

Summary
The tumor suppressor PTEN, a critical regulator for multiple cellular processes, is mutated or deleted frequently in various human cancers. Subtle reductions in PTEN expression levels have profound impacts on carcinogenesis. Here we show that PTEN level is regulated by ubiquitin-mediated proteasomal degradation, and purified its ubiquitin ligase as HECT-domain protein NEDD4-1. In cells, NEDD4-1 negatively regulates PTEN stability by catalyzing PTEN polyubiquitination. Consistent with the tumor suppressive role of PTEN, overexpression of NEDD4-1 potentiated cellular transformation. Strikingly, in a mouse cancer model and multiple human cancer samples where the genetic background of PTEN was normal but its protein levels were low, NEDD4-1 was highly expressed, suggesting aberrant upregulation of NEDD4-1 can posttranslationally suppress PTEN in cancers. Elimination of NEDD4-1 expression inhibited xenotransplanted tumor growth in a PTEN-dependent manner. Therefore, NEDD4-1 is a potential proto-oncogene that negatively regulates PTEN via ubiquitination, a paradigm analogous to that of Mdm2 and p53.
Introduction
The tumor suppressor PTEN plays critical roles in cell growth, migration, and death (Di Cristofano et al., 1998; Raftopoulou et al., 2004; Sansal and Sellers, 2004; Stambolic et al., 1998; Tamura et al., 1998). It is mutated or deleted with high frequency in various human cancer tissues to promote tumorigenesis (Li et al., 1997; Sansal and Sellers, 2004; Steck et al., 1997). Mouse models for human cancer revealed that PTEN is haploinsufficient (Kwabi-Addo et al., 2001; Trotman et al., 2003). Molecularly, PTEN is a phosphatase for second messenger phosphatidyl inositol-3,4,5-triphosphate (PIP3) (Maehama et al., 2001; Sansal and Sellers, 2004; Stambolic et al., 1998), which is required for activation of kinase AKT/PKB and the downstream cellular survival and growth responses (Luo et al., 2003).
The crucial function of PTEN in multiple cellular processes and its involvement in human diseases suggest that the enzyme needs to be deliberately regulated in vivo. This notion is supported by a recent genetic study showing quantitatively that subtle decreases of PTEN expression in mice predispose to tumorigenesis and accelerate cancer progression (Trotman et al., 2003). Gene deletion and mutation of PTEN occur frequently during cancer development. These genetic changes, however, are not necessarily the only ways to inactivate PTEN during tumorigenesis: a defect in posttranslational regulation might also be able to suppress this important tumor suppressor.
Previous reports indicate that PTEN is indeed regulated by multiple posttranslational mechanisms. For example, a repressor of PTEN named DJ-1 was genetically isolated recently from Drosophila (Kim et al., 2005). However, how this potential oncoprotein represses PTEN function biochemically is not clear. PTEN has been shown to be phosphorylated in cells, and the phosphorylation appears to affect PTEN function (Adey et al., 2000; Leslie and Downes, 2004; Torres and Pulido, 2001; Vazquez et al., 2000). Again, the mechanisms that regulate PTEN phosphorylation and dephosphorylation, and the exact mechanisms by which PTEN phosphorylation impacts its function are not clear.
In addition, PTEN might also be regulated by ubiquitin-mediated proteasomal degradation, a common mechanism to control protein levels posttranslationally (Ciechanover et al., 1984; Hershko and Ciechanover, 1998). PTEN has two canonical PEST motifs, a signature in many short-lived proteins degraded by the ubiquitin pathway (Rogers et al., 1986), and treatment of cells with proteasome inhibitors can cause an increase of PTEN protein level (Torres and Pulido, 2001; Wu et al., 2003). These results prompted us to investigate the role of ubiquitin-mediated proteasomal degradation in regulation of PTEN. In this study, we report identification of the ubiquitin ligase for PTEN and its roles in regulating PTEN and tumorigenesis.
Results
In vivo and In vitro PTEN Ubiquitination
We first examined whether PTEN is ubiquitinated in cells. 293T cells were cotransfected with plasmids encoding for His-tagged PTEN and HA-tagged ubiquitin. The expressed His-tagged PTEN was specifically pulled down from cell lysates by Ni2+-NTA beads, and nonspecific proteins and indirectly associated proteins were washed away using 6 M guanidine as reported (Treier et al., 1994). Subsequently, immunoblotting against HA-tag was performed to detect ubiquitinated PTEN (Figure 1A). We found that the overexpressed PTEN was polyubiquitinated, and the polyubiquitinated PTEN was the substrate for proteasomes, because treatment of cells with the proteasome inhibitor MG132 caused a robust increase of PTEN polyubiquitination (Figure 1A).
Figure 1. Polyubiquitination of PTEN in vivo and in vitro.
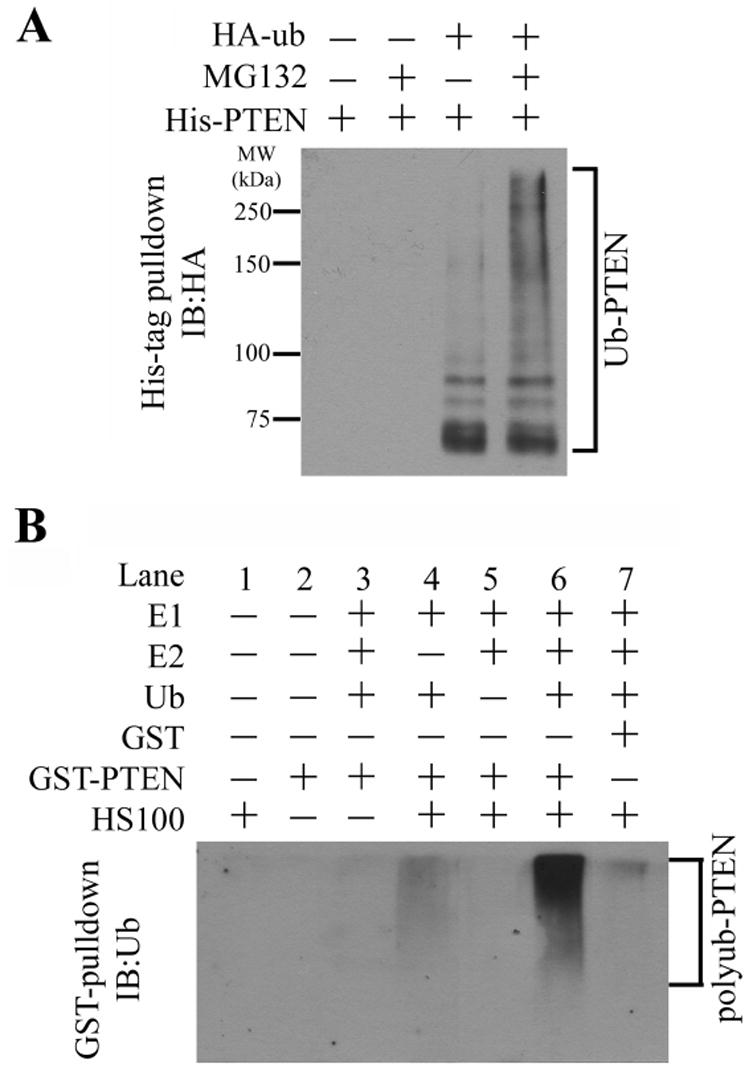
(A) Polyubiquitination of transfected PTEN. C-terminal His-tagged PTEN was co-transfected with HA-tagged ubiquitin (HA-Ub) in 293T cells as indicated. The cells were treated with or without 25 μM proteasome inhibitor MG132 for 4 hours and then harvested and lysed. His-tagged PTEN was pulled down with Ni2+ beads, washed with 6 M guanidine as described in Experimental Procedures, and subjected to immunoblotting against HA-tag to detect ubiquitinated PTEN.
