

Abstract
Magnaporthe grisea is responsible for a devastating fungal disease of rice called blast. Current control of this disease relies on resistant rice cultivars that recognize M. grisea signals corresponding to specific secreted proteins encoded by avirulence genes. The M. grisea ACE1 avirulence gene differs from others, since it controls the biosynthesis of a secondary metabolite likely recognized by rice cultivars carrying the Pi33 resistance gene. Using a transcriptional fusion between ACE1 promoter and eGFP, we showed that ACE1 is only expressed in appressoria during fungal penetration into rice and barley leaves, onion skin, and cellophane membranes. ACE1 is almost not expressed in appressoria differentiated on Teflon and Mylar artificial membranes. ACE1 expression is not induced by cellophane and plant cell wall components, demonstrating that it does not require typical host plant compounds. Cyclic AMP (cAMP) signaling mutants ΔcpkA and Δmac1 sum1-99 and tetraspanin mutant Δpls1::hph differentiate melanized appressoria with normal turgor but are unable to penetrate host plant leaves. ACE1 is normally expressed in these mutants, suggesting that it does not require cAMP signaling or a successful penetration event. ACE1 is not expressed in appressoria of the buf1::hph mutant defective for melanin biosynthesis and appressorial turgor. The addition of hyperosmotic solutes to buf1::hph appressoria restores appressorial development and ACE1 expression. Treatments of young wild-type appressoria with actin and tubulin inhibitors reduce both fungal penetration and ACE1 expression. These experiments suggest that ACE1 appressorium-specific expression does not depend on host plant signals but is connected to the onset of appressorium-mediated penetration.
Magnaporthe grisea species complex attacks a wide range of grasses, including wheat, barley, and rice (10, 26), and is a model organism for the study of plant fungal interactions (11, 42). The M. grisea infection cycle is characteristic of grass leaf spot diseases. After spore attachment and germination, the fungus differentiates an appressorium through the perception of physical and chemical surface parameters (hydrophobicity, hardness, and cuticle monomers) (21, 42). This differentiation is the result of a complex morphogenetic process that involves cyclic AMP (cAMP), mitogen-activated protein kinases, and calcium signaling pathways (7, 45, 50). Early stages of appressorium development are associated with the deposition of a melanin layer between the cell wall and plasma membrane (21), migration of lipid bodies from spore to appressorium, mobilization of glycogen, and the formation of a septum sealing the appressorium (5, 43). Maturation of the appressorium is characterized by the degradation of lipid bodies and glycogen (43) and the generation of a high turgor (22). Finally, a reorganization of the cytoskeleton is induced at the point of emergence of the penetration peg that penetrates the host cuticle and cell wall (5, 35). Inside the plant, M. grisea differentiates bulbous infectious hyphae (44) that colonize host tissues without visible damage for 4 to 5 days after penetration. Then the fungus rapidly expands and destroys colonized tissues, leading to small necrotic lesions producing spores spreading the disease.
Most M. grisea genes identified as essential for infection encode proteins involved in appressorium differentiation and appressorium-mediated penetration. They are involved in surface sensing, signaling, melanin/sugar/lipids metabolism, secretion, and membrane remodeling (42). M. grisea genes expressed in infected tissues (28) and appressoria (3, 11, 16, 23, 30, 41) were also identified using genomic tools (expressed sequence tags, arrays). Up to now, only a few of these genes are specifically expressed in appressoria or during infection. GAS1 and GAS2 encode related proteins of unknown function involved in penetration and specifically expressed in appressoria (48). PLS1 encodes a membrane protein from the tetraspanin superfamily required for penetration and specifically expressed in appressoria (9). CBP1 encodes a secreted chitin-binding protein that is not required for penetration and is specifically expressed in appressoria (25, 41). Two other genes identified as specifically expressed in appressoria encode a putative secreted protein (AI068463) (3) and a glucose dehydratase (AP3C19) (41). Yet their role in penetration is unknown. M. grisea avirulence (AVR) genes PWL2, AVR-PITA, and AVR1-CO39 (18, 34, 39) encode small cysteine-rich proteins with putative secretion signal peptides that are likely recognized by plants carrying the corresponding resistance gene (19, 24). AVR-PITA and PWL2 are specifically expressed during penetration, fungal colonization, and late infection (34, 29). ACE1 differs from previous AVR genes, as it encodes a cytoplasmic enzyme involved in secondary metabolism exclusively expressed in appressoria (4). Since Ace1 biosynthetic activity is required for avirulence, the signal recognized by rice plants carrying Pi33 resistance gene is supposed to be a secondary metabolite whose biosynthesis requires Ace1 (4).
In this report, we have studied the factors involved in ACE1 appressorium-specific expression. ACE1 expression was monitored during appressorial differentiation and penetration into plant tissues or artificial membranes using a transcriptional fusion between ACE1 promoter and eGFP, and quantitative reverse transcriptase PCR (RT-PCR). We showed that ACE1 is only expressed in appressoria during penetration of either leaves or cellophane-based membranes, but not on Mylar or Teflon artificial membranes. ACE1 expression was not induced by cellophane or plant cell wall components. Using M. grisea penetration-deficient mutants, we showed that ACE1 is expressed in cAMP or PLS1-deficient mutants but not in melanin-deficient mutant buf1::hph. Addition of actin or tubulin inhibitors reduces both ACE1 expression and fungal penetration into the host plant. Based on these results, we propose that the induction of ACE1 expression is connected to the initiation of appressorium-mediated penetration.
MATERIALS AND METHODS
Fungal strains, growth conditions, and transformation.
Guy11 is a fertile M. grisea field isolate pathogenic on rice (33). Phenotypes of Guy11 ΔcpkA mutant I27 (47) and Guy11 Δmac1 sum1-99 mutant DA99 (1) were recently redescribed by Thines et al. (43). buf1::hph (unpublished) and pls1::hph (9) were obtained by REMI mutagenesis using the P1.2 M. grisea strain pathogenic on rice. Fungal strains were grown and stored as described by Dioh et al. (14). Strains were grown under osmotic stress conditions on complete liquid medium (Tanaka minimal medium with yeast extract described in reference 14) containing either 0.4 M NaCl or 1 M sorbitol. M. grisea strains were transformed as described by Sweigard et al. (38) and modified as described by Böhnert et al. (4). For hygromycin selection, transformants were selected on complete medium containing 120 mg/liter hygromycin (Sigma-Aldrich, St. Louis, MO). For Basta and sulfonylurea selection, transformants were selected on the complex medium defined by Sweigard et al. (38) containing 35 mg/liter glufosinate or 100 mg/liter chlorimuron-ethyl (Cluzeau Info Labo, Ste. Foy la Grande, France), respectively. Transformants were purified by isolation of single spores.
Cloning procedures and plasmid constructions.
Escherichia coli strain DH5α (Bethesda Research Laboratories) was used for cloning. Molecular methods followed protocols described by Sambrook et al. (37). eGFP was fused to the promoter and the terminator of ACE1 (promACE1::eGFP) and introduced into a plasmid conferring resistance to hygromycin as already described by Böhnert et al. (4). promACE1::eGFP was digested by EcoRI, and the 3.75-kb fragment containing the ACE1 promoter, eGFP open reading frame, and ACE1 terminator was introduced into pCB1635 (40), a vector conferring resistance to glufosinate. The resulting vector was called pCB1635-promACE1::eGFP. A genomic fragment containing the BUF1 gene was obtained from M. Farman (17) and cloned into pCB1004 vector (40). The sulfonylurea resistance cassette from pCB1637 was introduced in this plasmid using SalI restriction sites, and the resulting vector, pCB1004-BUF1-SULFR, was used to complement our buf1::hph mutant.
