

Summary
Circadian rhythms are endogenous oscillations of diverse physiological and behavioral phenomena with a period length near 24 hours. We generated transgenic mice carrying the FASPS hPER2 S662G mutation and showed that these mice recapitulate the human phenotype. We present evidence that phosphorylation of residue 662 leads to increased PER2 transcription and hypothesize that phosphorylation at another site leads to degradation of PER2 protein. In vivo, PER2 is primarily a nuclear protein whose abundance is associated with circadian period length. The PER2 S662G mutation does not affect the level of CRY1 mRNA. Through alteration of CKIδ expression in S662 mutant mice, we provide in vivo evidence that CKIδ can regulate period through PER2. Focusing study on residue 662, based on a naturally occurring human variant, is yielding novel insights into PER2 regulation.
Introduction
A wealth of data from model organisms and cell based studies have elucidated circadian mechanisms involving interlocked feedback loops with transcriptional and post-translational regulations (Harms et al., 2004; Lowrey and Takahashi, 2004; Reppert and Weaver, 2002; Young and Kay, 2001). Circadian regulation of the clock and clock-related genes is complex and not fully understood (Ueda et al., 2005). Two routes have been proposed for Per2 regulation of circadian rhythms in mammals. The first involves a negative feedback loop in which four clock genes (Per1, Per2, Cry1, and Cry2) are activated by heteromeric complexes of CLOCK/BMAL1 acting via E-box elements in their promoter/enhancer regions. This activation is terminated ~6–8 hr. later as Per and Cry protein complexes accumulate in the nuclei to suppress transcription (Hastings and Herzog, 2004). Per2 has been shown to inhibit transcriptional activation by CLOCK/BMAL1 in vitro (Akashi et al., 2002; Jin et al., 1999; Kume et al., 1999). Per2 also acts as an activator of Bmal1 transcription. This is based on in vitro Bmal1 luciferase data (Yu et al., 2002) and Per2 knock-out mutant mice which showed lower amplitude, phase-advanced Bmal1 expression (Shearman et al., 2000).
Circadian period length is directly connected to PER stability in Drosophila (Edery et al., 1994). Whether this is true for mammals has not been established. The time of nuclear entry and length of time core clock proteins spend in the nucleus (prior to degradation) are thought to be critical in regulating the period of the circadian clock (Tamanini et al., 2005). These processes are assumed to be highly regulated and evidence has been provided with in vitro systems (Nawathean and Rosbash, 2004; Yagita et al., 2002). Although it is known that Per2 phosphorylation plays an important role in circadian regulation, the exact mechanism is unclear. Furthermore, the majority of the data regarding Per2 phosphorylation comes from in vitro and cell culture studies (Eide et al., 2005; Lowrey et al., 2000; Toh et al., 2001; Yagita et al., 2002).
Ultimately, translating this information to understanding of human clock is necessary. There is no doubt that many aspects of clock function are conserved. However, there are dramatic differences between drosophila and mammalian clocks. Even differences between mouse and human are certain to exist: one obvious phenotypic difference is that mice are nocturnal animals while humans are diurnal. Probing the genetic basis of sleep in humans first became available with the identification of a Mendelian phenotype of circadian clock function (Jones et al., 1999). Familial advanced sleep phase syndrome (FASPS) is an autosomal dominant, highly penetrant phenotype of early morning awakening and early sleep times. Two genes have now been identified that result in the FASPS phenotype (Toh et al., 2001; Xu et al., 2005). Study of such mutations is different from work done in forward screens or in knocking out genes in rodents since phenotypes are likely more subtle (since these families have not been selected against during evolution). Also, missense mutations like the ones that have been identified, having dominant effects, are likely to teach us about functional domains of these proteins and give insights that cannot be gleaned from knocking out genes in rodents. To understand human circadian rhythm genetics and biology, it will be most fruitful to look at naturally occurring human mutations and coupling human work with studies in cell culture and model organisms.
Results
Further biochemical characterization of hPER2 S662G
The first human genetic variant demonstrated to cause FASPS was in the human Period 2 homolog (hPER2) (Toh et al., 2001). The serine to glycine mutation resulted in the PER2 polypeptide being hypo-phosphorylated by casein kinase I (CKI) in vitro. Similar experiments after introducing a negatively charged residue to mimic a phosphoserine lead to restoration of robust phosphorylation by CKI.
The serine at position 662 in hPER2 is the first of 5 serines spaced three amino acids apart. This SxxSxxSxxSxxS motif is highly conserved in mammalian PER proteins (Toh et al., 2001). We hypothesized that phosphorylation of S662 could facilitate the phosphorylation of serines at amino acid positions 665, 668, 671, and 674 by CKI, thus regulating PER2 protein stability, nuclear translocation, and transcriptional repressor activity (Camacho et al., 2001; Nawathean and Rosbash, 2004; Yamamoto et al., 2005). To further test this idea, we carried out in vitro phosphorylation reactions with two peptides of the same sequence (encompassing residues 660–674 of PER2) either with or without a phosphate group covalently linked to S662 (Figure 1A). In addition, we selected another peptide from the adenomatous polyposis coli protein (APC, residues 1386 to 1402, a non-circadian and known substrate of CKI) that had a similar motif (SxxSxxSxxS). In vitro kinase assays showed that the PER2 peptide with a phosphate covalently linked to the first serine is phosphorylated at other residues by CKI. The peptide without a phosphate at S662 is not phosphorylated by CKI (Figure 1B). These data suggest that S662 is not phosphorylated by CKI and that a phosphate at S662 is required for CKI to phosphorylate other residues in the peptide. A quantitative assay showed that an additional 4 moles of phosphate were incorporated per mole of Pep-2P substrate and an additional 3 moles of phosphate were incorporated per mole of APC peptide (Figure 1C). We then carried out phospho-amino acid analysis and showed that the threonine at position 667 and the tyrosine at position 672 are not targets of CKI (Figure 1A, D). These results support the hypothesis that the phosphorylation of S662 plays a critical role in the regulation of PER2 by CKI via a cascade of phosphorylations that requires a priming event at residue 662.
Figure 1.