(B) In vitro ubiquitination of PTEN. The in vitro assay was performed as described in Experimental Procedures, with individual components added as indicated. GST-PTEN was used as the substrate, and HeLa S-100 (HS100, 5 μl and 5 mg protein/ml) was required to provide the PTEN ubiquitin ligase (E3) activity.
We developed a biochemical assay that can detect polyubiquitination of PTEN in vitro. We used recombinant GST-tagged PTEN as the substrate. After incubation with recombinant ubiquitin activating enzyme (E1), ubiquitin conjugating enzyme UbcH5C or UbcH7 (E2), ATP, ubiquitin, and HeLa cell cytosolic protein fraction (HeLa S-100) as indicated (Figure 1B), GST-PTEN was pulled-down by glutathione-Sepharose, and polyubiquitination of GST-PTEN was analyzed by immunoblotting against ubiquitin. In such an assay, we observed HeLa S-100-dependent polyubiquitination of GST-PTEN (Figure 1B, lane 6) but not of GST (lane 7). Requirement of HeLa S-100 indicates that HeLa S-100 provides an ubiquitin ligase activity for PTEN.
Purification and Identification of the Ubiquitin Ligase for PTEN
In the polyubiquitination process, the substrate specificity is conferred by ubiquitin ligases, also known as E3 (Ciechanover et al., 1984; Hershko and Ciechanover, 1998). Ubiquitin ligases are also the crucial targets for biological regulation of the process (Ciechanover et al., 1984; Hershko and Ciechanover, 1998). Therefore, we set out to purify and identify the ubiquitin ligase for PTEN from HeLa S-100 by using the in vitro assay we developed. After a seven-step purification concluding with Mono Q chromatography (Figure 2A), we obtained strong ubiquitin ligase activity for PTEN (fractions 9 and 10, Figure 2B) correlating with a single protein band stained with silver in SDS-PAGE (Figure 2C). The size of this protein is 110 to 120 kDa. The purified E3 activity could support polyubiquitination of recombinant PTEN with a C-terminal HA-tag (Figure 2D, detected by an anti-HA antibody), thus ruling out any possible artifact that might be caused by the bulky GST tag of GST-PTEN.
Figure 2. Purification and identification of the PTEN ubiquitin ligase as NEDD4-1.

(A) The purification scheme. The detailed procedure was described in Experimental Procedures.
(B) The PTEN E3 activity in fractions (Frac, 2 μl each) from the final Mono Q column was measured by the in vitro PTEN ubiquitination assay.
(C) The final Mono Q fractions (15 μl each) were resolved by SDS-PAGE and stained with silver.
(D) The purified PTEN E3 activity can ubiquitinate HA-tagged recombinant PTEN. The purified E3 (2 μl) or HS100 (5 μl) was used in the reactions as indicated. The ubiquitinated PTEN was detected by immunoblotting against HA-tag.
(E) The domain structure of NEDD4-1. Amino acid positions of individual domains are denoted by corresponding numbers.
(F) Interaction of NEDD4-1 with PTEN in 293T cells determined by coimmunoprecipitation analysis. PTEN and HA-tagged NEDD4-1 (N4-HA) were transfected as indicated. After immunoprecipitation using anti-HA antibody, PTEN and N4 in the precipitates were detected by immunoblotting (the upper two panels). The cell lysate (before immunoprecipitation) was blotted for N4, PTEN, and tubulin (the lower three panels) as controls.
(G) Direct physical interaction of PTEN with NEDD4-1 (N4) determined by a GST pull-down assay. GST-PTEN but not GST alone pulled down purified NEDD4-1.
Subsequently, we conducted mass spectrometry analysis (MALDI-TOF-MS/MS) to determine the identity of the purified PTEN E3. The Mass fingerprinting matched human protein NEDD4-1 (neural precursor cell expressed, developmentally downregulated-4-1), a putative ubiquitin ligase containing a HECT domain.
It appears that the NEDD4-1 cDNA used in earlier publications all missed the N terminus of the open reading frame (ORF). We thus deduced the complete ORF based on human genomic sequence, and subsequently cloned it by connecting the coding region for its N terminus (from HeLa cells by genomic PCR) with the overlapping C-terminal part (from KIAA0093 clone (Nagase et al., 1995)). The full-length NEDD4-1 protein contains several functional motifs (Figure 2E). In the N terminus, there are a proline-rich domain and a poly-serine motif followed by a C2 domain. NEDD4-1 also contains four WW domains in the middle, and the signature ubiquitin ligase domain, the HECT domain, in the C terminus.
NEDD4-1 can interact with PTEN in cells as shown by coimmunoprecipitation analysis (Figure 2F). Further, in an in vitro GST pull-down experiment, GST-PTEN but not GST alone pulled down purified NEDD4-1 (Figure 2G). Therefore, the interaction of NEDD4-1 with PTEN is direct.
NEDD4-1 but not NEDD4-2 can ubiquitinate PTEN
NEDD4-1 has a close homolog, NEDD4-2 (Figure S1, in Supplemental Data). The human genomic loci of these two homologs are different, 15q21 for NEDD4-1 and 18q21 for NEDD4-2. Although the mass fingerprinting did not match with NEDD4-2, there were two peptide sequences obtained from the MS analysis existing in both proteins (Figure S1). To check whether there was NEDD4-2 in our purified PTEN E3 fraction, we performed immunoblotting analysis using antibodies that can distinguish human NEDD4-1 and NEDD4-2 (Kamynina et al., 2001). Although both proteins could be clearly detected in HeLa S-100, only NEDD4-1 but not NEDD4-2 was detected in the purified PTEN E3 fraction (Figure 3A). Further, using an anion exchange Mono Q column, we separated NEDD4-1 from NEDD4-2 in HeLa S-100, and found that the NEDD4-1 fractions but not NEDD4-2 fractions contained ubiquitin ligase activity for PTEN (Figure 3B). Therefore, NEDD4-1 but not NEDD4-2 can function as the ubiquitin ligase for PTEN.
Figure 3. NEDD4-1 but not NEDD4-2 is an ubiquitin ligase for PTEN.

(A) NEDD4-1 but not NEDD4-2 was presented in the purified PTEN E3 fraction. Different volume of HeLa S-100 (HS100, 5 mg protein/ml) and purified PTEN E3 activity (E3) were subjected to immunoblot against NEDD4-1 and NEDD4-2 as indicated.