Nucleic acid extraction and analysis.
Genomic DNA was isolated from M. grisea by following the miniprep procedure (39) with modifications described by Böhnert et al. (4). Total RNA was extracted from M. grisea liquid cultures using the hot acid-phenol protocol (9) or using Trizol reagent (Invitrogen, Carlsbad, CA). RT-PCR was carried out with 6 μg of total RNA as starting material using ReadyToGo You-Prime first-strand beads (Amersham Biosciences, Little Chalfont, United Kingdom) according to the manufacturer's protocol. The following ACE1-specific primers used in this study hybridize on both sides of the ACE1 second intron: I30+, 5′-GCGACACACTGACGGCGACC-3′ (6,208 bp from ATG); I3O−, 5′-GGAGCCGTTGCCCATGATGC-3′ (7,124 bp from ATG); I3i+, 5′-CCGCCGTCGTCACTCCCACC-3′ (6,346 bp from ATG); I3i−, 5′-TGACAGAGGACAGGAAGACG-3′ (6,987 bp from ATG).
Real-time RT-PCR.
Reverse transcription was carried out using the Thermoscript RT-PCR system (Invitrogen, Carlsbad, CA) with 5 μg of total RNA extracted from infected barley leaves at 0 h, 8 h, 17 h, 24 h, 30 h, 48 h, 52 h, and 72 h and from mycelium grown in complete liquid agitated medium for 24 h (three biological replicates for each time point). Real-time PCR was carried out with a LightCycler 1.0 (Roche Diagnostics, Indianapolis, IN) using Fast-Start DNA master SYBR green I kit (Roche Diagnostics, Indianapolis, IN). The following primers were designed using Primer Express (Applied Biosystem, Foster City, CA): qACE1-F, 5′-AGACGATGCCATTGGCAAA-3′; qACE1-R, 5′-AGCCAGCATGGAGTCCAATC-3′; qILV5-F, 5′-CCAGCTCTACGACTCGGTCAA-3′; qILV5-R, 5′-AGTCGGGCTGGCTGTTGTAGT-3′. ACE1 expression is calculated relative to the transcript levels of the constitutively expressed gene ILV5 (MGG_01808) using the formula 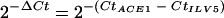 . StatBox V6.5 (Grimmersoft, Paris, France) was used for statistical analyses (critical threshold α = 0.05).
. StatBox V6.5 (Grimmersoft, Paris, France) was used for statistical analyses (critical threshold α = 0.05).
Phenotypic analysis and cytology.
Seedlings from barley cultivar Express were cultivated for 10 days (20°C during the day, 15°C at night). The detached barley leaf assay was carried out as described previously (4, 9). The onion skin assay was performed using the same protocol as the barley leaf assay. Appressoria were differentiated on artificial membranes composed of Teflon (Goodfellow, Cambridge, United Kingdom), Mylar (polyethylene terephtalate, Rhodia, Lyon, France), and PUDO-193 cellophane (gift from T. Bourett, DuPont de Nemours, Wilmington, DE) (5). Enhanced green fluorescent protein (eGFP) fluorescence was observed with a Nikon Optiphot fluorescence microscope equipped with a 488/DM510-550 eGFP-specific filter. Conidia and appressoria were observed on the leaf surface after a treatment for 1 min with a highly diluted calcofluor solution (fluorescent brightener 26 [Sigma-Aldrich], 0.01 mg/ml in water, pH 8), followed by water rinse. Cell wall calcofluor fluorescence was observed under UV light with a Nikon Optiphot fluorescence microscope. For melanin inhibition, 10 ppm tricyclazole (gift from E. Lilly Research Center Ltd., United Kingdom) (46) was added to spore suspensions before inoculation on membranes or leaves. Appressoria were treated with hyperosmotic solutions by replacing water droplets at 6 h after inoculation with solutes at the following concentrations: 0.6 M sucrose, 0.4 M NaCl, 1 M sorbitol, 0.15% polyethylene glycol (PEG), and 1 M glycerol. Similarly, 0.5% cellophane powder or plant cell wall components were added to appressoria by replacing water droplets at 8 h after inoculation (hai) with the following solutions: 0.5% cellulose, 25 mM cellobiose, 0.5% xylan (from beechwood or oat spelts), and 0.5% citrus pectin (Sigma-Aldrich, St. Louis, MO). Thirty, 100, and 300 ppm carbendazim (Ehrenstorfer GmbH, Augsburg, Germany) or 1, 3, and 10 μM cytochalasin A (Calbiochem, La Jolla, CA) were added at 8 hai on onion epidermis assays by replacing water droplets with inhibitor solutions.
Turgor assay using cytorrhysis.
M. grisea spore suspensions were deposited on artificial membranes or detached barley leaves. At selected times (8 to 24 hai), water droplets were replaced with increasing concentrations of KCl or PEG4000 (Sigma-Aldrich, St. Louis, MO) solutions. Following 15 min of incubation in the solute, appressoria collapses were observed under light microscope (magnification, ×100; Nikon Optiphot), and the percentage of cytorrhysis was determined for 100 appressoria in three independent droplets. This experiment was repeated at least twice. The 50% cytorrhysis was calculated from the solute dose-percent collapse curve and used as an estimate of appressorium turgor pressure using the relationships defined by Howard et al. (22), allowing a correspondence between the molarity of variable solutes and turgor pressure.
RESULTS
Expression of Magnaporthe grisea avirulence gene ACE1 during appressorium-mediated penetration.
ACE1 was previously shown to be exclusively transcribed in appressoria during early stages of plant infection (4). We monitored ACE1 transcription during barley leaf infection by quantitative RT-PCR relative to transcripts from the constitutively expressed gene ILV5 (MGG_01808, aceto-hydroxy-isomero-reductase, unpublished data) (Fig. 1). ACE1 transcripts were detected in trace amounts (2% of maximum expression) as soon as 8 hai, rapidly reaching a peak at 17 hai, followed by a decreased at 24 hai (20% of maximum) to 30 hai (5% of maximum). ACE1 transcripts were not detected in RNA from mycelium. To easily monitor ACE1 transcription, we constructed an expression vector corresponding to a transcriptional fusion between the eGFP reporter gene and ACE1 promoter and terminator sequences (promACE1::eGFP). This vector was introduced by transformation into M. grisea avirulent strain Guy11. Most transformants (18/25, 72%) displayed a strong appressorium-specific eGFP fluorescence. Two of these transformants carrying a single copy of promACE1::eGFP (data not shown) were used to monitor ACE1 transcription. These transformants did not display eGFP fluorescence in young (1 to 6 days) and old (7 to 15 days) mycelia grown in liquid (still/shake) or agar media (complete, minimal, or rice) nor in conidia produced from these cultures. A weak eGFP fluorescence was first observed in appressoria differentiated on barley leaves at 16 hai (Fig. 2A). This observation suggests that ACE1 transcripts that are already abundant at 17 hai (Fig. 1) are not yet efficiently translated into eGFP. ACE1 expression peaked at 24 hai, leading to a strong eGFP fluorescence of both appressoria and primary infectious hyphae (Fig. 2B). After 24 hai, ACE1 expression gradually decreased, and only 50% of appressoria were still fluorescent at 48 hai (Fig. 2C). As the ACE1 transcript is already at a very low level at 48 hai (Fig. 1), the eGFP fluorescence observed at that time likely results from the long half-life of this protein. eGFP fluorescence was never observed in secondary infectious hyphae formed inside infected epidermal cells at 30 to 48 hai nor later during infection, including sporulating lesions. We have previously shown that the Ace1-eGFP fusion protein was exclusively localized in appressoria and was not detected in primary infectious hyphae (4). This observation suggests that the eGFP fluorescence we detected in the primary infectious hyphae of transformants expressing the promACE1::eGFP transcriptional fusion results from the diffusion of soluble eGFP from appressoria to primary infectious hyphae, as these two structures are linked by the penetration peg. ACE1 displayed a similar expression pattern on onion epidermis (Table 1). These results show that ACE1 appressorium-specific expression is controlled at the transcriptional level and is restricted to a specific stage of infection (16 to 48 hai) corresponding to the penetration into host plant tissues (16 to 30 hai).