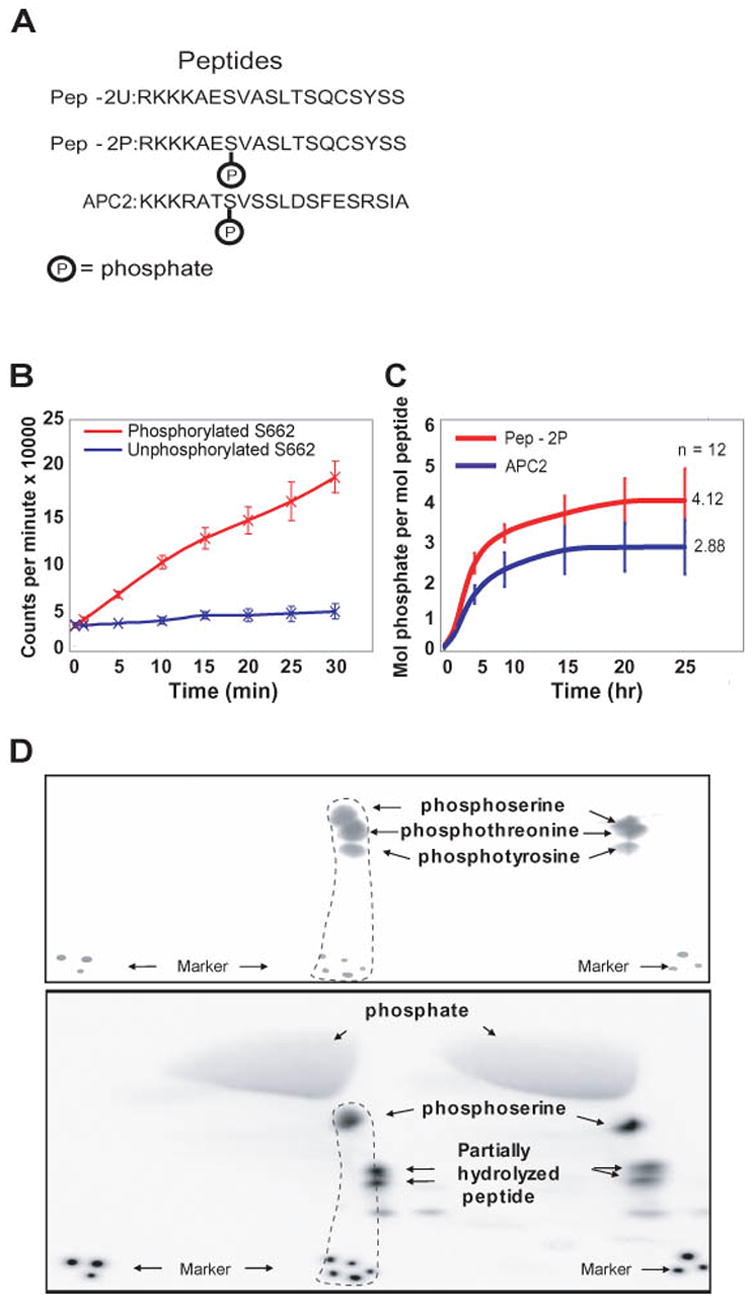
In vitro phosphorylation assay of synthetic peptides. (A) Phosphorylated (Pep-2P) and unphosphorylated (Pep-2U) peptides were used as substrates for the CKI in vitro assay. Another protein (Adenomatous polyposis coli, APC) with a similar SXXS motif was also studied. (B) A comparison of initial in vitro phosphorylation rates shows that peptide with a phosphate at S662 (Pep-2P, red) incorporates additional phosphate but Pep-2U (blue) does not. (C) Stoichiometric analysis of in vitro phosphorylation of Pep-2P (red) and APC2 (blue). The stoichiometry of phosphate incorporation is shown for each peptide as a function of time. (D) Phospho-amino acid analysis of Pep-2P, performed in duplicate. Both photographic (upper panel) and autoradiographic (lower panel) records are shown. Only the phospho-amino acid standards are at a sufficiently high concentration to be detected by ninhydrin staining in the photographic records. When the records are aligned with the aid of the markers at the bottom, phospho-serine is shown to be the only labeled phospho-amino acid.
Mouse Models of hPER2 S662 mutations
We generated mice with the PER2 S662G mutation to study its affect on circadian clock regulation in vivo. We also generated mice having an aspartate at position 662 as we previously showed that this reconstitutes the ability of the PER2 protein to be phosphorylated in vitro (Toh et al., 2001). Mice transgenic for wild-type hPER2 were generated as a control (S662WT). In each case, a human BAC was used since it carries the cis-acting genomic regulatory elements to faithfully recapitulate endogenous PER2 expression. One or 2 copies of the transgenes are typically inserted into the genome on a mouse background with 2 wild-type endogenous genes (Figure S1). We used a human transgene since it is highly homologous to the mouse gene and so that we could distinguish mRNA from the transgene vs. endogenous mouse loci. Furthermore, specific antibodies to mouse and human Per2 isoforms are available for comparing levels of transgene-encoded and endogenous protein.
Two knockout models of Per2 have been reported (Bae et al., 2001; Zheng et al., 1999). In both cases, the mice demonstrated a shorter period prior to becoming arrhythmic but there was huge variability in the number of days in constant darkness prior to animals becoming arrhythmic (Bae et al., 2001; Zheng et al., 1999). So that all our experiments could be performed on the same genetic background, we made the Per2ldc allele congenic on a C57BL/6J background (henceforth referred to as Per2−/−). Resulting mice had a normal period and did not become arrhythmic (data not shown). These mice were re-genotyped to make certain that they were Per2−/−. This demonstrates the dramatic affect of genetic background on the circadian phenotype.
The S662G transgenic mice showed a period (τ) ~2 hr. shorter than wild-type and, with the S662G transgene on a homozygous knock-out background, τ was even shorter (20.7 hr., Figure 2A, Table S1). Mice with the S662G transgene all showed a robust ~4 hr. phase advance of activity rhythms in LD12:12 regardless of the number of wild-type alleles (2 vs. 0) (Figure 2B). In contrast, the mice with the S662D transgene showed a longer τ (24.3 hr.) that was further lengthened on a homozygous knock-out background (24.7 hr., Figure 2A, Table S1). Mice from multiple lines for each transgene had identical phenotypes (data not shown). We next tested S662G and S662G/mPer2−/− mice in LD10.5:10.5 for 7 days. These mice were able to resume the normal “on-off” activity patterns as observed in other control mice (Figure 2B) suggesting that early onset of activity on/off time is due to the shortened endogenous τ of S662G mice.
Figure 2.
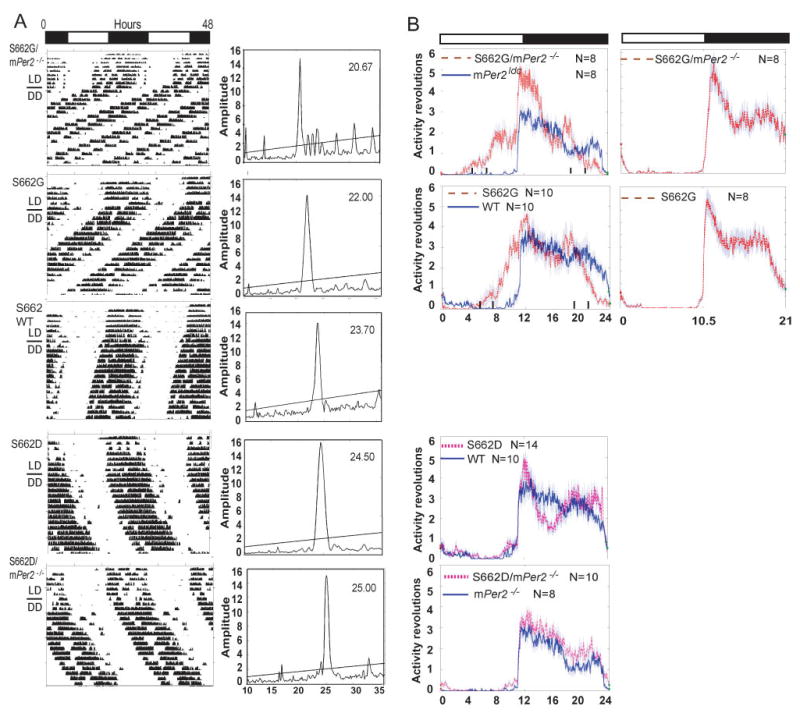
Circadian phenotypes of transgenic mice. (A) Locomotor activity recordings of representative mice (left column). Alternating white and dark bars indicate the LD cycles during entrainment prior to release in DD. τ analysis for each activity record is shown in the right column. (B) Activity patterns in LD12:12. Data was accumulated in 3 minute bins for ~7 days. Shadows indicate standard error of the mean. S662G/mPer2−/− and S662G mice show activity phase advance of ~4–6 hr. relative to mPer2−/− and WT. Activity onset is similar for S662D, WT, S662D/mPer2−/−, and mPer2−/− mice. The activity onset is at ‘lights out’ for S662G and S662G/mPer2−/− mice in LD10.5:10.5. (See Table S1 for population data)
Thus, despite being a nocturnal animal, mice carrying the hPER2 gene with the glycine mutation had a phenotype identical to that of a human FASPS with activity onset 4–6 hr. prior to lights out and activity offset before the lights came on. Furthermore, an aspartate at position 662, mimicking a constitutively phosphorylated serine, led to lengthening of τ (Figure 2A). Changing the charge on this single residue (glycine-uncharged, serine-partially charged due to phosphorylation, aspartate-mimicking a constitutively phosphorylated residue) was sufficient to dramatically alter the circadian τ from short to long. Both the S662G and the S662D mutations are dominant and produce a phenotype even with two wild-type mPer2 alleles in the background.