(B) NEDD4-1 fractions but not NEDD4-2 fractions possess PTEN E3 activity. NEDD4-1 and NEDD4-2 in HS100 were separated by Mono Q chromatography as shown by immunoblotting (top panel). The PTEN E3 activity in 5 μl of individual fractions (fractions 13 and 14 as NEDD4-1 fractions; 19 and 20 as NEDD4-2 fractions; fraction 11 as a negative control) was measured (bottom panel).
NEDD4-1 can ubiquitinate PTEN in Cells
We confirmed the role of NEDD4-1 as PTEN E3 in cells. 293T cells were cotransfected with His-tagged PTEN and HA-ubiquitin, in the presence or absence of NEDD4-1 overexpression. Overexpression of NEDD4-1 in 293T cells caused an increase of PTEN polyubiquitination in a NEDD4-1 dose-dependent manner (Figure 4A and Figure S2). As expected, a high level of NEDD4-1 overexpression also caused a decrease of endogenous PTEN protein in 293T cells, and such decrease of PTEN protein level can be blocked by proteasomal inhibitor MG132 (Figure 4B). Further, we showed that NEDD4-1 overexpression can stimulate polyubiquitination of endogenous PTEN in 293T cells (Figure 4C, lane 3), and the NEDD4-1-catalyzed PTEN polyubiquitination can be further enhanced by treatment of MG132 (Figure 4C, lane 4). These results indicate that NEDD4-1 can polyubiquitinate endogenous PTEN and thereby target PTEN to proteasomal degradation.
Figure 4. Regulation of PTEN ubiquitination and degradation by NEDD4-1 in cells.

(A) Overexpression of NEDD4-1 enhances polyubiquitination of PTEN. His-tagged PTEN, HA-Ub, and increasing amount of NEDD4-1 were co-transfected in 293T cells as indicated. The ubiquitination of His-tagged PTEN was detected as described in Figure 1A. Lower panel shows expression of NEDD4-1.
(B) Overexpression of NEDD4-1 causes a decrease in PTEN protein level. As indicated, NEDD4-1 with a C-terminal HA-tag or vector alone was transfected in 293T cells by electroporation. After 24 hours, the cells were treated with or without 50 μM MG132 for 4 hours as indicated. Subsequently, the cells were harvested and lysed, and the endogenous PTEN of individual samples was detected by immunoblotting. β-Tubulin was used as the loading control. PTEN levels (relative to Tubulin) were quantitated by densitometry.
(C) NEDD4-1 polyubiquitinates endogenous PTEN and targets it for proteasomal degradation. As described in Experimental Procedures, His-tagged ubiquitin and NEDD4-1-HA were transfected into 293T cells as indicated. The cells were treated with or without MG132 as indicated, and then harvested and lysed. Proteins modified with His-tagged ubiquitin were pulled down and subjected to immunoblotting against PTEN.
(D) RNAi of NEDD4-1. The efficacy of several human NEDD4-1 RNAi constructs (iN4, A–C) was examined by immunoblotting against HA-tag in HeLa cells co-transfected with NEDD4-1-HA and indicated RNAi constructs. A LacZ RNAi construct (iLacZ) was used as control.
(E) Elimination of NEDD4-1 by RNAi increases PTEN level in cells. Individual RNAi constructs were co-transfected into HeLa cells with PTEN-HA as indicated. The expressed PTEN-HA was detected by immunoblotting.
(F) NEDD4-1 overexpression decreases PTEN stability. As detailed in Experimental Procedures, PTEN plasmid was cotransfected with NEDD4-1 or control plasmid as indicated, cells were treated with cycloheximide, and harvest at indicated time points. Subsequently, immunoblotting against PTEN, NEDD4-1 (both HA-tagged), and tubulin control was performed.
(G) NEDD4-1 RNAi increases PTEN stability. PTEN was cotransfected with iN4A (NEDD4-1 RNAi) or control plasmid as indicated, cells were treated with cycloheximide, and harvest at indicated time points. Subsequently, immunoblotting against PTEN and tubulin control was performed.
Using RNAi approach, we demonstrated quantitatively that endogenous NEDD4-1 negatively regulates PTEN levels (Figures 4D and 4E). We generated several shRNA constructs (iN4 plasmids) against human NEDD4-1. Among them, iN4A and iN4B can strongly and modestly reduce NEDD4-1 expression respectively, while iN4C had no effect (Figure 4D). Consistently, iN4A and iN4B caused high and modest increase of PTEN protein level, but iN4C failed to increase PTEN in cells (Figure 4E).
We further showed that negative regulation of PTEN steady-state level by NEDD4-1 is indeed through modulating PTEN degradation. To examine the rate of PTEN degradation, PTEN was co-transfected with plasmids for NEDD4-1 overexpression or RNAi knockdown. Subsequently, protein translation in cells were inhibited by cycloheximide treatment, and PTEN protein levels were detected at the indicated times of treatment. As shown in Figures 4F and 4G, overexpression of NEDD4-1 and RNAi knockdown of NEDD4-1 caused an increase and decrease of PTEN degradation rate, respectively.
Taken together, we conclude that NEDD4-1 is a physiological ubiquitin ligase for PTEN that targets PTEN to proteasomal degradation.
NEDD4-1 regulates tumorigenesis
PTEN is a potent tumor suppressor that negatively regulates the PI3K-Akt pathway. Therefore, the ubiquitin ligase of PTEN, NEDD4-1, might function as an oncogene. To test this possibility, we first determined whether endogenous NEDD4-1 can regulate AKT signaling via PTEN (Figure 5A). Indeed, knockdown of NEDD4-1 by RNAi in 293 cells caused an increase in endogenous PTEN and a concurrent decrease of AKT phosphorylation. Further, we performed soft-agar colony formation assays in p53−/− primary mouse embryonic fibroblasts (MEFs) (Jacks et al., 1994; Pao et al., 2003) to measure the cell transforming ability of NEDD4-1. In Figure 5B, we confirmed that overexpression of NEDD4-1 could cause a decrease of PTEN in the p53−/− primary MEFs. In soft-agar colony formation experiments, although overexpression of NEDD4-1 alone did not induce transformation, it dramatically enhanced the oncogenic capability of oncogenic K-Ras, as shown by an increase of both number and size of cell colonies compared with that caused by K-Ras alone (Figure 5C). Intriguingly, NEDD4-1 coexpression with K-Ras reproducibly resulted in a larger number but smaller size of colonies than that caused by coexpression of Myc with Ras (data not shown). This suggests that cooperation of NEDD4-1 with Ras might provide greater survival advantage but less growth advantage than Myc-Ras cooperation.
Figure 5. NEDD4-1 potentiates cell transformation in a PTEN-dependent manner.
