

Abstract
Members of the nuclear hormone receptor superfamily function as key transcriptional regulators of inflammation and proliferation in cardiovascular diseases. In addition to the ligand-dependent peroxisome proliferator-activated receptors and liver X receptors, this family of transcription factors includes a large number of orphan receptors, and their role in vascular diseases remains to be investigated. The neuron-derived orphan receptor-1 (NOR1) belongs to the ligand-independent NR4A subfamily, which has been implicated in cell proliferation, differentiation, and apoptosis. In this study, we demonstrate NOR1 expression in vascular smooth muscle cells (SMC) of human atherosclerotic lesions. In response to mitogenic stimulation with platelet-derived growth factor (PDGF), SMC rapidly express NOR1 through an ERK-MAPK-dependent signaling pathway. 5′-Deletion analysis, site-directed mutagenesis, and transactivation experiments demonstrate that PDGF-induced NOR1 expression is mediated through a cAMP-response element-binding protein (CREB)-dependent transactivation of the NOR1 promoter. Consequently, short interfering RNA-mediated depletion of CREB abolished PDGF-induced NOR1 expression in SMC. Furthermore, PDGF induced Ser-133 phosphorylation of CREB and subsequent binding to the CRE sites of the endogenous NOR1 promoter. Functional analysis demonstrated that PDGF induces NOR1 transactivation of its consensus NGFI-B-response elements (NBRE) in SMC. We finally demonstrate that SMC isolated from NOR1-deficient mice exhibit decreased cell proliferation and characterize cyclin D1 and D2 as NOR1 target genes in SMC. These experiments indicate that PDGF-induced NOR1 transcription in SMC is mediated through CREB-dependent transactivation of the NOR1 promoter and further demonstrate that NOR1 functions as a key transcriptional regulator of SMC proliferation.
Atherosclerosis, the subsequent development of occlusive vascular diseases, and the failure of treatment approaches such as postangioplasty restenosis involve several interrelated processes (1, 2). In addition to endothelial dysfunction and inflammation, proliferation of smooth muscle cells (SMC)3 is considered to play a pivotal role in the pathogenesis of atherosclerosis and the failure of interventional approaches used to treat related occlusive vascular complications (2–4). With the evolving understanding of the mechanisms contributing to the development of vascular diseases, members of the nuclear hormone receptor superfamily of transcription factors have emerged as key transcriptional regulators of inflammation and cell proliferation (5, 6). Based on this evidence, elucidation of the molecular pathways utilized by nuclear receptors to regulate programs of gene expression is expected to facilitate the development of novel pharmacological approaches for the treatment of cardiovascular diseases. Nuclear receptors of the peroxisome proliferator-activated receptor (PPAR) and liver X receptor (LXR) subfamilies are expressed in SMC and inhibit their proliferation in response to mitogenic stimulation thereby limiting the development of postangioplasty restenosis (7–9). Although much attention has focused on the role of PPARs and LXRs as transcriptional regulators of genes involved in SMC proliferation, the nuclear receptor superfamily includes a large number of so-called orphan nuclear receptors, whose ligands, target genes, and physiological functions are unknown and remain to be discovered (10).
The neuron-derived orphan receptor-1 (NOR1) is a transcription factor belonging to the NR4A (also referred as NGFI-B) subfamily within the nuclear receptor superfamily (11, 12). Members of the NR4A subfamily transactivate target gene promoters through monomer binding to consensus NGFI-B-response elements (NBRE), homodimer binding to a palindromic NurRE sequence, or heterodimer binding with retinoid X receptor to specific DR5 elements of target genes (13–16). In contrast to other members of the nuclear hormone receptor superfamily, crystal structure and NMR data indicate that the ligand-binding pocket of the NR4A receptors is covered by hydrophobic residues, and these receptors have been demonstrated to function as ligand-independent constitutively active receptors (17, 18).
