP, requires a completely formed active site. In addition, the active site residues must be in the correct position to be catalytically active and for autophophorylation to be possible.
What is the structural basis for the correction of the folding defect by phosphorylation? The X-ray structure of the phosphorylated NDP kinase A is not known. Therefore we used the highly similar Drosophila NDP kinase structure phosphorylated on the active site histidine residue [4] as a model to detect interactions in the phosphorylated enzyme (Figure 6). The side-chain of Ser121 forms a hydrogen-bond with Glu130, which is part of the α4 helix. The phosphoryl group interacts with Tyr53, which belongs to a different part of the subunit (αA helix). The phosphorylation does not replace the missing interactions resulting from the absence of the Ser120 side-chain. We think that the correction of the folding defect by phosphorylation is due to the increase in the thermodynamic stability of phosphorylated M over that of I. We are currently searching for intragenic suppressors of the folding defect associated with the S120G mutation to confirm this hypothesis.
Figure 6. Structure of the Drosophila NDP kinase phosphorylated intermediate.

The PDB file 1NSQ co-ordinates were used to draw the figure. (A) View of the hexamer along the 3-fold symmetry axis (only the top subunits are shown). (B) Details of the phosphorylated active site histidine residue environment.
Finally, it should be mentioned that folding defects of natural mutants corrected by cellular metabolites may be quite general. This simple strategy allows marginally stable or unfolded proteins to be expressed as native proteins.
Acknowledgments
This work was supported in part by grants from the Association pour la Recherche sur le Cancer and the Conseil Régional d'Aquitaine (I.L.), and from Associazione Italiana per la Ricerca sul Cancro (A.G.). We thank Dr Marie-France Giraud and Dr Alain Dautant for the careful reading of the manuscript and for preparing Figure 6, and Dr. Constantin Craescu for the preliminary NMR experiments.
References
- 1.Janin J., Deville-Bonne D. Nucleoside-diphosphate kinase: structural and kinetic analysis of reaction pathway and phosphohistidine intermediate. Methods Enzymol. 2002;354:118–134. doi: 10.1016/s0076-6879(02)54009-x. [DOI] [PubMed] [Google Scholar]
- 2.Lascu I., Gonin P. The catalytic mechanism of nucleoside diphosphate kinases. J. Bioenerg. Biomembr. 2000;32:237–246. doi: 10.1023/a:1005532912212. [DOI] [PubMed] [Google Scholar]
- 3.Gilles A. M., Presecan E., Vonica A., Lascu I. Nucleoside diphosphate kinase from human erythrocytes. Structural characterization of the two polypeptide chains responsible for heterogeneity of the hexameric enzyme. J. Biol. Chem. 1991;266:8784–8789. [PubMed] [Google Scholar]
- 4.Morera S., Chiadmi M., LeBras G., Lascu I., Janin J. Mechanism of phosphate transfer by nucleoside diphosphate kinase: X-ray structures of the phosphohistidine intermediate of the enzymes from Drosophila and Dictyostelium. Biochemistry. 1995;34:11062–11070. [PubMed] [Google Scholar]
- 5.Timmons L., Shearn A. Role of AWD/nucleoside diphosphate kinase in Drosophila development. J. Bioenerg. Biomembr. 2000;32:293–300. doi: 10.1023/a:1005545214937. [DOI] [PubMed] [Google Scholar]
- 6.Postel E. H. Multiple biochemical activities of NM23/NDP kinase in gene regulation. J. Bioenerg. Biomembr. 2003;35:31–40. doi: 10.1023/a:1023485505621. [DOI] [PubMed] [Google Scholar]
- 7.Ma D., McCorkle J. R., Kaetzel D. M. The metastasis suppressor NM23-H1 possesses 3′–5′ exonuclease activity. J. Biol. Chem. 2004;279:18073–18084. doi: 10.1074/jbc.M400185200. [DOI] [PubMed] [Google Scholar]
- 8.Fan Z., Beresford P. J., Oh D. Y., Zhang D., Lieberman J. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell. 2003;112:659–672. doi: 10.1016/s0092-8674(03)00150-8. [DOI] [PubMed] [Google Scholar]
- 9.Cervoni L., Pietrangeli P., Chichiarelli S., Altieri F., Egistelli L., Turano C., Lascu I., Giartosio A. In vivo cross-linking of nm23/nucleoside diphosphate kinase to the PDGF-A gene promoter. Mol. Biol. Rep. 2003;30:33–40. doi: 10.1023/a:1022261009207. [DOI] [PubMed] [Google Scholar]