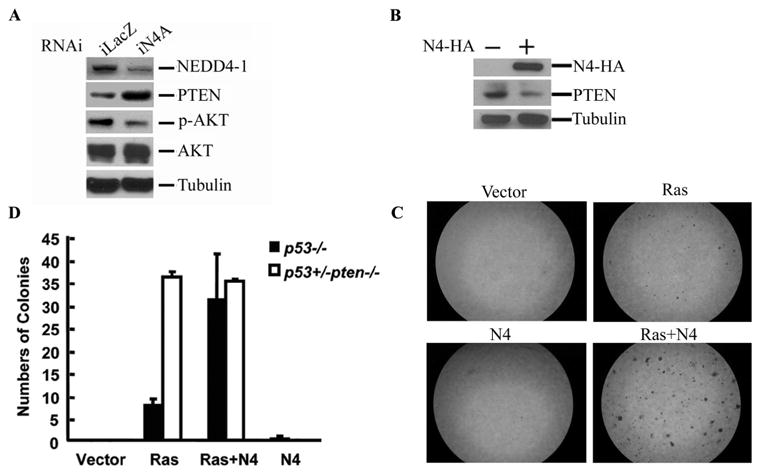
(A) NEDD4-1 regulates AKT phosphorylation. RNAi plasmid iLacZ or iN4A was transfected into 293 cells by electroporation. Immunoblotting against NEDD4-1, PTEN, phospho-AKT (p-Akt), total AKT, and β-tubulin was performed as indicated.
(B) Overexpression of NEDD4-1 in p53−/− primary MEFs decreased endogenous PTEN level. The MEFs were transfected with vector control or NEDD4-1 (N4-HA) as indicated by electroporation. The cells were lysed 24 hours later. N4-HA and endogenous PTEN were detected by immunoblotting. β-tubulin was blotted as the loading control.
(C) Overexpression of NEDD4-1 promotes Ras-induced cell transformation in p53−/− primary MEFs. The p53−/− primary MEFs were infected with retroviruses encoding for no ectopic protein (Vector), Ras, and/or NEDD4-1 (N4), as indicated. Soft-agar colony formation assays were performed as described in Experimental Procedures. The pictures showing the typical colony formation assay result.
(D) Overexpression of NEDD4-1 promotes cell transformation in a PTEN-dependent manner. The colony formation assays were performed in both p53−/− primary MEFs and p53+/−Pten−/− primary MEFs. The plot represents the results from three independent experiments with standard deviation.
To examine whether such oncogenic activity of NEDD4-1 is PTEN-dependent, we performed the same colony-formation assays in p53+/−Pten−/− primary MEFs (Chen et al., 2005). In such Pten-null background, NEDD4-1 overexpression failed to enhance the transforming activity of Ras (Figure 5D and Figure S3). Therefore, the oncogenic activity of NEDD4-1 is PTEN-dependent.
To determine whether the oncogenic function of NEDD4-1 is relevant physiologically to cancer development, we utilized a mouse model that has a hypomorphic allele and a null allele of Pten (Ptenhy/−) (Chen et al., 2005; Trotman et al., 2003). Prostates in Ptenhy/− mice have only 30% of PTEN protein level compared to that in wild-type mice, and display accelerated prostate tumorigenesis compared to Pten+/− mice (Trotman et al., 2003). Intriguingly, tumors consistently arise from single glands that contain lower PTEN protein levels than their noncancerous neighbors and proliferate more rapidly (Figure 6A). This further decrease of PTEN protein in the tumor lesions, likely an initiating oncogenic event, was not caused by loss of the hypomorphic Pten allele, because a low level of PTEN protein is always clearly detectable (Figure 6A). To test if it was caused by overexpression of NEDD4-1 and subsequent accelerated PTEN degradation, we performed immunohistochemistry (IHC) staining of such early prostate lesions. Neoplastic areas of Ptenhy/− prostates are easily identified by strong phospho-Akt staining (Figure 6A, p-Akt, tm) and these glands invariably display lower PTEN levels than the normal ones (Pten, tm versus. nl). Importantly, by using an antibody that is specific for NEDD4-1 (Figure S4), NEDD4-1 was found consistently higher in areas of low PTEN (Figure 6A, N4, tm vs. nl), strongly suggesting that NEDD4-1 dictates PTEN protein levels in these tissues and thus is a key regulator of tumorigenesis in prostate.
Figure 6. Reverse correlation of NEDD4-1 and PTEN levels in cancer samples.
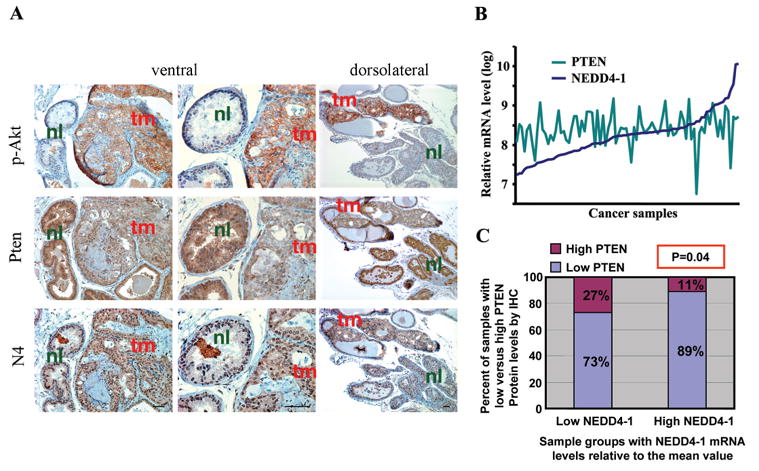
(A) High levels of NEDD4-1 inversely correlate further decrease of PTEN levels in a mouse prostate cancer model. The panel shows typical result of PTEN, NEDD4-1 (N4), and p-Akt IHC staining of Ptenhy/− mouse prostate tissues. The IHC shows that high NEDD4-1 expression levels always correlate with low PTEN levels which in turn correspond with high p-Akt and prostate tumour initiation (P-value < 0.001 calculated by Chi-square test). Glands with tumor (tm) and normal glands (nl) are indicated.
(B) The mRNA levels of NEDD4-1 and PTEN in a cohort of 87 invasive human bladder cancer samples. The mRNA levels were measured using Affymetrix 2.0 Plus Chip.
(C) Reverse correlation of NEDD4-1 mRNA levels with PTEN protein levels in the human bladder cancer samples. The 87 samples were classified into two groups (low NEDD4-1, 40 samples, and high NEDD4-1, 47 samples) based on the NEDD4-1 mRNA level of each sample relative to the mean NEDD4-1 mRNA value of the whole cohort. Then the PTEN IHC status of the two groups was plotted. The inverse correlation of NEDD4-1 mRNA levels with PTEN protein levels in these samples is statistically significant with a P-value of 0.04 calculated by both Chi-square and Mann-Whitney tests.
We then studied the clinical relevance of NEDD4-1 in human cancers. Previously we found that certain human bladder cancer carcinomas with normal PTEN mRNA levels displayed surprisingly low to undetectable PTEN protein expression. In light of the function of NEDD4-1 as PTEN ubiquitin ligase, we hypothesize that overexpression of NEDD4-1 might contribute to the low PTEN protein in these samples. Therefore, we examined a cohort of invasive human bladder cancer samples (87 cases) whose mRNA array results and PTEN IHC status were documented. These samples showed relatively normal PTEN mRNA levels (Figure 6B). Many of them have high NEDD4-1 mRNA levels, and a subset of these samples (~12%) expressed more than 5-fold higher NEDD4-1 mRNA levels above the median value (Figure 6B). At mRNA level, there was no correlation between NEDD4-1 and PTEN expression. However, when analyzing the mRNA array and PTEN IHC data of these samples, an inverse correlation between NEDD4-1 mRNA levels and PTEN protein levels was observed with statistic significance (Figure 6C). Further, when these samples were classified into two groups as those with high PTEN protein levels and those with low PTEN protein levels, the mean NEDD4-1 mRNA value of the latter group is two-fold of the mean value of the other group (data not shown). Therefore, we postulate that in these human bladder cancer samples, overexpression of NEDD4-1 contributes to the posttranslational downregulation of PTEN.