FIG. 1.

Quantification of ACE1 mRNA by real-time RT-PCR during infection of barley leaves. ACE1 expression was quantified by real-time RT-PCR using RNA extracted from infected barley leaves collected at different times after droplet inoculation of Guy11 spore suspensions. ACE1 expression is calculated relative to the transcript levels of the constitutively expressed gene ILV5 using the formula 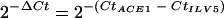 . Each data point is the average of results from three biological replicates. Standard deviations are indicated by error bars.
. Each data point is the average of results from three biological replicates. Standard deviations are indicated by error bars.
FIG. 2.

ACE1 appressorium-specific expression on leaves and cellophane membranes. M. grisea Guy11 transformants carrying the promACE1::eGFP vector were used to monitor ACE1 expression as eGFP fluorescence at 16 (A), 24 (B), and 48 (C) h after inoculation of spores on barley leaves and at 24 h after inoculation of spores on a cellophane membrane (D). Bar, 10 μm. Bright field (visible): observation with a microscope at ×40 magnification under bright light. Fluorescence (blue light): observation with a microscope at ×40 magnification under UV light with an eGFP-specific filter.
TABLE 1.
ACE1 expression in appressoria differentiated on host plants or artificial membranes
| Membrane or host plant | Appressorium differentiation (%) | Appressorial turgor (MPa)a | Penetration | Appressoria expressing ACE1 (%)b | Time (h) of maximum ACE1 expression |
|---|---|---|---|---|---|
| Teflon | 95 | 4 | − | 1-5 | 24 |
| Mylar | 95 | 5.5 | − | 1-5 | 24 |
| Cellophane | 90 | ND | + | 50-75 | 72 |
| Barley | 98 | 5.5 | + | 50-75 | 24 |
| Onion | 98 | ND | + | 50-75 | 24 |
Turgor was determined using a cytorrhysis assay. ND, not determined.
eGFP fluorescence of appressoria from M. grisea Guy11 transformants carrying the promACE1::eGFP vector was monitored after inoculation of spores on barley leaves, onion epidermis, or artificial membranes.
Effect of host plant on ACE1 expression.
We monitored ACE1 expression in appressoria differentiated on Teflon, Mylar artificial membranes, and PUDO-193 cellophane membranes (5) using Guy11 transformants expressing the promACE1::eGFP fusion. Appressoria differentiated on these membranes are similar to those observed on leaves and develop a similar turgor (Table 1). The frequency of appressoria expressing ACE1 was strongly reduced on Teflon or Mylar membranes compared to those formed on barley leaves, as only 1 to 5% of appressoria were fluorescent at their peak of expression (24 hai). On the contrary, ACE1 expression was detected in up to 75% of appressoria formed on cellophane membranes at 48 hai with a peak of fluorescence at 3 days postinoculation when the fungus penetrates this membrane (Fig. 2D). ACE1 was not expressed in pseudoinfectious hyphae produced within the cellophane membrane. Since such membranes contain cellulose and related oligosaccharides, we tested the effect of ground PUDO-193 cellophane membranes (0.5%) and several plant cell wall components (0.5% cellulose, 25 mM cellobiose, 0.5% xylan, and 0.5% pectin) on ACE1 expression. We have not observed the induction of eGFP fluorescence when these compounds were added to 8-h-old appressoria differentiated on Teflon. These results demonstrate that ACE1 expression is induced only in appressoria formed on host plants or cellophane membranes, but this induction does not involve the cellophane itself or host cell wall components.
Relationship between cAMP signaling and ACE1 expression.
ACE1 encodes a multifunctional enzyme involved in M. grisea secondary metabolism. Since the cAMP signaling pathway negatively regulates the expression of genes involved in secondary metabolism in Aspergillus nidulans (49), we investigated whether this pathway is involved in the control of ACE1 expression. In M. grisea, cAMP signaling is required for both the differentiation of appressoria on hydrophobic surfaces and for appressorium-mediated penetration (1, 32, 43, 47). Some cAMP pathway mutants are able to form appressoria but are impaired in penetration. Deletion of the CPKA gene that encodes the catalytic subunit of cAMP-dependent protein kinase A affects appressorium morphogenesis, leading to a delayed formation of smaller, nonfunctional appressoria (32, 47). Δcpka mutants are retarded for glycogen and lipid mobilization during appressorium formation (43). These mutants are highly reduced in pathogenicity, inducing rare lesions and producing defective penetration pegs on onion epidermis (35). Δmac1 sum1-99 is a suppressor of Δmac1 mutation corresponding to a deletion of M. grisea adenylate cyclase gene MAC1 (1). sum1-99 corresponds to a mutation of the cAMP-binding pocket from the protein kinase A regulatory subunit, which leads to a constitutive activation of the cAMP pathway (1). Although it displays an accelerated conidial germination and appressorium development, this mutant is impaired in penetration, as its glycogen and lipid degradation is accelerated and completed before the onset of penetration (43). The promACE1::eGFP vector was introduced into Δcpka and Δmac1-sum99 mutants and ACE1 expression was monitored as eGFP fluorescence. ACE1 was normally expressed in appressoria of ΔcpkA and Δmac1 sum1-99 mutants formed on barley leaves (Table 2). These results demonstrate that ACE1 appressorium-specific expression is independent of the cAMP signaling pathway.
TABLE 2.
ACE1 expression in appressoria from M. grisea penetration-deficient mutants
ACE1 expression in appressoria of M. grisea penetration-deficient mutants.
ACE1 expression is restricted to appressoria penetrating leaves or cellophane membranes. To assay if ACE1 expression requires a successful penetration of host tissues, we expressed promACE1::eGFP in penetration-defective mutants Δpls1::hph and buf1::hph (Table 2). The Δpls1::hph mutant, defective for Pls1 tetraspanin, differentiates melanized appressoria with normal turgor (Table 2) that are unable to penetrate host leaves and cellophane membranes (9). This mutant is likely blocked at a late stage of appressorial development, as it is unable to degrade its glycogen (9). The naphthalene reductase mutant buf1::hph differentiates nonmelanized appressoria that cannot build up turgor and are unable to penetrate intact leaves or cellophane membranes (8, 12, 20, 22). ACE1 was normally expressed in appressoria of the Δpls1::hph mutant (Table 2). This result demonstrates that ACE1 appressorium-specific expression is independent of the PLS1 pathway required for appressorium-mediated penetration and does not require a successful penetration event. In contrast, ACE1 was not expressed in unmelanized appressoria from the buf1::hph mutant differentiated on barley leaves or cellophane membranes (Table 2). The buf1::hph melanin-deficient mutant tested was obtained by REMI mutagenesis during the screening of nonpathogenic mutants (9). Since secondary mutations are frequently observed during REMI mutagenesis (2, 38), we tested whether this buf1::hph mutation was responsible for the lack of ACE1 expression or not. To obtain melanin-deficient appressoria independently of the BUF1 null mutation, we inhibited melanin biosynthesis using tricyclazole, a specific inhibitor of naphthalene reductase encoded by BUF1 (8, 46). Tricyclazole was added to spore suspensions of Guy11 transformants expressing promACE1::eGFP inoculated on barley leaves or cellophane membranes. Tricyclazole-treated appressoria were not melanized (Fig. 3A) and unable to penetrate plant tissues or cellophane (Fig. 3C). ACE1 was not expressed in tricyclazole-treated appressoria differentiated on barley leaves (Fig. 3B) or cellophane (data not shown). We also complemented our buf1::hph promACE1::eGFP transformants with a vector carrying a wild-type BUF1 allele (17). The resulting buf1::hph/BUF1 transformants differentiated melanized appressoria identical to the wild type, and their pathogenicity on barley and rice was restored (Fig. 4). These buf1::hph/BUF1 transformants displayed a normal ACE1 appressorium-specific expression (Fig. 4). These results demonstrate that the inhibition of melanin biosynthesis either genetically (buf1::hph mutant) or chemically (tricyclazole) abolishes ACE1 expression in appressoria.