PER2 nuclear abundance correlates with circadian period
Based on working models of the mammalian clock, it was possible that S662G exerted its effect by one or more of the following: a) altering PER2 stability, b) altering PER2 nuclear translocation, or c) changing the role of PER2 in regulating transcription. Having established the S662G transgenic mouse as a good model for human FASPS, we then set out to begin molecular characterization in these and the S662D mice. To examine the level of phosphorylation of hPER2 protein in S662WT, S662G, and S662D transgenic mice on null backgrounds, we prepared liver extracts at different time points. Human PER2 was found predominantly in the nuclear fractions (Lee et al., 2001) (Figure 3A). We were not able to observe PER2 in cytoplasmic fractions of liver extracts (data not shown). As expected, the S662G+; −/− mice showed maximal phosphorylation levels at CT20, whereas S662WT and S662D+; −/− mice showed a mobility shift beginning at CT12. The intensity of mobility shifted bands observed in S662WT is intermediate between those seen in S662G and S662D, particularly after the CT12 time point (Figure 3A) consistent with biochemical data showing that phosphorylation of 4 other downstream residues is modulated by the phosphorylation state of S662. The in vivo nuclear abundance of PER2 also correlates with the phosphorylation state of S662 (Figure 3B).
Figure 3.
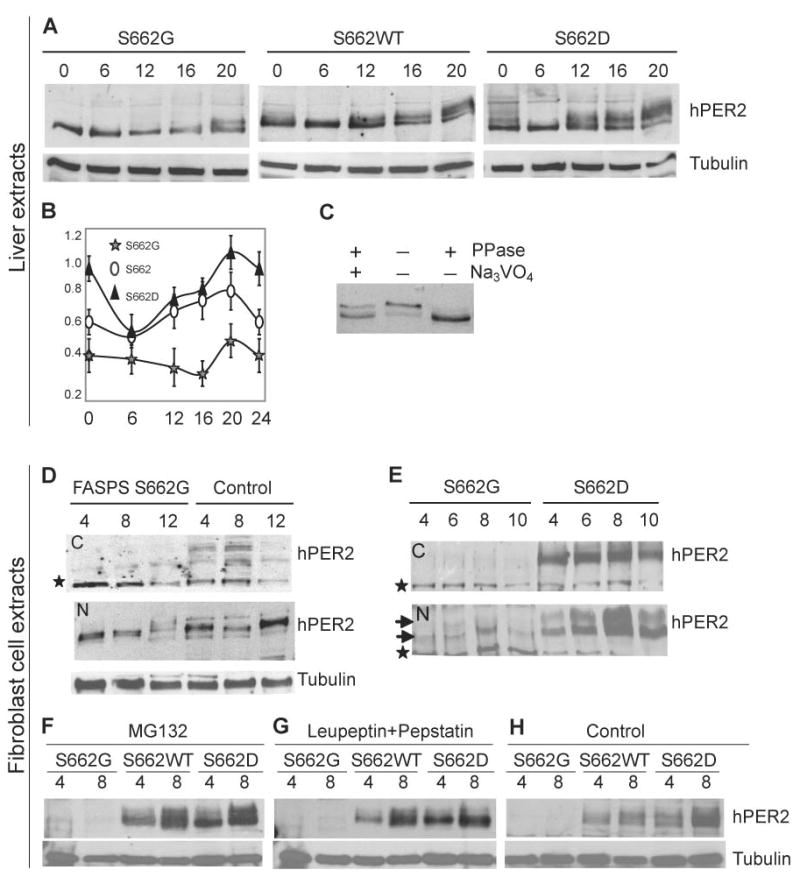
PER2 S662 phosphorylation results in increased PER2 protein. (A) hPER2 abundance and phosphorylation in S662G, S662WT, and S662D transgenic mouse liver extracts (nuclear fractions) at different CTs. (B) Quantification of protein from liver extracts in (A). Bars represent standard deviation for each time point in 3 independent experiments. Differences using the t-test were statistically significant (p<0.05) for S662G vs. S662D at all time points, for S662 vs. S662G at all time points except 6 hr., and for S662 vs. S662D at t=0, 20, and 24. (C) Immunoprecipitated complexes from S662D mouse liver at CT16 were untreated, or treated with either phosphatase alone or both phosphatase and sodium vanadate. Changes in PER2 mobility are due to changes in phosphorylation. (D) hPER2 from human S662G and control fibroblasts show that protein is primarily in the nuclear fractions (‘N’ vs. cytosolic ‘C’) and that levels of hPER2 protein were lower in S662G subjects vs. controls. The star represents a non-specific band that serves as an internal loading control. (E) Western blot analysis of cultured fibroblasts from S662G and S662D transgenic mice. Upper and lower arrows indicate low and high mobility hPER2 protein. hPER2 protein is undetectable in cytosolic fractions (‘C’) of S662G transgenic mice and are much higher (C and N) in S662D transgenic mice. (F) Fibroblasts were cultured, serum-shocked, and MG132 was added at t=0 and again at 4 hr. Cells were harvested at times 4 and 8 hr. The proteasome inhibitor MG132 does not lead to an increase in S662G hPER2 protein. (G) Lysosome inhibitors were added to cells 2 hr. after serum shock and 2hr. before cells were harvested at times 4 and 8 hr. The lysosome inhibitors leupeptin and pepstatin failed to increase S662G hPER2 protein. (H) Control cells were treated with vehicle (DMSO) at t=0 for the 4 hr. time point and at t=0 and t=4 for the 8 hr. time point.
Anti-human PER2 antibody was used to immunoprecipitate PER2 from liver lysates. These were treated with and without protein phosphatase 2A (PP2A) and sodium vanadate and run on polyacrylamide gels to assess phosphorylation status of PER2. A higher molecular weight PER2 band was present in samples that were either untreated or treated with both PP2A and the phosphatase inhibitor (Figure 3C) demonstrating that the upper band is phosphorylated PER2. We cultured fibroblasts from skin biopsies to determine whether PER2 is also less abundant in FASPS individuals harbouring the S662G mutation. Human PER2 was present in both the nuclear and cytoplasmic fractions at three time points in fibroblasts from control individuals (Figure 3D) with much higher levels in the nuclear fractions. Fibroblasts from FASPS individuals showed lower abundance and phosphorylation of PER2 than fibroblasts from control individuals, consistent with observations in transgenic mouse liver extracts.
We also examined protein abundance in cultured fibroblasts from transgenic mice after cells were synchronized by serum shock (Balsalobre et al., 1998). The level of PER2 is dramatically elevated at different time points in S662D vs. S662G (Figure 3E). Cytosolic PER2 was barely detectable in fibroblasts from both FASPS individuals and S662G transgenic mice (Figure 3D, E). Nuclear PER2 from FASPS fibroblasts and S662G mice showed reduced phosphorylation relative to fibroblasts from normal human individuals and S662D mice (Figure 3D, E). These data demonstrate that hPER2 is phosphorylated later and to a lower degree in S662G mice vs. wild-type or S662D.
PER2 mutations at position 662 alter transcriptional regulation of its own message
To examine the mRNA levels of endogenous mPer2 and the hPER2 transgene in vivo, we entrained mice of different genotypes to 12 hr. light and 12 hr. dark (LD12:12) and then released them into constant darkness (DD). mRNA levels in liver were assessed at different time points during the second day in DD. Both hPER2 and endogenous mPer2 mRNA levels peaked earlier for S662G and later for S662D when compared to wild-type (Figure 4A, B). In addition, the mRNA levels were lower for S662G and higher for S662D when compared to wild-type emphasizing again the dominant affect of these mutations. We then examined message levels of the hPER2 mRNA in fibroblasts from animals of both genotypes after serum shock and showed that S662D message levels were higher at all time points when compared to S662G (Figure 4C). Since both mPer2 and hPER2 message levels are decreased in the S662G mice, this argues for altered transcriptional activity rather than decreased S662G hPER2 mRNA stability. In addition, analysis of hPER2 pre-mRNA (Ripperger and Schibler, 2006) showed the same pattern as seen for hPER2 and mPer2 mRNA (Figure 4J). Thus, changing the charge at residue 662 alters the ability of PER2 to regulate its own transcription, presumably through interaction with other proteins since PER2 itself does not bind DNA (Reppert and Weaver, 2001).
Figure 4.