Given that NEDD4-1 possesses oncogenic function, we examined whether elimination of NEDD4-1 expression by RNAi could inhibit tumor growth. We used two human prostate cancer cell lines, DU-145 and PC3, for this purpose. After RNAi, the cells were injected into athymic nude mice, and the xenotransplanted tumor growth was measured. We found that when DU-145 cells, which are PTEN-positive, were used as the tumor donor, NEDD4-1 RNAi caused a clear inhibition of tumor growth compared with control LacZ RNAi (Figure 7A). However, when PC3 cells, which are PTEN-negative, were used as the tumor donor, NEDD4-1 RNAi had no effect on tumor growth (Figure 7A). Therefore, elimination of NEDD4-1 is effective in inhibiting tumor growth in a PTEN-dependent manner. This result suggests that NEDD4-1 might be a valuable therapeutic target in treating PTEN-positive cancers.
Figure 7. NEDD4-1 RNAi inhibited xenotransplanted tumor growth in a PTEN-dependent manner.

(A) The human prostate cancer cell lines, DU-145 (PTEN-positive) and PC3 (PTEN-negative), were stably infected with NEDD4-1 RNAi and the control LacZ RNAi retroviruses. The infected cells were subcutaneously injected into athymic nude mice and the xenotransplanted tumor growth was measured, as described in Experimental Procedures.
(B) A Model for the potential function of NEDD4-1 in prostate tumorigenesis. After loss of one allel of PTEN, aberrant upregulation of NEDD4-1 function will further decrease PTEN protein level and thus accelerates cancer development. Upregulation of NEDD4-1 might lead to malignancy more rapidly than a complete loss of PTEN gene, because the latter will trigger INK4a and p53-dependent senescence (Chen et al., 2005). NEDD4-1 offers a new entry point for therapeutic intervention that could arrest tumorigenesis in cooperation with the senescence response.
Discussion
In this study, we have identified NEDD4-1 as an ubiquitin ligase for the tumor suppressor PTEN. NEDD4-1 can polyubiquitinate PTEN and thus targets PTEN for proteasomal degradation. This finding opens a new avenue to study the roles of PTEN in apoptosis, tumorigenesis, and other important processes.
NEDD4-1 is a HECT-domain ubiquitin ligase. It should be noted that NEDD4-1 has been reported to be ubiquitin ligase for multiple viral Gag proteins (Blot et al., 2004; Strack et al., 2000), which are exogenous proteins. The close homolog of NEDD4-1, NEDD4-2, cannot ubiquitinate PTEN; on the other hand, NEDD4-2 but not NEDD4-1 is the ubiquitin ligase for epithelial and cardiac voltage-gated sodium channels (Kamynina et al., 2001; Rougier et al., 2005). How these two highly homologous enzymes possess distinct substrate specificity is of interest for future study. Further, there are additional NEDD4-like ubiquitin ligases with distinct functions in mammals as well as in other organisms (Ingham et al., 2004). For example, NEDD4 family proteins have been shown to downregulate Notch signaling in Drosophila (Fostier et al., 1998), and ΔNp63α, a p53-related transcriptional factor, in zebrafish (Bakkers et al., 2005).
Importantly, our study indicates that NEDD4-1 can potentiate tumorigenesis, and aberrant activation of NEDD4-1 can downregulate PTEN function. Thus, the relationship of NEDD4-1 with PTEN is similar to that of Mdm2 versus p53, in which the oncoprotein Mdm2 is an ubiquitin ligase for the tumor suppressor p53 (Bond et al., 2005; Brooks and Gu, 2003; Michael and Oren, 2002). Interestingly, posttranslational modification of both Mdm2 and p53, such as phosphorylation and acetylation, can regulate the binding and activity of Mdm2 toward p53 (Bond et al., 2005; Brooks and Gu, 2003; Michael and Oren, 2002). Similar modes of regulation might also exist for NEDD4-1 and PTEN. For example, PTEN can be phosphorylated, and its phosphorylation has been implicated in its function and stability (Adey et al., 2000; Leslie and Downes, 2004; Torres and Pulido, 2001; Vazquez et al., 2000). Therefore, PTEN phosphorylation might somehow modulate its own ubiquitination by NEDD4-1. Further, NEDD4-1 possesses multiple domain structures, suggesting that its enzymatic activity could also be closely regulated.
Nevertheless, regulation of PTEN by NEDD4-1 may also prove to be fundamentally different from that of p53 by Mdm2. It is well known that p53 is a fast turnover protein while PTEN is a relatively stable protein. Thus a cellular requirement for higher p53 levels is met by inhibition of its degradation. We propose that PTEN degradation is modulated in the opposite manner: PTEN is by default a stable protein and its degradation is accelerated when needed. This could be achieved by posttranslational activation of NEDD4-1 function, in addition to increase of its expression level, as suggested by many of our observations. For example, in certain cell lines, we observed stable PTEN levels despite high levels of NEDD4-1, and only a very robust overexpression of NEDD4-1 could reduce PTEN protein levels; NEDD4-1-mediated PTEN degradation appears to be deliberately affected by cell adhesion and growth condition; notably, the potent PTEN ubiquitination activity of purified NEDD4-1 can be dramatically inhibited by HeLa cell extracts in vitro, suggesting dominant posttranslational regulation of NEDD4-1 (X.W. and X.J., unpublished data). Furthermore, NEDD4-1 can also cause PTEN nuclear transport via catalyzing PTEN mono-ubiquitination (Trotman et al, in press). Intriguingly, nuclear transport of PTEN may protect PTEN from being further polyubiquitinated by NEDD4-1 (a predominantly cytoplasmic protein) and subsequently degraded by proteasome. Therefore, NEDD4-1 might modulate PTEN function both positively and negatively, depending on the amount, activation, and localization of NEDD4-1. Taken together, while we have identified NEDD4-1 as the ubiquitin ligase for PTEN, this finding in turn raised many other important questions, such as, how is NEDD4-1 activity regulated in vivo? And what physiological triggers can induce NEDD4-1-mediated PTEN downregulation versus nuclear translocation?
Interestingly, while p53-loss or Mdm2 hyperactivation or amplification could result in comparable outcomes favoring tumor progression, NEDD4-1 might play a more complex role in PTEN-loss-driven tumorigenesis: in contrast to p53-loss, although reductions in PTEN levels have dramatic consequences on carcinogenesis (Trotman et al., 2003), the complete loss of PTEN triggers a cell-autonomous senescence response, which antagonizes tumor growth (Chen et al., 2005). Thus, strong NEDD4-1 activity might be promoting tumorigenesis in a PTEN-heterozygous background more effectively than complete PTEN-loss, because NEDD4-1 could attenuate PTEN function and thereby enhance proliferation without triggering senescence. Considering that a large fraction of primary human cancers, for example, about 70% of prostate cancers (Gray et al., 1998; Whang et al., 1998), display loss of only one PTEN allele, we propose that they are at risk of carcinogenesis driven by high NEDD4-1 activity (Figure 7B, red arrows). Conversely, these cancers may benefit significantly from therapies specifically targeting NEDD4-1 (Figure 7B, green arrows).