FIG. 3.
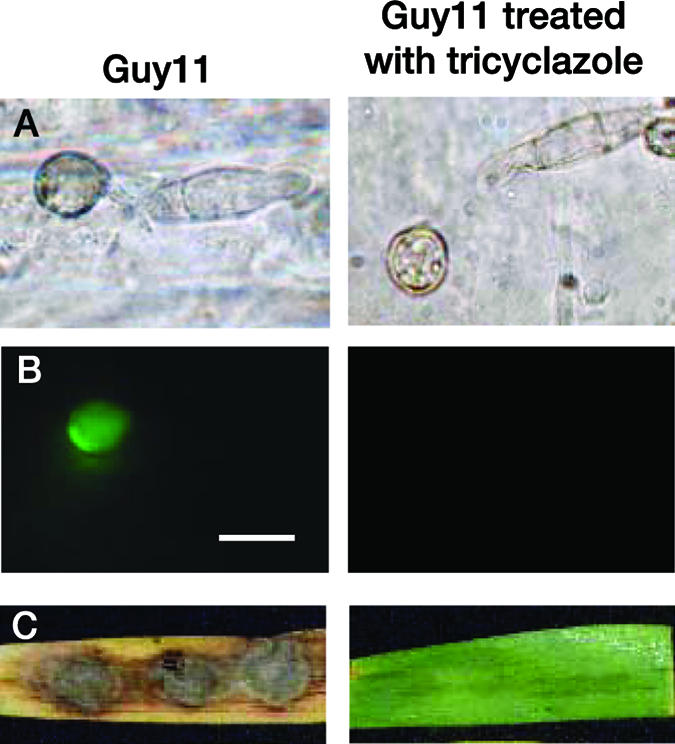
Effect of tricyclazole on ACE1 appressorium-specific expression. (A) Inhibition of appressorium melanization by tricyclazole. Microscopic observations were performed at ×100 magnification under bright light. (B) Inhibition of ACE1 expression in appressoria differentiated on barley leaves treated with tricyclazole 24 h after inoculation. M. grisea Guy11 transformants carrying the promACE1::eGFP vector were used to monitor ACE1 expression as eGFP fluorescence. Microscopic observations were performed at ×100 magnification under UV light with an eGFP-specific filter. (C) Lack of pathogenicity on barley leaves of Guy11 transformants carrying promACE1::eGFP vector treated with tricyclazole 5 days after inoculation. Bar, 10 μm.
FIG. 4.
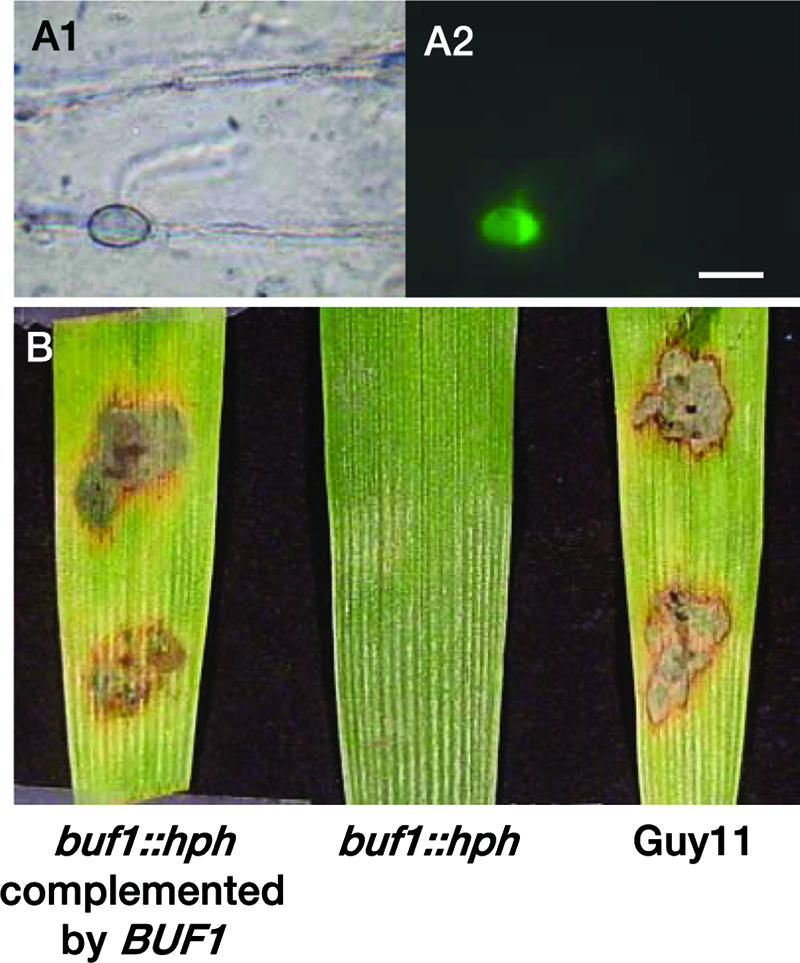
Complementation of buf1::hph mutant with BUF1 restores ACE1 expression. (A) M. grisea P1.2 buf1::hph/BUF1 transformants carrying promACE1::eGFP vector were used to monitor ACE1 expression as eGFP fluorescence. Microscopic observations were performed at ×100 magnification under bright light (A1) or UV light with an eGFP-specific filter (A2). Bar, 10 μm. (B) Pathogenicity on barley leaves of the P1.2 buf1::hph mutant (buf1::hph) and P1.2 buf1::hph mutant complemented with the BUF1 wild-type allele (buf1::hph/BUF1) and wild-type Guy11 5 days after inoculation.
Effect of hyperosmotic solutes on ACE1 expression in buf1::hph melanin-deficient appressoria.