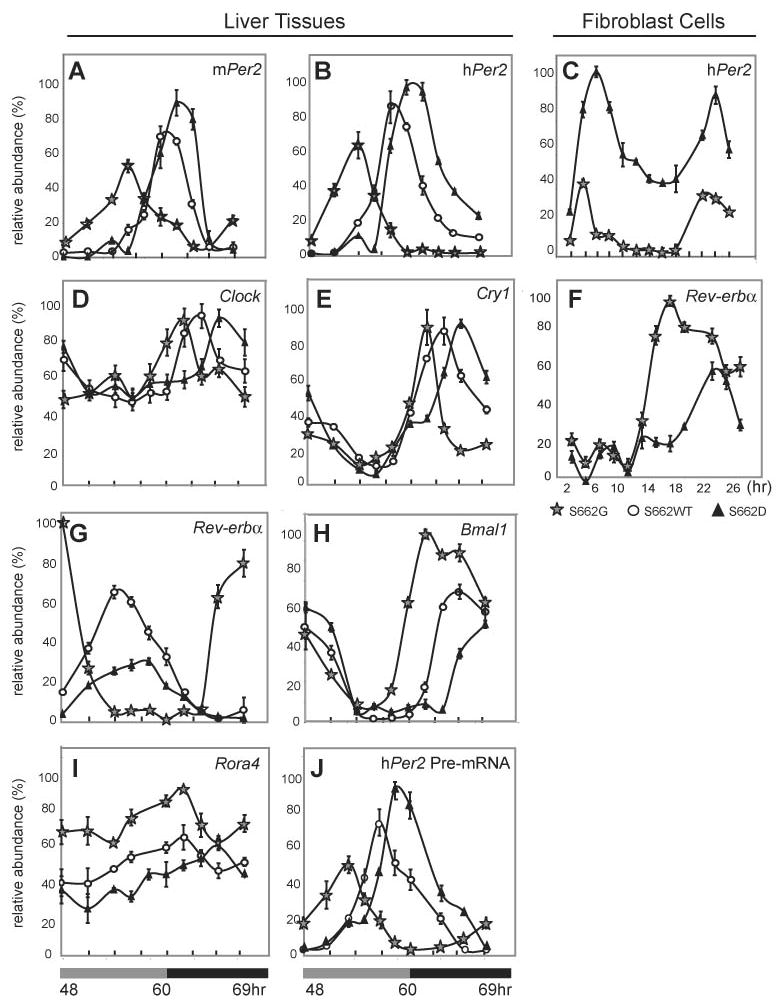
Expression levels of clock genes in S662G, S662WT, and S662D transgenic mice in liver (A–B, D–E, G–J) and in fibroblasts (C, F). Both mPer2 (A) and hPER2 (B) mRNAs peak earlier for S662G and later for S662D than for wild-type in liver from animals that are entrained in LD12:12 and released into DD. (C) hPER2 mRNA in synchronized fibroblast cultures was significantly higher at all time points for S662D vs. S662G. Transcript levels of (D) mClock and (E) mCry1 at different time points in liver are not different in amplitude but peak earlier for S662G and later for S662D relative to wild-type. Each value represents the mean of four independent experiments and was normalized to the corresponding Gapdh levels. (F) Rev-erbα mRNA in fibroblasts was significantly lower in the dark phase for S662D vs. S662G. Transcript levels of (G) mRev-erbα, (H) mBmal1, and (I) mRora4 in liver peaked earlier and at higher levels in S662G vs. S662D. (J) hPER2 pre-mRNA levels mirrored both hPER2 and mPer2 mRNA profiles in liver.
S662G does not affect PER2 degradation or nuclear localization
In order to test whether protein degradation contributes to these protein level differences, a proteasome inhibitor (MG132) or lysosome inhibitors (leupeptin and pepstatin) were added to fibroblast cultures from S662G, S662WT and S662D mice. While PER2 was abundant both before and after treatment with either proteasome or lysosome inhibitors in both S662WT and S662D, S662G PER2 could not be visualized, even after application of the inhibitors. Thus, the reduced PER2 levels in S662G fibroblasts did not result from alterations in protein degradation (Figure 3F–H).
We then set out to test whether the S662G or S662D mutations affected nuclear translocation of PER2. Initially, we began using transfection of cultured HEK293 cells with hPER2 cDNAs driven by ubiquitous promoters and showed that a large amount of over expressed protein was present but that there was no difference in subcellular localization among the different genotypes (Figure S2 A–C). Because of concern that overexpression of hPER2 in cells may not reflect normal PER2 cellular localization, we used the hPER2 promoter (15 kb upstream of the ATG start site) to drive expression of wild-type, S662G, and S662D PER2 cDNAs as fusion proteins with GFP. Using the hPER2 promoter, PER2 is largely (if not exclusively) a nuclear protein and no changes in localization were recognized among the genotypes (Figure S2 D-O). Quantification of data is shown for CMV (Figure S2-P) and hPER2 promoters (Figure S2-Q). We cultured fibroblast cells from Cry1/Cry2 double knockout mice to examine whether PER2 nuclear entry requires CRY. Nuclear localization was observed in 95% of cells when transfected with hPER2 (Figure S2-R). These data demonstrate that changing the amino acid at position 662 does not affect PER2 cellular localization or nuclear translocation and that PER2 nuclear entry is independent of CRY1 and 2.
Changing CKIδ dosage modulates phenotype of S662 mutants but not wild-type Per2
Neither CKIδ +/− animals nor CKIδ WT transgenic mice have an altered circadian τ (Figure 5). We next crossed the PER2 transgenic mice with both CKIδ WT transgenic and heterozygous CKIδ knock-out mice. The S662G allele leads to a short τ (~22 hr., Figure 2A). When S662G is crossed onto a background with a WT CKIδ transgene, τ gets even shorter (20.8 hr., Figure 5, Table S2). Crossing S662G onto a CKIδ heterozygous null background partially corrects τ shortening of S662G (22.5 hr., Figure 5, Table S2). The τ of S662D transgenic mice becomes even longer on a CKIδ +/− background (25 hr., Figure 5, Table S2).
Figure 5.
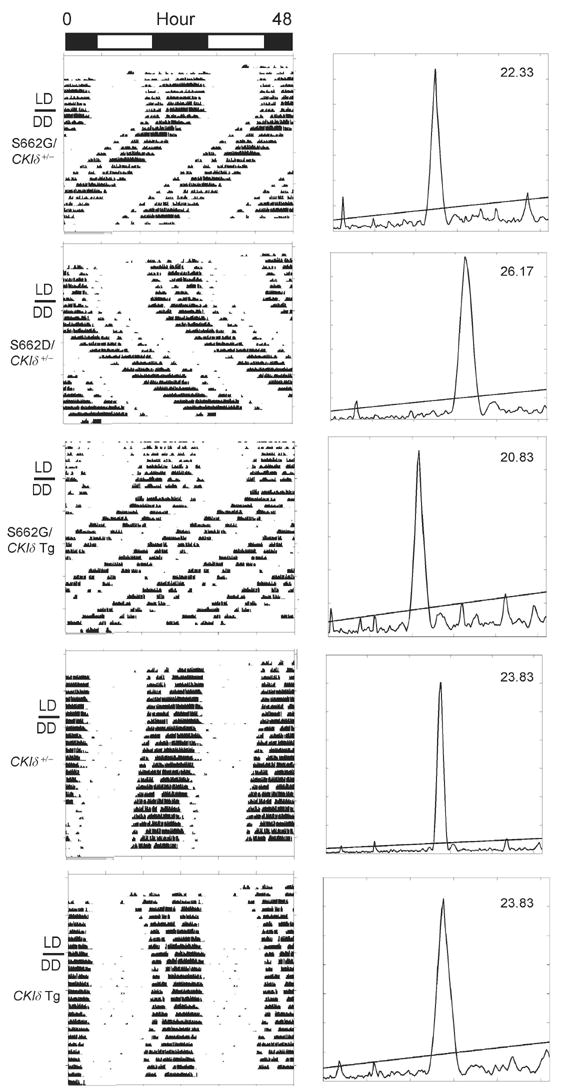
Altered CKIδ dose modifies expression of S662G and S662D circadian phenotypes. Representative locomotor activity records of representative mice with the indicated genotypes are shown on the left. The right panel shows τ of these mice as determined by chi-square periodgram analysis. (See Table S2 for population data)
In vitro transfection assays of wild-type and mutant Per2 phosphorylation by CKI have previously shown that the region immediately C-terminal of S662 is not the sole CKI phosphorylation site on Per2 (Eide et al., 2005). Since the phosphorylation cascade downstream of S662 leads to increased Per2 levels (via increased Per2 transcription), and the S662G mutation leads to decreased Per2 levels, we propose that phosphorylation of another site on Per2 by CKI targets Per2 for degradation. Another theoretical possibility is that CKIδ is mediating this effect indirectly through phosphorylation of another substrate. We favor the former model based on work in Drosophila and in cell culture showing that phosphorylation of Per leads to Per degradation.