Experimental Procedures
Preparation of Recombinant Proteins
The Plasmid pT7-UbcH5c and pET28a-PTEN-HA (C-terminal HA-tagged) were transformed into E. Coli strain BL21(DE3). The recombinant proteins were expressed and purified by Ni-NTA affinity chromatography according to standard procedures. Recombinant PTEN was further purified using a 1-ml HiTrap SP column (Amersham), and dialyzed against Buffer A (20 mM Tris-HCl, pH 7.5, 50 mM NaCl, 1 mM DTT). The purified proteins were stored at −80°C in aliquots. The plasmid pGEX4T1-PTEN-HA was used to express recombinant GST-PTEN in BL21 strain according to standard procedures. For purification with glutathione-Sepharose, after the recombinant protein was bound to the column equilibrated with Buffer A, the column was washed with 10 x column volume of Buffer A supplemented with 500 mM NaCl. After washing again with Buffer A, the recombinant protein was eluted with Buffer A containing 15 mM reduced glutathione. The eluant was further purified with a 1-ml HiTrap Q column (Amersham). The purified GST-PTEN-HA was dialyzed against Buffer A, stored at −80°C in small aliquots.
In vitro Ubiquitination of PTEN
The reaction was carried out at 30°C for 1 hour in 15 μl reaction buffer (40 mM Tris-HCl, pH 7.5, 2 mM DTT, 5 mM MgCl2) containing the following components: 10 μg of ubiquitin (Boston Biochem, # U-100), 20 nM human E1 (Boston Biochem, # E302), 500 nM of recombinant UbcH5c, 2 μM ubiquitin aldehyde (Boston Biochem, #U-201), 5 mM ATP or ATPγS, and 150 ng of GST-PTEN. HeLa S-100 or different chromatographic fractions were added as E3 source as indicated in individual experiments. The reaction was terminated by adding 0.8 ml Pull-down Buffer (20 mM Tris-HCl, pH 7.5, 500 mM NaCl, 1% Triton X-100, 0.02% BSA, and 5 mM β-Mercaptoethanol). After addition of 8 μl of Glutathione Sepharose-4B, the samples were rotated at room temperature for 40 minutes. The beads were washed with 1 ml of Pull-down Buffer for 5 times. The proteins bound to beads were released by boiling in 30 μl of 2 x SDS-PAGE sample buffer for 5 minutes. The samples were then resolved by 8% SDS-PAGE followed by immunoblot analysis using a monoclonal anti-ubiquitin antibody (BD Biosciences, #550944). In the reactions using C-terminal HA-tagged recombinant PTEN instead of GST-PTEN as substrate, all the components were the same as described above except that 50 ng of recombinant PTEN-HA was added to replace GST-PTEN. After ubiquitination reaction, the samples were directly boiled with SDS-PAGE loading buffer and analyzed by immunoblotting against HA-tag (Babco, HA.11).
Purification and Identification of PTEN E3 Activity from HeLa S-100
All purification steps were carried out at 4°C, and chromatography was performed with an Amersham FPLC system. HeLa cell S-100 (HS100) in Buffer A (20 mM Tris-HCl, pH 7.5, 50 mM NaCl, 1 mM DTT) supplemented with protease inhibitors was prepared from large-scale cell culture purchased from National Cell Culture Center. For the purification, 315 ml of HS100 (~1.5 g total protein) was applied to a Q-Sepharose column (60-ml bed volume) equilibrated with Buffer A. After washing the column with Buffer A containing 0.15 M NaCl, the PTEN ubiquitin ligase activity was eluted into 120 ml of Buffer A containing 0.35 M NaCl (480 mg protein). The activity was then subject to 20%–50% (saturation) ammonium sulfate precipitation. The protein pellet, which contained the activity, was dissolved in 40 ml Buffer A (199 mg protein) and then ammonium sulfate concentration was adjusted to 15%. The activity was subject to two runs through a 5-ml HiTrap Phenyl-Sepharose HP column (Amersham) equilibrated with 15% ammonium sulfate in Buffer A. After washing the column with 10% ammonium sulfate, the activity was eluted with a 10%–0% gradient of ammonium sulfate in 80 ml Buffer A, followed by 20 ml Buffer A. Fractions of 4 ml were collected, dialyzed, and assayed for activity. The fractions containing the activity peak were pooled (3.85 mg protein) and further fractionated with a 2-ml Hydroxyapatite (Bio-Rad) column by using a 3–200 mM phosphate gradient in 30 ml Buffer B (20 mM HEPES, pH 7.5, 10 mM KCl, 1 mM DTT). Fractions of 2 ml were collected, dialyzed, and assayed for activity. Then the activity fractions were combined (1.07 mg protein) and fractionated with a 1-ml HiTrap Heparin-Sepharose HP column. The activity was eluted by a gradient of 0–300 mM NaCl in 20 ml Buffer A. Fractions of 1 ml were collected, dialyzed, and assayed for activity. The active fractions were combined and underwent multiple runs through a 25-ml Superdex 200 gel-filtration column in Buffer A plus 50 mM NaCl. The activity fractions were combined and resolved by a Mono Q column with a 150–350 mM NaCl gradient in 20 ml Buffer A. Fractions of 1 ml were collected. After dialysis, activity assay were performed using 2 μl of each fraction, and SDS-PAGE followed by silver-staining was performed using 15 μl of each fraction. A single protein band correlating to the PTEN ubiquitin ligase activity was identified. The whole Mono Q fractions containing the purified activity were concentrated, resolved by SDS-PAGE, and stained with Coomassie blue. The protein band was subject to protein identity determination by performing mass spectrometry analysis (MALDI-TOF-MS/MS). The activity was identified as human NEDD4-1.
Cloning of Full-Length NEDD4-1
HeLa cell genomic DNA was prepared as described in Molecular Cloning manual book. Using it as template, nest PCR were performed to obtain DNA sequence containing the N-terminal fragment of NEDD4-1, a GC-rich sequence (First round PCR used primers AATATTAAAAAGCATGATTATTCC and TCGTCCTCCAGGAGCCCGA. The second round PCR used primers ATAACCTTATTCCACAGGAGG and ACACCTCC ACCGCGCAAGT). The C-terminal fragment of NEDD4-1 was amplified by PCR from clone KIAA0093 using primers CTCCACAGTTGCCTGCCCT and GCCCTCGAGCTA ATCAACTCCATCAAAGCC (3’-XhoI). The two PCR products overlap with each other for 66 bp. Then by mixing the N-terminal and C-terminal PCR fragments as template and using Primer 5’-EcoRI (CCGGAATTCTATGGCACAAAGCTTACGATTGC) and Primer 3’-XhoI, the full-length NEDD4-1 was amplified by PCR and cloned into pCDNA3.1 using EcoRI and XhoI sites. For NEDD4-1-HA construct, a HA-tag was inserted at the C-terminus by PCR. All constructs were verified by DNA sequencing.