During appressorium maturation, a high internal turgor pressure (4 to 8 Mpa) (22) is built up as a result of the accumulation of an osmolyte thought to be glycerol (12). Melanin-deficient mutants are unable to retain osmolytes accumulated in appressoria (8, 12, 20) and are therefore unable to build up appressorial turgor. We hypothesized that addition of hyperosmotic solutes to buf1::hph appressoria could mimic the high solute concentration reached in wild-type appressoria and induce ACE1 expression. We applied different hyperosmotic solutes to appressoria of buf1::hph transformants carrying promACE1::eGFP. We performed the same experiments with appressoria of Guy11 transformants expressing eGFP under the control of either ACE1 or MPG1 promoters (27) as controls. Six hours after inoculation of spores from Guy11 wild-type or buf1::hph promACE1::eGFP transformants on barley leaves or Teflon, young appressoria were fully differentiated. Residual water drops covering appressoria were replaced by hyperosmotic solutes, and eGFP fluorescence was monitored for 48 h. The concentrations of these solutes were chosen to generate an osmotic potential equivalent to those of wild-type appressoria (0.15 M PEG, 0.6 M sucrose, 1 M glycerol, 1 M sorbitol) (22) or to induce a hyperosmotic stress, as already observed for M. grisea mycelia (0.4 M NaCl) (15). Glycerol slightly decreased the proportion of eGFP fluorescent appressoria (60%) of Guy11 transformants formed on barley leaves compared to water (95%), whereas PEG, NaCl, sucrose, and sorbitol had no obvious effect on ACE1 expression (80 to 85%). Addition of PEG or glycerol did not restore ACE1 expression in buf1::hph appressoria differentiated on barley, while addition of NaCl, sucrose, or sorbitol significantly restored ACE1 expression (Table 3). Fifty-three percent of buf1::hph appressoria differentiated on barley leaves strongly expressed ACE1 when treated with NaCl, 58% with sorbitol, and 67% with sucrose. Interestingly, buf1::hph appressoria differentiated secondary hyphae on the surface of barley leaves or Teflon following NaCl, sucrose, or sorbitol treatment (Fig. 5). Similar secondary hyphae were not observed with wild-type appressoria treated with hyperosmotic solutes nor with untreated buf1::hph appressoria.
TABLE 3.
Effect of hyperosmotic solutes on ACE1 expression in appressoria from buf1::hph melanin-deficient mutants
| Molarity | Osmotic solutea | Appressoria expressing ACE1 (%) fromb:
|
|
|---|---|---|---|
| Guy11 | buf1::hph | ||
| Water | 95 ± 7 | 0 | |
| 0.15 | PEG | 83 ± 2 | 0 |
| 1.00 | Glycerol | 63 ± 16 | 0 |
| 0.40 | NaCl | 84 ± 7 | 53 ± 14 |
| 0.60 | Sucrose | 81 ± 12 | 67 ± 7 |
| 1.00 | Sorbitol | 81 ± 2 | 58 ± 8 |
Spores from Guy11 wild-type transformant and P1.2 buf1::hph transformant expressing promACE1::eGFP vector were inoculated on barley leaves. After 6 h, water droplets were replaced with the osmotic solute.
Appressoria from M. grisea Guy11 or P1.2 buf1::hph transformants were monitored for eGFP fluorescence 24 h after inoculation of spores on barley leaves.
FIG. 5.

Effect of hyperosmotic solute (0.4 M NaCl) on ACE1 expression in a buf1::hph mutant. Spores from a P1.2 buf1::hph transformant expressing promACE1::eGFP vector were inoculated on barley leaves. After 6 h, water droplets were replaced by 0.4 M NaCl and leaves were observed under a microscope 24 h after inoculation. (A) Microscopic observation was performed at ×100 magnification under UV light after staining with calcofluor. A spore (Sp), an appressorium (Ap), and a secondary hypha (Sh) originating from the appressorium are visible through the bright blue fluorescence of their cell walls. (B) Microscopic observation was performed at ×100 magnification under UV light with an eGFP-specific filter. eGFP fluorescence was detected only in the appressorium (Ap) and in the secondary hypha (Sh). Bar, 10 μm.
Although ACE1 expression was restored in 6-h-old buf1::hph appressoria by a treatment with hyperosmotic solutes, this expression was observed only 18 h after this treatment (24 hai). To analyze the effect of the application time on ACE1 expression, we treated buf1::hph appressoria with 0.6 M sucrose at 15 hai. Forty percent of appressoria expressed ACE1, but eGFP fluorescence was only observed at 48 hai (no fluorescence at 24 hai). We conclude that treatments with hyperosmotic solutes restore ACE1 expression in 6- to 15-h-old buf1::hph appressoria after a delay of 18 to 24 h. We also tested if ACE1 was expressed in mycelia following treatment with hyperosmotic solutes. We could not detect eGFP fluorescence in mycelia of Guy11 transformants expressing promACE1::eGFP grown in liquid medium containing either 0.4 M NaCl or 1 M sorbitol. ACE1-specific RT-PCR was performed on total RNA extracted from Guy11 mycelia grown under these hyperosmotic conditions for 15, 30, 60, or 120 min. An ACE1 RT-PCR product was detected 60 min after treatment with NaCl and 120 min after treatment with sorbitol (data not shown). Quantification of these transcripts by quantitative RT-PCR showed that only a very low level of transcript was produced (around 1/1,000 of the maximum transcript level in appressoria). These experiments suggest that ACE1 expression can be induced in mycelia grown under hyperosmotic conditions, but this expression level is very low compared to ACE1 appressorium expression.
Effect of cytoskeleton inhibitors on ACE1 expression.
The onset of appressorium-mediated penetration is associated with important reorganizations of actin and tubulin cytoskeleton associated with the formation of the penetration peg (5, 35). We speculated that the inhibition of these cytoskeleton modifications by inhibitors of actin and tubulin could inhibit appressorium-mediated penetration and consequently ACE1 expression. We used carbendazim, which induces the depolymerization of microtubules, and cytochalasin A, which represses actin polymerization (36). Eight-hours-old appressoria differentiated on onion epidermis were treated with carbendazim (30, 100, or 300 ppm) and cytochalasin A (1, 3, or 10 μM) to avoid any interference with appressorium differentiation and to specifically inhibit penetration peg formation. Carbendazim treatment reduced the penetration of the fungus into onion epidermis in a dose-dependent relationship starting from 21% inhibition at 30 ppm to a complete inhibition at 300 ppm (Table 4). This high concentration also completely inhibited ACE1 expression. At lower carbendazim concentrations, the expression of ACE1 was reduced to the same extent (30 ppm) or more (100 ppm) than penetration. Cytochalasin A treatments reduced the penetration of the fungus into onion epidermis in a dose-dependent relationship, as observed for carbendazim. For example, 3 μM cytochalasin A reduced penetration by 29% while 10 μM cytochalasin A strongly reduced penetration (67%). At these two concentrations, the expression of ACE1 was reduced to the same extent as penetration. In these experiments, we have observed that ACE1 is only expressed in appressoria penetrating into host tissues. Since penetration and ACE1 expression were coupled, we conclude that ACE1 expression depends on the initiation of penetration.
TABLE 4.
Effect of cytoskeleton polymerization inhibitors on ACE1 appressorium-specific expression
| Treatmenta | Appressorium differentiation (%) | Penetration (%)b | Appressoria expressing ACE1 (%)b | ACE1 expression inhibition rate (%) |
|---|---|---|---|---|
| Water | 100 | 57 ± 14* | 52 ± 8* | 0 |
| Carbendazim | ||||
| 30 ppm | 100 | 37 ± 7† | 41 ± 9† | 21 |
| 100 ppm | 100 | 41 ± 16† | 14 ± 7‡ | 73 |
| 300 ppm | 100 | 1 ± 1§ | 0§ | 100 |
| Cytochalasin A | ||||
| 1 μM | 100 | 54 ± 7* | 40 ± 13† | 23 |
| 3 μM | 100 | 46 ± 15† | 37 ± 13† | 29 |
| 10 μM | 84 ± 12 | 22 ± 12‡ | 17 ± 6‡ | 67 |
Inhibitors were applied 8 h after inoculation. eGFP fluorescence of appressoria from M. grisea Guy11 transformants carrying the promACE1::eGFP vector was monitored 24 h after inoculation of spores on onion epidermis.
Statistical groups indicated by symbols (*, †, ‡, and §) are based on mean comparisons using Student or Mann-Whitney tests (critical threshold α/2 = 0.025).
DISCUSSION
Induction of ACE1 appressorium-specific expression is independent of host plant.