Taken together, these data support a model where CKI, through different phosphorylation sites, is playing opposing roles in the regulation of Per2 levels (Figure 6A). Per2 protein levels are balanced by opposing actions of CKI in increasing Per2 degradation (left side) vs. increasing Per2 transcription (and thus, protein levels, right side). Data from S662D, showing increased Per2 message suggests that phosphorylation of this motif leads to activation of Per2 expression. Changes in CKIδ copy number affect both regulatory actions in opposite directions and to approximately equal extents thus causing no change in τ (Figures 6B, C). The S662G mutation interferes with the downstream phosphorylation cascade, resulting in lower rates of Per2 transcription and less Per2 protein (Figure 6D). Phosphorylation by CKI at another site continues to target Per2 for degradation thus disrupting the balance toward lower Per2 levels and shorter τ. Increased CKIδ dosage further accelerates Per2 degradation of S662G leading to an even shorter τ (Figure 6F) while CKIδ haploinsufficiency partially corrects τ shortening caused by this mutation (Figure 6E).
Figure 6.
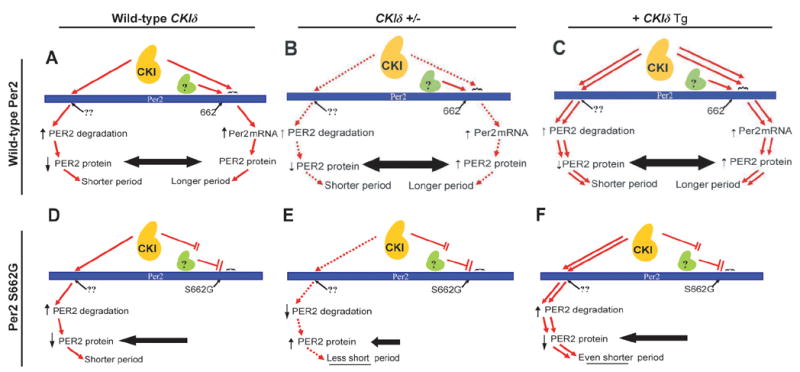
Model of CKIδ regulation of Per2. (A) The kinase (CKI) acts on at least two Per2 sites. Phosphorylation at one (??) leads to degradation of Per2. After phosphorylation of S662 by a priming kinase (green), CKI is able to phosphorylate a series of downstream serines. This leads to increased Per2 mRNA and protein. The equilibrium results in Per2 levels that dictate period length. (B) Removal of one copy of CKIδ by homologous recombination (CKIδ +/−), or (C) Addition of 1–2 genomic copies of a CKIδ transgene does not change the period because of maintenance of the balance of these opposing affects. (D) The inability of the priming kinase to phosphorylate S662G leads to hypophosphorylation of the downstream residues by CKI. The net effect is decreased Per2 and shortening of the period. (E) Crossing the S662G transgene onto a heterozygous CKIδ knock out partially corrects the shortening conferred by S662G by decreasing phosphorylation mediated Per2 degradation. (F) In the S662G mouse, addition of 1–2 CKIδ genomic transgenes now shortens period further as increased degradation is not opposed by transcription regulation from phosphorylation downstream of residue 662.
This model also fits with a recent report demonstrating that all CKI short-period mutations tested enhance the phosphorylation of Per proteins in cells but that not all of them accelerated the degradation of Per (Gallego et al., 2006). It is interesting to speculate that the CKI mutations leading to increased degradation may result in increased phosphorylation of one Per2 site and that those not affecting degradation have affects on phosphorylation of another site(s). They postulated distinct regulatory sites that fit with our model (Figure 6A). Gain-of-function mutations in the kinase at a site leading to degradation would produce similar downstream effects (decreased Per2 levels) to a mutation decreasing the ability of another phosphorylation site on the substrate (downstream of S662G, for example) to mediate increases in Per2 expression and protein.
Cycling of other clock genes
The expression profile of clock related genes (mCry1, mBmal1, mClock, mRora, and mRev-erbα) was analyzed to investigate the molecular mechanism underlying τ alterations (Figure 4). Clock and Cry1 expression were rhythmic and followed the same altered pattern of peak expression: S662G shifted forward and S662D shifted backward when compared to S662WT (Figure 4D, E), with a 4 hr. phase difference between S662G and S662D. The RNA oscillations of these clock genes are consistent with previous reports (Albrecht et al., 1997; Lee et al., 2001; Zylka et al., 1998) and the expression levels are similar in S662WT, S662G, and S662D transgenic mice.
PER2 has been postulated to be a negative regulator of mRev-erbα expression (Preitner et al., 2002). The peak expression of Rev-erbα is significantly higher in S662G than in S662D and the phase is advanced at least 8hr. in S662G mice when compared to S662D mice (Figure 4F, G). Given the dominant role of REV-ERBα in suppressing Bmal1 transcription (Preitner et al., 2002), Bmal1 expression should have been strongly inhibited in S662G mice (with low PER2 levels and high Rev-erbα expression) compared with S662D mice (high PER2 levels and low expression of Rev-Erbα). Unexpectedly, Bmal1 expression showed modest reduction in S662D mice compared with S662G, although robust molecular oscillations persist in both transgenic lines (Figure 4H). The trough level of Bmal1 was prolonged in the S662D and shortened in S662G compared with S662WT mice. We monitored expression of Rora in S662WT, S662G and S662D mice since RORA is thought to activate Bmal1 transcription in competition with REV-ERBα transcriptional repression (Emery and Reppert, 2004; Sato et al., 2004). As described previously (Ueda et al., 2002), Rora displayed no obvious cycling in liver. Rora expression levels were also lower in S662D mice than in S662G mice (Figure 4I). Expression levels of Rev-erbα, Bmal1, and Rora were different (S662G vs. S662WT vs. S662D, Figure 4G–I) while levels of Clock and Cry1 were unchanged (Figure 4D, E). This suggests that abundance and/or phosphorylation of Per2 plays a role in the regulation of the transcriptional loop involving Rev-erbα, Bmal1, and Rora with Per2 potentially acting as a negative regulator of Rora and Rev-erbα expression.
Discussion
The discovery of the S662G mutation causing human FASPS was direct evidence that PER2 plays an essential role in the human circadian clock. Mice transgenic for S662G recapitulate the short τ FASPS phenotype seen in humans. Mice with a transgene encoding an acidic residue at position 662 showed τ elongation. The fact that a single amino acid change can produce such profoundly different affects on τ and phase angle of entrainment suggests that this residue plays a central regulatory role in period length determination. Identification of the priming kinase is an important future direction since this enzyme plays a key PER2 regulatory role.
These mouse models provide exciting opportunities to probe changes in circadian regulation in vivo. It is intriguing that the mPer2ldc allele on the C57BL/6J background did not become arrhythmic, at least for the 2–3 weeks these animals were maintained in DD. Genotypes were double checked to confirm that these mice are homozygous for the knocked out allele. In addition, western analysis demonstrated no Per2 protein in tissue from these mice. Finally, period changes in S662 mutant transgenic mice were further exaggerated when crossed onto the Per2−/−background.