- 10.Steeg P. S., Ouatas T., Halverson D., Palmieri D., Salerno M. Metastasis suppressor genes: basic biology and potential clinical use. Clin. Breast Cancer. 2003;4:51–62. doi: 10.3816/cbc.2003.n.012. [DOI] [PubMed] [Google Scholar]
- 11.Leone A., Flatow U., King C. R., Sandeen M. A., Margulies I. M., Liotta L. A., Steeg P. S. Reduced tumor incidence, metastatic potential, and cytokine responsiveness of nm23-transfected melanoma cells. Cell. 1991;65:25–35. doi: 10.1016/0092-8674(91)90404-m. [DOI] [PubMed] [Google Scholar]
- 12.D'Angelo A., Garzia L., Andre A., Carotenuto P., Aglio V., Guardiola O., Arrigoni G., Cossu A., Palmieri G., Aravind L., Zollo M. Prune cAMP phosphodiesterase binds nm23-H1 and promotes cancer metastasis. Cancer Cell. 2004;5:137–149. doi: 10.1016/s1535-6108(04)00021-2. [DOI] [PubMed] [Google Scholar]
- 13.Chang C. L., Zhu X. X., Thoraval D. H., Ungar D., Rawwas J., Hora N., Strahler J. R., Hanash S. M., Radany E. Nm23-H1 mutation in neuroblastoma. Nature. 1994;370:335–336. doi: 10.1038/370335a0. [DOI] [PubMed] [Google Scholar]
- 14.Almgren M. A., Henriksson K. C., Fujimoto J., Chang C. L. Nucleoside diphosphate kinase A/nm23-H1 promotes metastasis of NB69-derived human neuroblastoma. Mol. Cancer Res. 2004;2:387–394. [PubMed] [Google Scholar]
- 15.Kim Y. I., Park S., Jeoung D. I., Lee H. Point mutations affecting the oligomeric structure of Nm23-H1 abrogates its inhibitory activity on colonization and invasion of prostate cancer cells. Biochem. Biophys. Res. Commun. 2003;307:281–289. doi: 10.1016/s0006-291x(03)01195-1. [DOI] [PubMed] [Google Scholar]
- 16.Forus A., D'Angelo A., Henriksen J., Merla G., Maelandsmo G. M., Florenes V. A., Olivieri S., Bjerkehagen B., Meza-Zepeda L. A., del Vecchio Blanco F., et al. Amplification and overexpression of PRUNE in human sarcomas and breast carcinomas: a possible mechanism for altering the nm23-H1 activity. Oncogene. 2001;20:6881–6890. doi: 10.1038/sj.onc.1204874. [DOI] [PubMed] [Google Scholar]
- 17.Otero A. S., Doyle M. B., Hartsough M. T., Steeg P. S. Wild-type NM23-H1, but not its S120 mutants, suppresses desensitization of muscarinic potassium current. Biochim. Biophys. Acta. 1999;1449:157–168. doi: 10.1016/s0167-4889(99)00009-9. [DOI] [PubMed] [Google Scholar]
- 18.Lascu I., Schaertl S., Wang C., Sarger C., Giartosio A., Briand G., Lacombe M. L., Konrad M. A point mutation of human nucleoside diphosphate kinase A found in aggressive neuroblastoma affects protein folding. J. Biol. Chem. 1997;272:15599–15602. doi: 10.1074/jbc.272.25.15599. [DOI] [PubMed] [Google Scholar]
- 19.Schaertl S., Geeves M. A., Konrad M. Human nucleoside diphosphate kinase B (Nm23-H2) from melanoma cells shows altered phosphoryl transfer activity due to the S122P mutation. J. Biol. Chem. 1999;274:20159–20164. doi: 10.1074/jbc.274.29.20159. [DOI] [PubMed] [Google Scholar]
- 20.Chang C. L., Strahler J. R., Thoraval D. H., Qian M. G., Hinderer R., Hanash S. M. A nucleoside diphosphate kinase A (nm23-H1) serine 120→glycine substitution in advanced stage neuroblastoma affects enzyme stability and alters protein-protein interaction. Oncogene. 1996;12:659–667. [PubMed] [Google Scholar]
- 21.Giraud M. F., Georgescauld F., Lascu I., Dautant A. Crystal structures of S120G mutant and wild type of human nucleoside diphosphate kinase A in complex with ADP. J. Bioenerg. Biomembr. 2006;38:261–264. doi: 10.1007/s10863-006-9043-0. [DOI] [PubMed] [Google Scholar]
- 22.Erent M., Gonin P., Cherfils J., Tissier P., Raschella G., Giartosio A., Agou F., Sarger C., Lacombe M.-L., Konrad M., Lascu I. Structural and catalytic properties and homology modelling of the human nucleoside diphosphate kinase C, product of the DRnm23 gene. Eur. J. Biochem. 2001;268:1972–1981. doi: 10.1046/j.1432-1327.2001.2076.doc.x. [DOI] [PubMed] [Google Scholar]
- 22a.Gonin P. Ph.D. Thesis. Bordeaux, France: University of Bordeaux-2; 2000. Nucleoside Diphosphate Kinases: Studies on the Catalytic Mechanism and Use as Hexamerization Domain in Protein Engineering. [Google Scholar]
- 23.Biggs J., Tripoulas N., Hersperger E., Dearolf C., Shearn A. Analysis of the lethal interaction between the prune and Killer of prune mutations of Drosophila. Genes Dev. 1988;2:1333–1343. doi: 10.1101/gad.2.10.1333. [DOI] [PubMed] [Google Scholar]