We monitored ACE1 expression during fungal development and host plant infection using quantitative RT-PCR and a transcriptional fusion between the ACE1 promoter and eGFP reporter gene. We showed that ACE1 was only transcribed in mature appressoria, reaching a maximum at 17 hai and decreasing after 24 hai (real-time RT-PCR). ACE1 expression monitored by eGFP fluorescence followed the same kinetics, although with a 6-h delay. ACE1 appressorium-specific expression was also observed on artificial membranes, although at different degrees, ranging from 1 to 5% of appressoria on Mylar and Teflon to 75% on cellophane. A major difference between cellophane and Teflon/Mylar membranes is their chemical nature, with cellophane containing cellulose also found in plant cell walls, while Teflon and Mylar are inert chemical polymers. We tested different plant cell wall components (xylan, cellulose, pectin) and ground PUDO-193 cellophane on ACE1 expression. These compounds are unable to induce ACE1 expression in spores, germ tubes, and appressoria formed on a Teflon membrane, demonstrating that ACE1 is not induced by cellophane components. Appressoria formed on Teflon or Mylar could differ in their physiology from those produced on the leaf surface, although they reach turgor levels similar to appressoria on host leaves (Table 1). Indeed, appressoria formed on Mylar or Teflon are unable to pierce these membranes, and only a few of these appressoria are able to differentiate penetration pegs (22). The small number of appressoria expressing ACE1 on Teflon and Mylar may correspond to the few appressoria initiating a penetration peg. Overall, these observations show that ACE1 is exclusively expressed in mature appressoria during penetration into a leaf or an artificial cellophane membrane. This induction is independent of compounds from the cellophane membrane or plant cell wall.
ACE1 is not expressed in melanin-deficient mutant appressoria.
Early stages of appressorial development are characterized by the deposition of a melanin layer between the fungal membrane and cell wall that is required for turgor buildup (20, 21). This melanin layer acts as a semipermeable membrane, retaining solutes such as glycerol (12) accumulated in appressoria and allowing the buildup of a high internal hydrostatic pressure as water flows into this cell (20, 21). This turgor is required for appressorium-mediated penetration (8, 22). Melanin-deficient mutants do not retain appressorial solutes and are unable to penetrate leaves or cellophane (8, 12, 20). We clearly showed that ACE1 was not expressed in the melanin-deficient mutant buf1::hph. Since the major defect of this mutant is the lack of turgor, we first hypothesized that appressorial turgor is needed for the induction of ACE1 appressorium-specific expression. This is obviously not the case, as ACE1 was not expressed in appressoria formed on Teflon and Mylar that generate a normal turgor. Treatments of wild-type appressoria with external hyperosmotic solutes reduce internal turgor dramatically (22). These treatments did not reduce ACE1 expression, confirming that turgor is not required for ACE1 expression. The other major defect of melanin-deficient mutant buf1::hph is the absence of accumulation of solutes. Therefore, we hypothesized that the addition of hyperosmotic solutes to buf1::hph appressoria could mimic the high solute concentration reached in wild-type appressoria and induce ACE1 expression. Indeed, addition of hyperosmotic NaCl, sorbitol, or sucrose solutions to buf1::hph appressoria restored ACE1 expression in mature mutant appressoria, while it did not induce its expression in mycelium, spores, germ tubes, and young appressoria. The wide range of compounds able to restore ACE1 expression in buf1::hph appressoria suggests that this induction is not a consequence of the presence of a particular solute at a high concentration. NaCl, sorbitol, and sucrose that induce ACE1 expression are not accumulated in wild-type appressoria (12). On the contrary, glycerol that is normally accumulated in appressoria during turgor buildup (12) did not induce ACE1 expression. These results suggest that the restoration of ACE1 expression in buf1::hph appressoria is a direct or indirect consequence of the hyperosmotic stress induced by these solutes.
When buf1::hph appressoria were treated with hyperosmotic solutes, we always observed the restoration of ACE1 expression after a delay of at least 12 h. Treatment of M. grisea mycelia with hyperosmotic solutes induces the transcription of target genes after a short delay of 1 to 2 h (13, 15). This short delay in the direct transcriptional response of fungal cells to hyperosmotic stress suggests that the induction of ACE1 expression in buf1::hph appressoria is not the result of a direct response to osmotic stress. The same osmotic stress did not induce ACE1 expression in spores, young appressoria, or mycelia, even though a very small induction was observed at the mRNA level in stressed mycelia. Alternatively, buf1::hph appressoria could be blocked at an early stage of appressorial development. Hyperosmotic solutes could reinitiate appressorial development in buf1::hph appressoria, allowing them to reach the developmental stage required to induce ACE1 expression. This hypothesis is strengthened by the fact that buf1::hph appressoria treated by hyperosmotic solutes reach a novel developmental stage associated with the differentiation of secondary hyphae formed at the base of the appressorium. These secondary hyphae likely arise from penetration pegs, suggesting that treated buf1::hph appressoria reach the penetration stage, although they are unable to pierce host cell wall.
ACE1 expression is connected to the onset of appressorium-mediated penetration.
M. grisea penetration-deficient mutants were used to assess whether a successful penetration was required for ACE1 appressorium-specific expression or not. ACE1 was normally expressed in the ΔcpkA and Δmac1 sum1-99 mutants deficient for or with a constitutively active cAMP signaling pathway, respectively (1, 32, 43, 47). These observations demonstrate that the control of ACE1 expression is independent of the cAMP signaling pathway. Additionally, ACE1 was also normally expressed in appressoria of the mutant Δpls1::hph (9) unable to penetrate host tissues, demonstrating that its expression does not require a successful penetration event. We have previously shown that ACE1 is not fully expressed in wild-type appressoria formed on Teflon and Mylar membranes that do not allow penetration peg formation (22). These apparently contradictory observations suggest that ΔcpkA and Δpls1::hph appressoria reach a developmental stage connected to ACE1 expression, while appressoria formed on Teflon and Mylar do not. This developmental stage corresponds to the onset of appressorium-mediated penetration, as ΔcpkA mutant appressoria are still able to differentiate penetration pegs and attempt to penetrate the host cell wall (35).
To test this hypothesis, we used actin and tubulin inhibitors that should disturb the reorganization of cytoskeleton observed at an early stage of penetration peg formation (5, 35) and consequently inhibit penetration. Both carbendazim and cytochalasin A inhibited the penetration of M. grisea into onion epidermal cells in a dose-dependent manner. These treatments also inhibited ACE1 expression quantitatively, and the only appressoria expressing ACE1 were those which penetrated successfully into onion epidermis. These results show that the inhibition of cytoskeleton reorganization in the appressorium and, consequently, penetration peg formation also abolish ACE1 expression.
Overall, these experiments demonstrate that the induction of ACE1 expression in appressoria is connected to a specific appressorial developmental stage associated with penetration peg formation. ACE1 expression is therefore a landmark of this early stage of appressorium-mediated penetration.
ACE1 is a secondary metabolism gene with a novel expression pattern.
ACE1 encodes a putative hybrid polyketide synthase (PKS)-nonribosomal peptide synthetase (NRPS) (4). Expression of fungal PKS- and NRPS-encoding genes is frequently induced during stationary phase (49) and is affected by environmental and nutritional factors such as temperature, pH, carbon and nitrogen sources, and lipids (6, 31). These genes are also frequently repressed during mycelial growth and induced during sporulation (49). We have shown that ACE1 expression is specifically connected to the onset of appressorium-mediated penetration. The ACE1 expression pattern therefore confirms the general assumption that genes from secondary metabolism are expressed at particular developmental stages. The regulatory networks involved in the tight appressorial expression of ACE1 remain to be discovered, since it is independent of appressorial signaling pathways identified so far. The identification of these appressorium-specific regulatory networks will be very helpful to understand the early stages of appressorium-mediated penetration.