Mice with 1 or 2 copies of an hPER2 S662G transgene had an average circadian τ that was significantly shorter than the one circadian τ (23.3 hr.) so far reported from a heterozygous S662G human (Jones et al., 1999). The relative endogenous periods of mice and humans carrying the same circadian mutation have not previously been reported and therefore the reasons for this discrepancy are not known with certainty. Possible explanations include differences in human vs. mouse circadian function or in experimental design. As one example of the latter, we note that the one τ measurement in a S662G gene carrier was obtained from a paradigm (Jones et al., 1999) that is different from the mouse experiments and considered by some to over-estimate τ (Campbell, 2000; Czeisler et al., 1999; Klerman et al., 1996). More comparisons of human and murine circadian phenotypes under the influence of the same human clock genes are needed to enhance the applicability of murine models to human circadian disorders.
The changes in τ seen in S662G and S662D mice with 2 endogenous wild-type alleles (but not the wild-type transgenics) demonstrate that the S662G and S662D mutations are dominant. In vivo experiments in these mice showed PER2 mRNA and protein to be decreased in S662G and increased in S662D when compared to wild-type. These phenotypes were even more exaggerated when the mutant transgenes were crossed onto Per2−/−. Having ruled out increased degradation and altered nuclear translocation of PER2 in S662G mice as the cause of decreased mRNA and protein, the remaining possibility is that the mutation affects transcriptional regulation of hPER2 and mPer2 with S662G leading to decreased Per2 expression and S662D leads to increased Per2 expression. We can not rule out the possibility that the mutation also alters RNA stability but the trans- effect that the S662G hPer2 protein has on expression of the endogenous mPer2 locus shows that this is the relevant consequence for the circadian phenotype. Our data also support differential regulation of Per2 and Crys as different levels of PER2 in S662G and S662D were associated with altered timing, but not amplitude, of mCry mRNA.
An important future direction is to elucidate the mechanism whereby charge changes at PER2 residue 662 regulating downstream phosphorylation can change transcriptional activity. PER2 does not bind DNA itself and thus, must exert these affects through other partners. This effect must be through CLOCK/BMAL, either directly or indirectly.
One plausible model involves the cell sensing PER2 levels changing over time. A new cycle begins as PER2 levels drop below a critical threshold. PER2 gene expression is activated followed 4–6 hr. later by activation of CRY. PER2 and CRY accumulate until CRY levels reach a threshold at which PER2 expression is turned off. PER2 protein degrades and a cycle ends when PER2 levels drop below the threshold thus initiating the next cycle. In S662G individuals or mice, the resulting alteration in transcription leads to production of less PER2 mRNA and protein. Phosphorylation at another site targeting PER2 for degradation is unaffected (Figure 6). Thus, PER2 levels are lower at all time points and in the latter half of a cycle, fall below the threshold earlier than wild-type, leading to activation of transcription earlier and resulting in a shorter τ. There is a higher abundance of PER2 in the nuclei of S662D mice resulting from increased transcriptional activation due to hyperphosphorylation downstream of S662. Thus, cellular machinery takes longer to degrade the protein to levels below the threshold leading to a delay in activation of expression in the next cycle relative to wild-type (and thus, to a longer τ). This model explains the co-dominance of both mutations with wild-type. Consistent with the data, this model predicts that CRY repression of PER2 transcription would be dominant over the transcriptional activator(s) of PER2. Wild-type PER2 results in higher PER2 mRNA and protein levels in vitro and in vivo (and S662D in even higher levels). Thus, it is clear that PER2 must also be involved in transcriptional activation of PER2, likely through regulation of PER2 abundance and/or regulation of particular PER2 phosphorylation states.
Methods
Synthetic Peptides
Pep-2U and Pep-2P incorporate residues 659 to 674 of hPER2 and includes basic amino acids at their N-terminals. S662 is phosphorylated in Pep-2P. APC2 is peptide incorporating residues 1386 to 1402 of the human APC protein. All peptides were purified with high performance liquid chromatography (HPLC) and confirmed with mass spectrometry.
In vitro phosphorylation
Peptides at 10 μM concentration were phsophorylated in buffer containing 30 mM HEPES pH7.5, 7 mM MgCl2, 250 μM ATP, 0.5 mM DTT, 0.1 mg/ml bovine albumin and a small amount of γ32P-ATP. In initial experiments comparing Pep-2U and Pep-2P phosphorylation, his-tagged full length CKIε was used at a concentration of 0.4 ng/μl. In the experiments to determine phosphorylation stoichiometry, a his-tagged truncated version of CKIε lacking the autoregulatory C-terminal tail was used at a concentration of 1.5 ng/μl to ensure complete peptide phosphorylation over the longer periods needed for these experiments. At selected timepoints, 20 μl aliquots were removed and mixed with 50 μl 30 % acetic acid to inactivate the kinase. The mixtures are spotted onto Whatman P81 paper, allowed to dry, and then washed with 74 mM orthophosphoric acid three times for 5 minutes. The Cerenkov radiation of the labeled peptides was counted.
To calculate the stoichiometry of the phosphorylations, serial dilutions of the kinase reaction mixes were prepared at the end of each experiment and spotted onto P81 circles. The unwashed circles were placed in scintillation vials and the Cerenkov radiation was counted. Based on the concentrations of unlabelled ATP (present in excess) and peptide, the specific activity of the ATP and the moles of phosphate transferred per mole of peptide were then calculated. These experiments were performed in triplicate.
Phosphoaminoacid Analysis
Reaction mixes containing peptides that had been phosphorylated for 24 hr. were passed through Microcon 10 (Amicon) filter devices. The radioactive species in the filtrate consisted only of labeled peptides (molecular mass less than 3 KD) and unincorporated γ32P-ATP. 30 μl aliquots of the filtrate were spotted onto P81 circles, allowed to dry and washed with o-phosphoric acid to remove the ATP. The washed circles were air-dried and the labeled peptides eluted with freshly prepared 50 mM NH4HCO3. The peptides were dried and washed once with distilled water and once with pH1.9 buffer (0.58 M formic acid, 1.36 M acetic acid). The peptides were dried after each wash with Speed vac concentrator.
The washed peptides were re-suspended in 6 M HCl and incubated at 110°C to hydrolyze them to their constituent amino acids. After cooling to room temperature, the samples were transferred to fresh tubes, dried, and the Cerenkov radiation counted before the peptides were dissolved in pH1.9 buffer. Equal counts of hydorlysate were spotted onto thin layer cellulose (TLC) plastic sheets (EM Science) with a solution containing 2 mg/ml of phosphoserine, phosphothreonine and phosphotyrosine standards and electrophoresed in two dimensions with pH1.9 and pH3.5 buffer, respectively, using a previously described protocol (Boyle et al., 1991). (pH3.5 buffer contained 0.87 M acetic acid, 0.5% v/v pyridine and 0.5 mM EDTA.) The sheets were dried, sprayed with 0.2% ninhydrin in ethanol solution (Sigma) and baked at 80 °C for 10 minutes to visualize the phosphor-amino acid standards. Radiolabelled alignment markers were spotted onto the dried sheets and the radioactive spots containing γ32P-ATP, phosphoaminoacids and partially hydrolyzed peptides were visualized with PhosphorImager screens scanned with Scanner Control SI software (Molecular Dynamics) after overnight exposure. These experiments were performed in duplicate.
Transgenesis, breeding, and animal behavior analysis
Animal studies were approved by the Animal Care and Use Committee at UCSF. Generation of transgenic animals and locomotor activity and activity pattern analysis were performed as described previously (Xu et al., 2005). Since Per2−/−, S662WT, S662G, S662D, CKIδ wild-type transgenic, and CKIδ +/− animals were all on a C57BL/6J background, animals could be crossed without requiring generations of backcrossing to generate a homogeneous background. Animals were genotyped using standard protocols.