NEDD4-1 RNAi Constructs
The shRNA constructs against human NEDD-1 were generated using pSuperRetro vector according to manufacturer’s procedure (OligoEngine). The NEDD4-1 DNA sequences used in the RNAi constructs are: 5’TGGCGATTTGTAAACCGAA3’ (iN4A); 5’TGCAGAA CAGGCTGAGGAA3’ (iN4B); and ATGAAACTTCGCCGAGCAA (iN4C).
Transfection and in vivo PTEN-Ubiquitination Assay
Cell culture transfection was performed using polyethylenimine (PEI) reagent, or, where indicated, electroporation approach (Amaxa) for high transfection efficiency (>65%). For in vivo PTEN-ubiquitination assay, PEI transfection was performed when 293T cells in 10-cm plates reached ~ 50% confluence. Plasmids encoding for PTEN, NEDD4-1, and HA- or His-tagged ubiquitin were used in transfection as indicated in individual experiments. Twenty four hours after transfection, cells were treated with or without 25 μM proteasome inhibitor MG132 (CalBiochem) for 6 hours. The cells were washed with PBS, pelleted, and lysed in 0.1 ml of PBS plus 1% NP-40, 25 μM MG132, 2 μM ubiquitin aldehyde, and protease inhibitor cocktail. The lysates were centrifuged to obtain cytosolic proteins. Subsequently, we follow a published protocol (Treier et al., 1994) with minor modifications. Briefly, individual samples were diluted with 1 ml of Guanidine Buffer (6 M guanidine, 0.1 M sodium phosphate, pH 8.0, 0.2% Titon-X100, 10 mM imidazole) before addition of 20 μl TALON Cobalt transition metal beads (BD Biosciences) beads pre-washed with Guanidine Buffer (GB). After rotating at 4°C for 3 hour, the beads were washed twice with 1ml of GB followed by two washes with 1 ml of GB/TI, which composes 1:3 (vol) of GB vs. buffer TI (20 mM Tris-HCl, pH 6.8, 0.2% Titon-X100, 20 mM imidazole, 0.2% SDS). Then the beads were washed twice with TI followed by two washes with TI plus 1 M NaCl. After another wash with TI, the supernatant was completely removed with thin tips. The proteins were released from the beads by boiling in 40 μl of 2x SDS-PAGE sample buffer plus 200 mM imidazole for 10 min. Twenty-five μl of the samples were subjected to immunoblot against antibodies as specified in individual experiments.
Cycloheximide chase experiments
To examine the effect of NEDD4-1 overexpression on PTEN degradation, three 10-cm plates of HeLa cells were co-transfected with 2μg/plate of PTEN plasmid and 6μg/plate of NEDD4-1 plasmid or control plasmid. To examine the effect of NEDD4-1 RNAi on PTEN degradation, three 10-cm plates of HeLa cells were co-transfected with 0.8 μg/plate of PTEN plasmid and 3.2 μg/plate of RNAi construct against human NEDD4-1 (iN4A) or control plasmid. Ten hours after transfection, the cells from the triplicate plates were pooled together and re-plated into four 6-cm plates to minimize the variation in transfection among the plates. Twelve hours later after re-plating, 50 μg/ml of cycloheximide was added to the 6-cm plates, and the cycloheximide treatment was terminated at 0, 3, 6, and 9 hours time points as indicated. The cell lysates were made, and protein concentration was determined. Subsequently, twenty μg of total protein from each sample, except 60 μg of total protein from the RNAi vector control samples, was analyzed by immunoblotting as indicated.
Soft-Agar Colony Formation Assay
Retroviruses were generated using pBabe-puro vector alone, pBabe-puro-NEDD4-1, and pBabe-puro-Ras. Primary MEFs (passage 3) were plated in 10-cm plates (2 x 105/plate). After overnight, they were infected by three rounds of infection (4 hours each round) with indicated retroviruses. Forty-eight hours after infection, cells were allowed to grow for three days in medium containing 2 μg/ml puromycin. After recovering from puromycin selection for 12 hours in puromycin-free medium, viral-infected cells were trypsinized and plated for soft-agar colony formation assay. For each assay, 5 x 105 viral-infected cells were plated with soft-agar medium to individual wells in 6-well plates. The plates were incubated for two to three weeks, and phase-contrast microscopic pictures were taken for each sample using a digital camera coupled to a microscope to show colony formation. Quantification and standard deviation were obtained from results of three independent experiments.
Human Cancer Tissues and Microarray Analysis
Tumor tissues and clinical data were obtained through an IRB-approved protocol at Memorial Sloan-Kettering Cancer Center. Tumors were microdissected to ensure that at least 70% of cells present were tumor. RNA was extracted using Trizol (Invitrogen, Carlsbad CA) followed by cleanup with Rneasy columns (Qiagen, Valencia CA). Double stranded cDNA was produced using a ds cDNA synthesis kit (Invitrogen) followed by production of labelled RNA probe using an in vitro transcription kit (Enzo Diagnostics, Farmingdale NY). Hybridization, washing and imaging of Affymetrix U133 Plus v. 2.0 microarrays (Affymetrix, Santa Clara CA) were performed according to manufacturer's protocols. Microarray data were normalized, background subtracted and log transformed using the robust multi-array average (RMA) method (Irizarry et al., 2003) within bioconductor for R (Gentleman et al., 2004).
Immunohistochemistry of mouse prostates
IHC was carried out with the automated staining processor (Ventana Medical Systems, Arizona) for mouse prostates as previously described (Trotman et al., 2003) using the following antibodies: rabbit anti-phospho-Akt (Ser473-IHC specific, Cell signalling # 9277), rabbit anti-PTEN (Ab-2, NeoMarkers), anti-Nedd4 (Upstate, # 07-049). For correlation of PTEN and NEDD4-1 status in Ptenhy/− mice, single prostate glands on a total of 9 adjacent sections from 2 animals (6-month old) were classified for NEDD4-1 and PTEN high or low staining. A total of 16 glands were scored, yielding an inverse correlation for PTEN and NEDD4-1 level as following: no gland with PTEN high/NEDD4-1 high or PTEN low/NEDD4-1 low; 11 glands with PTEN high/NEDD4-1 low (normal glands); 5 glands with PTEN low/NEDD4-1 high (tumor glands, p-Akt-positive). The statistical significance of this inverse correlation was calculated using a chi-square analysis (P < 0.001).
Xenotransplantation
DU-145 cells and PC3 cells were infected with iN4A or iLacZ pSuper Retrovirus, selected with 2 μg/ml puromycin for 4 days, and allowed to recover in the absence of puromycin. Subsequently, 106 DU-145 or PC3 tumor cells, bearing NEDD4-1 RNAi or LacZ RNAi retroviruses as indicated, were suspended in 100 μl 10% FCS and 100 μl Matrigel (BD Biosciences), and subcutaneously injected into the right flank of 6-week-old athymic nude mice (NCRNU-M, Taconic Farms Inc.) in triplicates. Every 3 days tumor size was measured with a caliper, and volumes were calculated as L × W2 × 0.52, where L is the longest and W is the shortest diameter.