Acknowledgments
This work was supported by EU grant ICA4-CT-2000-30021 (INCO-DEV project “RESIDIV”), including a Ph.D. fellowship to I.F., and by the French Ministère de la Recherche with the MENRT Ph.D. fellowship to J.C.
We thank Jin-Rong Xu (Purdue University) for ΔcpkA-I27 and Δmac1 sum1-99 mutants and Mark Farman (University of Kentucky) for the BUF1 wild-type gene. We also thank Tim Bourret (E. I. Du Pont de Nemours, Wilmington, DE) for PUDO-193 cellophane membrane, Ronald De Vries (Utrecht University, The Netherlands) for helpful advices on cell wall components, and Bernard Dumas (CNRS-UPS, Toulouse, France) for critical reading of the manuscript.
Footnotes
Published ahead of print on 1 December 2006.
REFERENCES
- 1.Adachi, K., and J. E. Hamer. 1998. Divergent cAMP signaling pathways regulate growth and pathogenesis in the rice blast fungus Magnaporthe grisea. Plant Cell 10:1361-1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balhadère, P. V., and N. J. Talbot. 2001. PDE1 encodes a P-type ATPase involved in appressorium-mediated plant infection by the rice blast fungus Magnaporthe grisea. Plant Cell 13:1987-2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banno, S., M. Kimura, T. Tokai, S. Kasahara, A. Higa-Nishiyama, N. Takahashi-Ando, H. Hamamoto, M. Fujimura, B. J. Staskawicz, and I. Yamaguchi. 2003. Cloning and characterization of genes specifically expressed during infection stages in the rice blast fungus. FEMS Microbiol. Lett. 222:221-227. [DOI] [PubMed] [Google Scholar]
- 4.Böhnert, H. U., I. Fudal, W. Dioh, D. Tharreau, J. L. Notteghem, and M. H. Lebrun. 2004. A putative polyketide synthase/peptide synthetase from Magnaporthe grisea signals pathogen attack to resistant rice. Plant Cell 16:2499-2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bourett, T. M., and R. J. Howard. 1990. In vitro development of penetration structures in the rice blast fungus Magnaporthe grisea. Can. J. Bot. 68:329-342. [Google Scholar]
- 6.Cary, J. W., J. E. Linz, and D. Bhatnagar. 2000. Aflatoxins: biological significance and regulation of biosynthesis, p. 317-361. In J. W. Cary, J. E. Linz, and D. Bhatnagar (ed.), Microbial foodborne diseases: mechanisms of pathogenesis and toxin synthesis. Technomic, Lancaster, PA.
- 7.Choi, W., and R. A. Dean. 1997. The adenylate cyclase gene MAC1 of Magnaporthe grisea controls appressorium formation and other aspects of growth and development. Plant Cell 9:1973-1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chumley, F., and B. Valent. 1990. Genetic analysis of melanin-deficient, nonpathogenic mutants of Magnaporthe grisea. Mol. Plant-Microbe Interact. 3:135-143. [Google Scholar]
- 9.Clergeot, P. H., M. Gourgues, J. Cots, F. Laurans, M. P. Latorse, R. Pepin, D. Tharreau, J. L. Notteghem, and M. H. Lebrun. 2001. PLS1, a gene encoding a tetraspanin-like protein, is required for penetration of rice leaf by the fungal pathogen Magnaporthe grisea. Proc. Natl. Acad. Sci. USA 98:6963-6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Couch, B. C., I. Fudal, M. H. Lebrun, D. Tharreau, B. Valent, P. van Kim, J. L. Notteghem, and L. M. Kohn. 2005. Origins of host-specific populations of the blast pathogen Magnaporthe oryzae in crop domestication with subsequent expansion of pandemic clones on rice and weeds of rice. Genetics 170:613-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dean, R. A., N. J. Talbot, D. J. Ebbole, M. L. Farman, T. K. Mitchell, M. J. Orbach, M. Thon, R. Kulkarni, J. R. Xu, H. Pan, N. D. Read, Y. H. Lee, I. Carbone, D. Brown, Y. Y. Oh, N. Donofrio, J. S. Jeong, D. M. Soanes, S. Djonovic, E. Kolomiets, C. Rehmeyer, W. Li, M. Harding, S. Kim, M. H. Lebrun, H. Böhnert, S. Coughlan, J. Butler, S. Calvo, L. J. Ma, R. Nicol, S. Purcell, C. Nusbaum, J. E. Galagan, and B. W. Birren. 2005. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434:980-986. [DOI] [PubMed] [Google Scholar]
- 12.de Jong, J. C., B. J. McCormack, N. Smirnoff, and N. J. Talbot. 1997. Glycerol generates turgor in rice blast. Nature 389:244-245. [Google Scholar]
- 13.de Vries, R. P., S. J. Flitter, P. J. van de Vondervoort, M. K. Chaveroche, T. Fontaine, S. Fillinger, G. J. Ruijter, C. d'Enfert, and J. Visser. 2003. Glycerol dehydrogenase, encoded by gldB is essential for osmotolerance in Aspergillus nidulans. Mol. Microbiol. 49:131-141. [DOI] [PubMed] [Google Scholar]
- 14.Dioh, W., D. Tharreau, J. L. Notteghem, M. Orbach, and M. H. Lebrun. 2000. Mapping of avirulence genes in the rice blast fungus, Magnaporthe grisea, with RFLP and RAPD markers. Mol. Plant-Microbe Interact. 13:217-227. [DOI] [PubMed] [Google Scholar]
- 15.Dixon, K. P., J. R. Xu, N. Smirnoff, and N. J. Talbot. 1999. Independent signaling pathways regulate cellular turgor during hyperosmotic stress and appressorium-mediated plant infection by Magnaporthe grisea. Plant Cell 11:2045-2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ebbole, D. J., Y. Jin, M. Thon, H. Pan, E. Bhattarai, T. Thomas, and R. Dean. 2004. Gene discovery and gene expression in the rice blast fungus, Magnaporthe grisea: analysis of expressed sequence tags. Mol. Plant-Microbe Interact. 17:1337-1347. [DOI] [PubMed] [Google Scholar]
- 17.Farman, M. L. 2002. Meiotic deletion at the BUF1 locus of the fungus Magnaporthe grisea is controlled by interaction with the homologous chromosome. Genetics 160:137-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farman, M. L., Y. Eto, T. Nakao, Y. Tosa, H. Nakayashi, S. Mayama, and S. A. Leong. 2002. Analysis of the structure of the AVR1-CO39 avirulence locus in virulent rice-infecting isolates of Magnaporthe grisea. Mol. Plant-Microbe Interact. 15:1-16. [DOI] [PubMed] [Google Scholar]
- 19.Hammond-Kosack, K. E., and J. E. Parker. 2003. Deciphering plant-pathogen communication: fresh perspectives for molecular resistance breeding. Curr. Opin. Biotechnol. 14:177-193. [DOI] [PubMed] [Google Scholar]
- 20.Howard, R. J., and M. A. Ferrari. 1989. Role of melanin in appressorium function. Exp. Mycol. 13:403-418. [Google Scholar]
- 21.Howard, R. J., and B. Valent. 1996. Breaking and entering: host penetration by the fungal rice blast pathogen Magnaporthe grisea. Annu. Rev. Microbiol. 50:491-512. [DOI] [PubMed] [Google Scholar]
- 22.Howard, R. J., M. A. Ferrari, D. H. Roach, and N. P. Money. 1991. Penetration of hard substrates by a fungus employing enormous turgor pressures. Proc. Natl. Acad. Sci. USA 88:11281-11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irie, T., H. Matsumura, R. Terauchi, and H. Saitoh. 2003. Serial analysis of gene expression (SAGE) of Magnaporthe grisea: genes involved in appressorium formation. Mol. Genet. Genomics 270:181-189. [DOI] [PubMed] [Google Scholar]
- 24.Jia, Y., S. A. McAdams, G. T. Bryan, H. P. Hershey, and B. Valent. 2000. Direct interaction of resistance gene and avirulence gene products confers rice blast resistance. EMBO J. 19:4004-4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamakura, T., S. Yamaguchi, K. I. Saitoh, T. Teraoka, and I. Yamaguchi. 2002. A novel gene, CBP1, encoding a putative extracellular chitin-binding protein, may play an important role in the hydorphobicity surface sensing of Magnaporthe grisea during appressorium differentiation. Mol. Plant-Microbe Interact. 15:437-444. [DOI] [PubMed] [Google Scholar]
- 26.Kato, H., M. Tamamoto, T. Yamaguchi-Ozaki, H. Kadouchi, Y. Iwamoto, H. Nakayashiki, Y. Tosa, S. Mayama, and N. Mom. 2000. Pathogenicity, mating ability and DNA restriction fragment length polymorphisms of Pyricularia populations isolated from Gramineae, Bambusideae and Zingiberaceae plants. J. Gen. Plant Pathol. 66:30-47. [Google Scholar]
- 27.Kershaw, M. J., G. Wakley, and N. J. Talbot. 1998. Complementation of the Mpg1 mutant phenotype in Magnaporthe grisea reveals functional relationships between fungal hydrophobins. EMBO J. 17:3838-3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim, S., I. P. Ahn, and Y. H. Lee. 2001. Analysis of genes expressed during rice-Magnaporthe grisea interactions. Mol. Plant-Microbe Interact. 14:1340-1346. [DOI] [PubMed] [Google Scholar]
- 29.Lagorce, A., A. Darchis, F. Munier, J. B. Morel, R. De Rose, R. Beffa, and M. H. Lebrun. 2005. Transcriptional analysis of the pathogenic fungus Magnaporthe grisea during rice infection, abstr. 175. XXIII Fungal Genetics Conference, Pacific Grove, CA.