Western blotting and immunoprecipitation (IP)
Fresh liver extracts from designated circadian times were prepared according to nuclear extraction kit (Actif Motif, http://www.activemotif.com/home) manufacturer’s protocol with the following modification. Briefly, 100mg fresh liver was washed with 5 ml ice-cold PBS/Phosphatase Inhibitors and transferred to 500 μl ice-cold 1 X Hypotonic buffer supplemented with 5 μl 1M DTT and 5 μl provided Detergent and homogenized. After centrifugation for 10 min at 850 X g, cells were gently resuspended in 500 μl 1 X Hypotonic buffer for another 15 min and centrifuged as before. The nuclear pellet was resuspended in 50 μl complete lysis buffer with 10 times protein inhibitor cocktail. The Active Motif protocol was then followed through extraction of nuclear protein. All procedures were performed in 4°C. Cytoplasmic and nuclear fractions were quantified with the Bicinchoninic Acid protein assay kit (Pierce) and aliquoted in liquid nitrogen. For fibroblast cells, cells were harvested from 90 mm dishes at the indicated times after serum shock. In order to increase the visibility of S662G PER2 in Figure 3E, this procedure was modified as follows: 10 times less volume of hypotonic buffer and nuclear lysis buffer with 10 times protein inhibitors were employed to purify cytoplasmic and nuclear fractions. The cytoplasmic and nuclear fractions were mixed with 4 × sample buffer and boiled for 3 min. Proteins were separated by electrophoresis through sodium dodecyl sulfate (SDS)-6% polyacrylamide gels (acrylamide 29.6g: bisacrylamide 0.4 g) and then transferred to nitrocellulose membranes. Membranes were blocked with 3% nonfat dry milk in Tris-buffered saline containing 0.05% Tween 20 and then incubated with hPER2 antibody (Cosmo Bio, Japan) (Miyazaki et al., 2004). Immunoreactive bands were detected using anti-rabbit immunoglobulin G secondary antisera and ECL (Amersham). These experiments were performed in triplicate.
Primary fibroblast culture from humans and mice
Skin biopsies were obtained on a protocol approved by the IRBs at the University of Utah and UCSF. A standard 4 mm skin punch biopsy was performed on the lower leg. Biopsy tissue was cut into several pieces and allowed to adhere onto 35 mm culture dishes for 10 min. 2 ml 10% FCS of DMEM with 5 mM glutamine was added to the dish and incubated at 37°C in a humidified 5% CO2 atmosphere. Explants were monitored for appearance of cells along the edges of tissue pieces every 2 days and media was changed weekly. Outgrowth of spindle-shaped fibroblasts usually occurred within the first 10 days. Keratinocytes appeared prior to fibroblasts but then differentiated and died in passage. When fibroblast outgrowth reached a size of about 1 cm2 (2–4 weeks), tissue pieces were removed gently into a new dish and were cultured again as before. 0.3 ml 0.25% trypsin was added to dishes until cells began to detach. 1.5 ml of DMEM (10% FCS) was added and cells were placed into new 35 mm dishes. Primary cultured fibroblasts were grown to confluence in 10 mm dishes and starved of serum for 24 hr. before synchronization. Cells were harvested at indicated time points. Cytoplasmic and nuclear fractions were analyzed on Western blots. The mouse embryonic fibroblasts were isolated from embryos at day E13.5 following standard procedures (Abbondanzo et al., 1993).
Protein degradation and stability
Confluent cells were cultured with serum-free DMEM for 24 hr. and then were stimulated with 50% horse serum for 2 hr. Proteasome inhibitor (MG132, 50 μM, Calbiochem) was added at t=0 and added again at 4 hr. Lysosome inhibitors (leupeptin, 100 μM and pepstatin, 100 μM, Roche) were added to cells 2 hr. after serum shock and 2hr. before cells were harvested. After treatment with inhibitors, cell extracts were prepared as described above at the times indicated. The Bicinchoninic Acid protein assay kit (Pierce) was applied to adjust for protein amount. Antibody to Tubulin was used as loading control. Control cells were treated with vehicle alone (DMSO) 2 hr. after serum shock. These experiments were performed in triplicate.
Quantitative RT-PCR
Total RNA was extracted using Trizol (Invitrogen) and random hexamers were used to prime reverse-transcription reactions with Superscript III (Invitrogen). Real-time quantitative PCR was performed using an ABI 7700 with SYBR green I reagents. The RT-PCR primers were as previously reported (Yamamoto et al., 2005). Efficiency of amplification and detection by all primers was validated by determining the slope of Ct versus dilution series. Transcript levels for each gene were normalized to Gapdh mRNA levels according to standard procedures. For hPER2 Pre-mRNA accumulation, primers 5’ GGGCGATTCTCTTTTGGGA3’, 5’ CCTTGGTTTCCTTGGCCTTC3’ and probe FAM-CCCTCCCTCCCCTCGCATGG-TAMRA were used to amplify nuclear RNA by real time RT-PCR according to gene specific primer (GSP) protocol (Invitrogen) (Ripperger and Schibler, 2006). These experiments were performed in triplicate.
Plasmid Constructs
The coding region of hPER2 was amplified from human cDNA by PCR using Platinum Taq polymerase and cloned into pCMV-Tag 2B (Promega). QuikChange site-Directed mutagenesis was used to construct hPER2S662G and S662D vectors (Stratagene). The vectors were further modified by adding EGFP. The hPER2 promoter was generated by applying Red/ET recombinant technology and cut off from BAC clone.
Transient transfection and nuclear localization assays
HEK293 cells were plated in 35 mm dishes at a density of 3X105 for 24 hr. before transfected by PolyFect (Qiagen). The fibroblast cells were transfected by LipofectAmine 2000 with Plus reagent (Invitrogen). At indicated time points, cells were fixed with 4% paraformaldehyde, washed with PBS, and mounted with DAPI mounting medium (VECTOR). A random population of 100 cells from each coverslip was examined by confocal laser scanning microscope (Zeiss).
Supplementary Material
Acknowledgments
We thank A. Rothenfluh, Q. Padiath, D. Virshup, P. O’Farrell, and members of the Rosbash Lab for helpful discussions and insightful comments. We also thank S. Reppert and D. Weaver for providing the mPer2ldc mice, A. Sancar for Cry1/Cry2 knockout mice and K. Miyazaki for helpful advice regarding the hPER2 antibody. We thank members of the Fu and Ptáček labs for discussions and technical help. This work was supported in part by a National Institutes of Health grant (to Y.-H.F. & L.J.P.) and a Sandler Neurogenetics grant (to Y.-H.F.). L.J.P. is the John C. Coleman Distinguished Professor of Neurology and an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbondanzo SJ, Gadi I, Stewart CL. Derivation of embryonic stem cell lines. Methods Enzymol. 1993;225:803–823. doi: 10.1016/0076-6879(93)25052-4. [DOI] [PubMed] [Google Scholar]
- Akashi M, Tsuchiya Y, Yoshino T, Nishida E. Control of intracellular dynamics of mammalian period proteins by casein kinase I epsilon (CKIepsilon) and CKIdelta in cultured cells. Mol Cell Biol. 2002;22:1693–1703. doi: 10.1128/MCB.22.6.1693-1703.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht U, Sun ZS, Eichele G, Lee CC. A differential response of two putative mammalian circadian regulators, mper1 and mper2, to light. Cell. 1997;91:1055–1064. doi: 10.1016/s0092-8674(00)80495-x. [DOI] [PubMed] [Google Scholar]
- Bae K, Jin X, Maywood ES, Hastings MH, Reppert SM, Weaver DR. Differential functions of mPer1, mPer2, and mPer3 in the SCN circadian clock. Neuron. 2001;30:525–536. doi: 10.1016/s0896-6273(01)00302-6. [DOI] [PubMed] [Google Scholar]
- Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998;93:929–937. doi: 10.1016/s0092-8674(00)81199-x. [DOI] [PubMed] [Google Scholar]
- Boyle WJ, Smeal T, Defize LH, Angel P, Woodgett JR, Karin M, Hunter T. Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell. 1991;64:573–584. doi: 10.1016/0092-8674(91)90241-p. [DOI] [PubMed] [Google Scholar]