Supplementary Material
Acknowledgments
We thank Drs. Olivier Staub and Daniela Rotin for providing specific antibodies against NEDD4-1 and NEDD4-2, Dr. James Zhijian Chen for ubiquitin plasmids, Drs. Harold Varmus and William Pao for primary p53−/− MEFs, and Dr. K.O. Manova-Todorova, B. Altayoz, and S. Jiao for help with IHC and pathology analysis. We also thank Drs. Joan Massague and Harold Varmus for discussion and reading of the manuscript. Large scale HeLa cell culture was from National Cell Culture Center (Minnesota).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adey NB, Huang L, Ormonde PA, Baumgard ML, Pero R, Byreddy DV, Tavtigian SV, Bartel PL. Threonine phosphorylation of the MMAC1/PTEN PDZ binding domain both inhibits and stimulates PDZ binding. Cancer Res. 2000;60:35–37. [PubMed] [Google Scholar]
- Bakkers J, Camacho-Carvajal M, Nowak M, Kramer C, Danger B, Hammerschmidt M. Destabilization of DeltaNp63alpha by Nedd4-mediated ubiquitination and Ubc9-mediated sumoylation, and its implications on dorsoventral patterning of the zebrafish embryo. Cell Cycle. 2005;4:790–800. doi: 10.4161/cc.4.6.1694. [DOI] [PubMed] [Google Scholar]
- Blot V, Perugi F, Gay B, Prevost MC, Briant L, Tangy F, Abriel H, Staub O, Dokhelar MC, Pique C. Nedd4.1-mediated ubiquitination and subsequent recruitment of Tsg101 ensure HTLV-1 Gag trafficking towards the multivesicular body pathway prior to virus budding. J Cell Sci. 2004;117:2357–2367. doi: 10.1242/jcs.01095. [DOI] [PubMed] [Google Scholar]
- Bond GL, Hu W, Levine AJ. MDM2 is a central node in the p53 pathway: 12 years and counting. Curr Cancer Drug Targets. 2005;5:3–8. doi: 10.2174/1568009053332627. [DOI] [PubMed] [Google Scholar]
- Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol. 2003;15:164–171. doi: 10.1016/s0955-0674(03)00003-6. [DOI] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Finley D, Varshavsky A. The ubiquitin-mediated proteolytic pathway and mechanisms of energy-dependent intracellular protein degradation. J Cell Biochem. 1984;24:27–53. doi: 10.1002/jcb.240240104. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- Fostier M, Evans DA, Artavanis-Tsakonas S, Baron M. Genetic characterization of the Drosophila melanogaster Suppressor of deltex gene: A regulator of notch signaling. Genetics. 1998;150:1477–1485. doi: 10.1093/genetics/150.4.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray IC, Stewart LM, Phillips SM, Hamilton JA, Gray NE, Watson GJ, Spurr NK, Snary D. Mutation and expression analysis of the putative prostate tumour-suppressor gene PTEN. Br J Cancer. 1998;78:1296–1300. doi: 10.1038/bjc.1998.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Ingham RJ, Gish G, Pawson T. The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene. 2004;23:1972–1984. doi: 10.1038/sj.onc.1207436. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Kamynina E, Tauxe C, Staub O. Distinct characteristics of two human Nedd4 proteins with respect to epithelial Na(+) channel regulation. Am J Physiol Renal Physiol. 2001;281:F469–477. doi: 10.1152/ajprenal.2001.281.3.F469. [DOI] [PubMed] [Google Scholar]
- Kim RH, Peters M, Jang Y, Shi W, Pintilie M, Fletcher GC, DeLuca C, Liepa J, Zhou L, Snow B, et al. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell. 2005;7:263–273. doi: 10.1016/j.ccr.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Kwabi-Addo B, Giri D, Schmidt K, Podsypanina K, Parsons R, Greenberg N, Ittmann M. Haploinsufficiency of the Pten tumor suppressor gene promotes prostate cancer progression. Proc Natl Acad Sci U S A. 2001;98:11563–11568. doi: 10.1073/pnas.201167798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie NR, Downes CP. PTEN function: how normal cells control it and tumour cells lose it. Biochem J. 2004;382:1–11. doi: 10.1042/BJ20040825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257–262. doi: 10.1016/s1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- Maehama T, Taylor GS, Dixon JE. PTEN and myotubularin: novel phosphoinositide phosphatases. Annu Rev Biochem. 2001;70:247–279. doi: 10.1146/annurev.biochem.70.1.247. [DOI] [PubMed] [Google Scholar]
- Michael D, Oren M. The p53 and Mdm2 families in cancer. Curr Opin Genet Dev. 2002;12:53–59. doi: 10.1016/s0959-437x(01)00264-7. [DOI] [PubMed] [Google Scholar]
- Nagase T, Miyajima N, Tanaka A, Sazuka T, Seki N, Sato S, Tabata S, Ishikawa K, Kawarabayasi Y, Kotani H, et al. Prediction of the coding sequences of unidentified human genes. III. The coding sequences of 40 new genes (KIAA0081–KIAA0120) deduced by analysis of cDNA clones from human cell line KG-1. DNA Res. 1995;2:37–43. doi: 10.1093/dnares/2.1.37. [DOI] [PubMed] [Google Scholar]
- Pao W, Klimstra DS, Fisher GH, Varmus HE. Use of avian retroviral vectors to introduce transcriptional regulators into mammalian cells for analyses of tumor maintenance. Proc Natl Acad Sci U S A. 2003;100:8764–8769. doi: 10.1073/pnas.1133333100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftopoulou M, Etienne-Manneville S, Self A, Nicholls S, Hall A. Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science. 2004;303:1179–1181. doi: 10.1126/science.1092089. [DOI] [PubMed] [Google Scholar]
- Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234:364–368. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- Rougier JS, van Bemmelen MX, Bruce MC, Jespersen T, Gavillet B, Apotheloz F, Cordonier S, Staub O, Rotin D, Abriel H. Molecular determinants of voltage-gated sodium channel regulation by the Nedd4/Nedd4-like proteins. Am J Physiol Cell Physiol. 2005;288:C692–701. doi: 10.1152/ajpcell.00460.2004. [DOI] [PubMed] [Google Scholar]
- Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22:2954–2963. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- Strack B, Calistri A, Accola MA, Palu G, Gottlinger HG. A role for ubiquitin ligase recruitment in retrovirus release. Proc Natl Acad Sci U S A. 2000;97:13063–13068. doi: 10.1073/pnas.97.24.13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280:1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001;276:993–998. doi: 10.1074/jbc.M009134200. [DOI] [PubMed] [Google Scholar]
- Treier M, Staszewski LM, Bohmann D. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell. 1994;78:787–798. doi: 10.1016/s0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Van Dyke T, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression as revealed by inherited mutation. Cell. doi: 10.1016/j.cell.2006.11.040. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20:5010–5018. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whang YE, Wu X, Suzuki H, Reiter RE, Tran C, Vessella RL, Said JW, Isaacs WB, Sawyers CL. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc Natl Acad Sci U S A. 1998;95:5246–5250. doi: 10.1073/pnas.95.9.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Wang X, Zhang W, Reed W, Samet JM, Whang YE, Ghio AJ. Zinc-induced PTEN protein degradation through the proteasome pathway in human airway epithelial cells. J Biol Chem. 2003;278:28258–28263. doi: 10.1074/jbc.M303318200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.