- 30.Lu, J. P., T. B. Liu, and F. C. Lin. 2005. Identification of mature appressorium-enriched transcripts in Magnaporthe grisea, the rice blast fungus, using suppression subtractive hybridization. FEMS Microbiol. Lett. 245:131-137. [DOI] [PubMed] [Google Scholar]
- 31.Mayorga, M., and W. Timberlake. 1992. The developmentally regulated Aspergillus nidulans wA gene encodes a polypeptide homologous to polyketide and fatty acid synthases. Mol. Gen. Genet. 235:205-212. [DOI] [PubMed] [Google Scholar]
- 32.Mitchell, G. F., and R. A. Dean. 1995. The cAMP-dependent protein kinase catalytic subunit is required for appressorium formation and pathogenesis by the rice blast pathogen Magnaporthe grisea. Plant Cell 7:1869-1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Notteghem, J. L., D. Tharreau, D. Silue, and E. Roumen. 1994. Present knowledge on rice resistance genetics and strategies for analysis of M. grisea pathogenicity and avirulence genes, p. 155-166. In R. Zeigler, S. A. Leong, and P. S. Teng (ed.), Rice blast disease. Commonwealth Agricultural Bureau International, Wallingford, United Kingdom.
- 34.Orbach, M. J., L. Farrall, J. A. Sweigard, F. G. Chumley, and B. Valent. 2000. A telomeric avirulence gene determines efficacy for the rice blast resistance gene Pi-ta. Plant Cell 12:2019-2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park, G., K. S. Bruno, C. J. Staiger, N. J. Talbot, and J. R. Xu. 2004. Independent genetic mechanisms mediate turgor generation and penetration peg formation during plant infection in the rice blast fungus. Mol. Microbiol. 53:1695-1707. [DOI] [PubMed] [Google Scholar]
- 36.Parsons, A. B., A. Lopez, I. E. Givoni, D. E. Williams, C. A. Gray, J. Porter, G. Chua, R. Sopko, R. L. Brost, C. H. Ho, J. Wang, T. Ketela, C. Brenner, J. A. Brill, G. E. Fernandez, T. C. Lorenz, G. S. Payne, S. Ishihara, Y. Ohya, B. Andrews, T. R. Hughes, B. J. Frey, T. R. Graham, R. J. Andersen, and C. Boone. 2006. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell. 126:611-625. [DOI] [PubMed] [Google Scholar]
- 37.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 38.Sweigard, J. A., A. M. Carroll, L. Farrall, F. G. Chumley, and B. Valent. 1998. Magnaporthe grisea pathogenicity genes obtained through insertional mutagenesis. Mol. Plant-Microbe Interact. 11:404-412. [DOI] [PubMed] [Google Scholar]
- 39.Sweigard, J. A., A. M. Carroll, S. Kang, L. Farrall, F. G. Chumley, and B. Valent. 1995. Identification, cloning, and characterization of PWL2, a gene for host species specificity in the rice blast fungus. Plant Cell 7:1221-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sweigard, J. A., F. G. Chumley, A. M. Carroll, L. Farrall, and B. Valent. 1997. A series of vectors for fungal transformation. Fungal Genet. Newsl. 44:52-53. [Google Scholar]
- 41.Takano, Y., W. Choi, T. K. Mitchell, T. Okuno, and R. Dean. 2003. Large scale parallel analysis of gene expression during infection-related morphogenesis of Magnaporthe grisea. Mol. Plant Pathol. 4:337-346. [DOI] [PubMed] [Google Scholar]
- 42.Talbot, N. J. 2003. On the trail of a cereal killer: exploring the biology of Magnaporthe grisea. Annu. Rev. Microbiol. 57:177-202. [DOI] [PubMed] [Google Scholar]
- 43.Thines, E., R. W. Weber, and N. J. Talbot. 2000. MAP kinase and protein kinase A-dependent mobilization of triacylglycerol and glycogen during appressorium turgor generation by Magnaporthe grisea. Plant Cell 12:1703-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Urban, M., T. Bhargarva, and J. E. Hamer. 1999. An ATP-driven efflux pump is a novel pathogenicity factor in rice blast disease. EMBO J. 18:512-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Viaud, M. C., P. V. Balhadère, and N. J. Talbot. 2002. A Magnaporthe grisea cyclophilin acts as a virulence determinant during plant infection. Plant Cell 14:917-930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woloshuk, C. P., H. D. Sisler, and E. L. Vigil. 1983. Action of the antipenetrant, tricyclazole, on appressoria of Pyricularia oryzae. Physiol. Plant Pathol. 22:245-259. [Google Scholar]
- 47.Xu, J. R., M. Urban, J. A. Sweigard, and J. E. Hamer. 1997. The CPKA gene of Magnaporthe grisea is essential for appressorial penetration. Mol. Plant-Microbe Interact. 10:187-194. [Google Scholar]
- 48.Xue, C., G. Park, W. Choi, L. Zheng, R. A. Dean, and J. R. Xu. 2002. Two novel fungal virulence genes specifically expressed in appressoria of the rice blast fungus. Plant Cell 14:2107-2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu, J. H., and N. Keller. 2005. Regulation of secondary metabolism in filamentous fungi. Annu. Rev. Phytopathol. 43:437-458. [DOI] [PubMed] [Google Scholar]
- 50.Zhao, X., Y. Kim, G. Park, and J. R. Xu. 2005. A mitogen-activated protein kinase cascade regulating infection-related morphogenesis in Magnaporthe grisea. Plant Cell 17:1317-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]