- Camacho F, Cilio M, Guo Y, Virshup DM, Patel K, Khorkova O, Styren S, Morse B, Yao Z, Keesler GA. Human casein kinase Idelta phosphorylation of human circadian clock proteins period 1 and 2. FEBS Lett. 2001;489:159–165. doi: 10.1016/s0014-5793(00)02434-0. [DOI] [PubMed] [Google Scholar]
- Campbell S. Is there an intrinsic period of the circadian clock? Science. 2000;288:1174–1175. [PubMed] [Google Scholar]
- Czeisler CA, Duffy JF, Shanahan TL, Brown EN, Mitchell JF, Rimmer DW, Ronda JM, Silva EJ, Allan JS, Emens JS, et al. Stability, precision, and near-24-hour period of the human circadian pacemaker. Science. 1999;284:2177–2181. doi: 10.1126/science.284.5423.2177. [DOI] [PubMed] [Google Scholar]
- Edery I, Zwiebel LJ, Dembinska ME, Rosbash M. Temporal phosphorylation of the Drosophila period protein. Proc Natl Acad Sci U S A. 1994;91:2260–2264. doi: 10.1073/pnas.91.6.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide EJ, Woolf MF, Kang H, Woolf P, Hurst W, Camacho F, Vielhaber EL, Giovanni A, Virshup DM. Control of mammalian circadian rhythm by CKIepsilon-regulated proteasome-mediated PER2 degradation. Mol Cell Biol. 2005;25:2795–2807. doi: 10.1128/MCB.25.7.2795-2807.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery P, Reppert SM. A rhythmic Ror. Neuron. 2004;43:443–446. doi: 10.1016/j.neuron.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Gallego M, Eide EJ, Woolf MF, Virshup DM, Forger DB. An opposite role for tau in circadian rhythms revealed by mathematical modeling. Proc Natl Acad Sci U S A. 2006;103:10618–10623. doi: 10.1073/pnas.0604511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms E, Kivimae S, Young MW, Saez L. Posttranscriptional and posttranslational regulation of clock genes. J Biol Rhythms. 2004;19:361–373. doi: 10.1177/0748730404268111. [DOI] [PubMed] [Google Scholar]
- Hastings MH, Herzog ED. Clock genes, oscillators, and cellular networks in the suprachiasmatic nuclei. J Biol Rhythms. 2004;19:400–413. doi: 10.1177/0748730404268786. [DOI] [PubMed] [Google Scholar]
- Jin X, Shearman LP, Weaver DR, Zylka MJ, de Vries GJ, Reppert SM. A molecular mechanism regulating rhythmic output from the suprachiasmatic circadian clock. Cell. 1999;96:57–68. doi: 10.1016/s0092-8674(00)80959-9. [DOI] [PubMed] [Google Scholar]
- Jones CR, Campbell SS, Zone SE, Cooper F, DeSano A, Murphy PJ, Jones B, Czajkowski L, Ptacek LJ. Familial advanced sleep-phase syndrome: A short-period circadian rhythm variant in humans [see comments] Nat Med. 1999;5:1062–1065. doi: 10.1038/12502. [DOI] [PubMed] [Google Scholar]
- Klerman EB, Dijk DJ, Kronauer RE, Czeisler CA. Simulations of light effects on the human circadian pacemaker: implications for assessment of intrinsic period. Am J Physiol. 1996;270:R271–282. doi: 10.1152/ajpregu.1996.270.1.R271. [DOI] [PubMed] [Google Scholar]
- Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, Maywood ES, Hastings MH, Reppert SM. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell. 1999;98:193–205. doi: 10.1016/s0092-8674(00)81014-4. [DOI] [PubMed] [Google Scholar]
- Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107:855–867. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- Lowrey PL, Shimomura K, Antoch MP, Yamazaki S, Zemenides PD, Ralph MR, Menaker M, Takahashi JS. Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science. 2000;288:483–492. doi: 10.1126/science.288.5465.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrey PL, Takahashi JS. Mammalian circadian biology: elucidating genome-wide levels of temporal organization. Annu Rev Genomics Hum Genet. 2004;5:407–441. doi: 10.1146/annurev.genom.5.061903.175925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki K, Nagase T, Mesaki M, Narukawa J, Ohara O, Ishida N. Phosphorylation of clock protein PER1 regulates its circadian degradation in normal human fibroblasts. Biochem J. 2004;380:95–103. doi: 10.1042/BJ20031308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawathean P, Rosbash M. The doubletime and CKII kinases collaborate to potentiate Drosophila PER transcriptional repressor activity. Mol Cell. 2004;13:213–223. doi: 10.1016/s1097-2765(03)00503-3. [DOI] [PubMed] [Google Scholar]
- Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, Schibler U. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110:251–260. doi: 10.1016/s0092-8674(02)00825-5. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. Molecular analysis of mammalian circadian rhythms. Annu Rev Physiol. 2001;63:647–676. doi: 10.1146/annurev.physiol.63.1.647. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- Ripperger JA, Schibler U. Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nat Genet. 2006;38:369–374. doi: 10.1038/ng1738. [DOI] [PubMed] [Google Scholar]
- Sato TK, Panda S, Miraglia LJ, Reyes TM, Rudic RD, McNamara P, Naik KA, FitzGerald GA, Kay SA, Hogenesch JB. A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron. 2004;43:527–537. doi: 10.1016/j.neuron.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Shearman LP, Sriram S, Weaver DR, Maywood ES, Chaves I, Zheng B, Kume K, Lee CC, van der Horst GT, Hastings MH, et al. Interacting molecular loops in the mammalian circadian clock [see comments] Science. 2000;288:1013–1019. doi: 10.1126/science.288.5468.1013. [DOI] [PubMed] [Google Scholar]
- Tamanini F, Yagita K, Okamura H, van der Horst GT. Nucleocytoplasmic shuttling of clock proteins. Methods Enzymol. 2005;393:418–435. doi: 10.1016/S0076-6879(05)93020-6. [DOI] [PubMed] [Google Scholar]
- Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, Ptacek LJ, Fu YH. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science. 2001;291:1040–1043. doi: 10.1126/science.1057499. [DOI] [PubMed] [Google Scholar]
- Ueda HR, Chen W, Adachi A, Wakamatsu H, Hayashi S, Takasugi T, Nagano M, Nakahama K, Suzuki Y, Sugano S, et al. A transcription factor response element for gene expression during circadian night. Nature. 2002;418:534–539. doi: 10.1038/nature00906. [DOI] [PubMed] [Google Scholar]
- Ueda HR, Hayashi S, Chen W, Sano M, Machida M, Shigeyoshi Y, Iino M, Hashimoto S. System-level identification of transcriptional circuits underlying mammalian circadian clocks. Nat Genet. 2005;37:187–192. doi: 10.1038/ng1504. [DOI] [PubMed] [Google Scholar]
- Xu Y, Pdaiath Q, Shapiro R, Jones CR, Wu SM, Saigoh N, Saitgoh K, Ptacek L, Fu Y. Functional consequences of a CKId mutation causing familial advanced sleep phase syndrome. Nature. 2005 doi: 10.1038/nature03453. [DOI] [PubMed] [Google Scholar]
- Yagita K, Tamanini F, Yasuda M, Hoeijmakers JH, van der Horst GT, Okamura H. Nucleocytoplasmic shuttling and mCRY-dependent inhibition of ubiquitylation of the mPER2 clock protein. Embo J. 2002;21:1301–1314. doi: 10.1093/emboj/21.6.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Yagita K, Okamura H. Role of cyclic mPer2 expression in the mammalian cellular clock. Mol Cell Biol. 2005;25:1912–1921. doi: 10.1128/MCB.25.5.1912-1921.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MW, Kay SA. Time zones: a comparative genetics of circadian clocks. Nat Rev Genet. 2001;2:702–715. doi: 10.1038/35088576. [DOI] [PubMed] [Google Scholar]
- Yu W, Nomura M, Ikeda M. Interactivating feedback loops within the mammalian clock: BMAL1 is negatively autoregulated and upregulated by CRY1, CRY2, and PER2. Biochem Biophys Res Commun. 2002;290:933–941. doi: 10.1006/bbrc.2001.6300. [DOI] [PubMed] [Google Scholar]
- Zheng B, Larkin DW, Albrecht U, Sun ZS, Sage M, Eichele G, Lee CC, Bradley A. The mPer2 gene encodes a functional component of the mammalian circadian clock. Nature. 1999;400:169–173. doi: 10.1038/22118. [DOI] [PubMed] [Google Scholar]
- Zylka MJ, Shearman LP, Levine JD, Jin X, Weaver DR, Reppert SM. Molecular analysis of mammalian timeless. Neuron. 1998;21:1115–1122. doi: 10.1016/s0896-6273(00)80628-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.