- 24.Lascu I., Chaffotte A., Limbourg-Bouchon B., Véron M. A Pro/Ser substitution in nucleoside diphosphate kinase of Drosophila melanogaster (mutation Killer of prune) affects stability but not catalytic efficiency of the enzyme. J. Biol. Chem. 1992;267:12775–12781. [PubMed] [Google Scholar]
- 25.Lascu I., Pop R. D., Porumb H., Presecan E., Proinov I. Pig heart nucleosidediphosphate kinase: phosphorylation and interaction with Cibacron blue 3GA. Eur. J. Biochem. 1983;135:497–503. doi: 10.1111/j.1432-1033.1983.tb07679.x. [DOI] [PubMed] [Google Scholar]
- 26.Wei Y. F., Matthews H. R. Identification of phosphohistidine in proteins and purification of protein-histidine kinases. Methods Enzymol. 1991;200:388–414. doi: 10.1016/0076-6879(91)00156-q. [DOI] [PubMed] [Google Scholar]
- 27.Gill S. C., von Hippel P. H. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 28.Lascu I., Deville-Bonne D., Glaser P., Veron M. Equilibrium dissociation and unfolding of nucleoside diphosphate kinase from Dictyostelium discoideum. J. Biol. Chem. 1993;268:20268–20275. [PubMed] [Google Scholar]
- 29.Schneider B., Biondi R., Sarfati R., Agou F., Guerreiro C., Deville-Bonne D., Veron M. The mechanism of phosphorylation of anti-HIV D4T by nucleoside diphosphate kinase. Mol. Pharmacol. 2000;57:948–953. [PubMed] [Google Scholar]
- 30.Cervoni L., Lascu I., Xu Y., Gonin P., Morr M., Merouani M., Janin J., Giartosio A. Binding of nucleotides to nucleoside diphosphate kinase: a calorimetric study. Biochemistry. 2001;40:4583–4589. doi: 10.1021/bi002432s. [DOI] [PubMed] [Google Scholar]
- 31.Shi L., Palleros D. R., Fink A. L. Protein conformational changes induced by 1,1′-bis(4-anilino-5-naphthalenesulfonic acid): preferential binding to the molten globule of DnaK. Biochemistry. 1994;33:7536–7546. doi: 10.1021/bi00190a006. [DOI] [PubMed] [Google Scholar]
- 32.Lascu I., Giartosio A., Ransac S., Erent M. Quaternary structure of nucleoside diphosphate kinases. J. Biomembr. Bioenerg. 2000;32:227–236. doi: 10.1023/a:1005580828141. [DOI] [PubMed] [Google Scholar]
- 33.Lascu I. Nm23-H1/NDP kinase folding intermediates and cancer: a hypothesis. J. Bioenerg. Biomembr. 2006;38:265–268. doi: 10.1007/s10863-006-9042-1. [DOI] [PubMed] [Google Scholar]
- 34.Cervoni L., Egistelli L., Mocan I., Giartosio A., Lascu I. Quaternary structure of Dictyostelium discoideum nucleoside diphosphate kinase counteracts the tendency of monomers to form a molten globule. Biochemistry. 2003;42:14599–14605. doi: 10.1021/bi035273w. [DOI] [PubMed] [Google Scholar]
- 35.Ellis J. R. Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 2001;26:597–604. doi: 10.1016/s0968-0004(01)01938-7. [DOI] [PubMed] [Google Scholar]
- 36.James L. C., Tawfik D. S. Conformational diversity and protein evolution: a 60-year-old hypothesis revisited. Trends Biochem. Sci. 2003;28:361–368. doi: 10.1016/S0968-0004(03)00135-X. [DOI] [PubMed] [Google Scholar]
- 37.Vamvaca K., Vogeli B., Kast P., Pervushin K., Hilvert D. An enzymatic molten globule: efficient coupling of folding and catalysis. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12860–12864. doi: 10.1073/pnas.0404109101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uversky V. N., Kutyshenko V. P., Protasova N. Yu., Rogov V. V., Vassilenko K. S., Gudkov A. T. Circularly permuted dihydrofolate reductase possesses all the properties of the molten globule state, but can resume functional tertiary structure by interaction with its ligands. Protein Sci. 1996;5:1844–1851. doi: 10.1002/pro.5560050910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bollen Y. J., Nabuurs S. M., van Berkel W. J., van Mierlo C. P. Last in, first out: the role of cofactor binding in flavodoxin folding. J. Biol. Chem. 2005;280:7836–7844. doi: 10.1074/jbc.M412871200. [DOI] [PubMed] [Google Scholar]
- 40.Andersson D., Hammarstrom P., Carlsson U. Cofactor-induced refolding: refolding of molten globule carbonic anhydrase induced by Zn(II) and Co(II) Biochemistry. 2001;40:2653–2661. doi: 10.1021/bi000957e. [DOI] [PubMed] [Google Scholar]
- 41.Bushmarina N. A., Blanchet C. E., Vernier G., Forge V. Cofactor effects on the protein folding reaction: acceleration of α-lactalbumin refolding by metal ions. Protein Sci. 2006;15:659–671. doi: 10.1110/ps.051904206. [DOI] [PMC free article] [PubMed] [Google Scholar]