NOR1 was originally identified from primary cultured rat fetal forebrain cells undergoing apoptosis (19). Subsequent studies characterized NOR1 as an early response gene rapidly induced by a variety of extracellular stimuli (12, 20). NOR1 is highly expressed in the central nervous system during embryonic development (21). Our studies using previously generated NOR1-deficient mice demonstrated that NOR1 plays a critical role in neuronal survival, axonal guidance, and is required for the continuous proliferative growth of the semicircular canals (22, 23). In addition, experiments using overexpression of NOR1 revealed that constitutive NOR1 expression in thymocytes results in apoptosis (24). These studies suggest that NOR1 may function as important regulator of key cellular mechanisms, including apoptosis, proliferation, and migration.
Recently, NOR1 expression has been identified in atherosclerotic lesions (25, 26). NOR1 expression is induced by oxidized low density lipoprotein in macrophages (27) and has been further identified as the most potently induced gene in endothelial cells treated with vascular endothelial growth factor (28). In addition, NOR1 is rapidly expressed after mitogenic stimulation of SMC (25). However, the molecular mechanisms governing NOR1 expression in response to growth factor stimulation, its biological function in SMC, and its role in the development of cardiovascular diseases remain to be investigated.
In this study, we analyzed the transcriptional regulation of NOR1 expression in response to stimulation with platelet-derived growth factor (PDGF), a key growth factor mediating SMC proliferation in atherosclerosis and postangioplasty restenosis (29). To establish an essential physiological function of NOR1 for SMC proliferation, we further analyzed cell proliferation of SMC isolated from NOR1-deficient mice. These experiments demonstrate that PDGF-induced NOR1 expression is mediated through a cAMP-response element-binding protein (CREB)-dependent transactivation of the NOR1 promoter and further outline an essential role for NOR1 to mediate SMC proliferation via an induction of cyclin D1 and D2 expression. Based on these observations, we propose that NOR1 functions as a key transcriptional regulator of SMC proliferation.
EXPERIMENTAL PROCEDURES
Immunohistochemistry
To demonstrate specificity of the employed NOR1 antibody, NOR1 was overexpressed in HEK293 cells. HEK293 cells seeded on 6-well plates were transfected with 1 μg of expression vector overexpressing NOR1 (pCMV-NOR1) (30) or empty vector (pCMV) using Lipofectamine 2000. 24 h after transfection, cells were fixed in acetone and stained with anti-NOR1 antibody (IMG-71915; Imgenex, Inc.) followed by Texas Red-conjugated goat anti-rabbit IgG (sc-2780; Santa Cruz Biotechnology). Cells were counterstained with 4′,6′-diamidinophenylindole and visualized using confocal microscopy.
For immunohistochemical analysis of NOR1 expression in human atherosclerotic lesions, segments of normal human coronary arteries or arteries containing atherosclerotic lesions at various stages were harvested from explanted hearts during cardiac transplantation. Tissues were fixed in 4% phosphate-buffered formaldehyde and embedded in paraffin. Transverse sections (8 μm) were collected, and immunohistochemical analysis of atherosclerotic lesions was performed as described previously (31). Paraffin-embedded sections were incubated with the rabbit polyclonal NOR1 antibody (IMG-71915; Imgenex, Inc.) followed by biotinylated goat anti-rabbit IgG (BA-1000; Vector Laboratories). SMC co-staining of serial sections was performed using an anti-mouse monoclonal anti-smooth muscle α-actin antibody (Ab18147; Abcam, Inc.) followed by biotinylated goat anti-mouse IgG (BA-9200; Vector Laboratories).
Cell Culture
Rat aortic vascular SMC were isolated and cultured as described previously (32). Cells were grown to 60–70% confluency, serum-deprived in 0.1% FBS for at least 48 h, and subjected to stimulation with rat PDGF-BB (Sigma) at a final concentration of 25 ng/ml. For all data shown, individual experiments were repeated at least three times with different preparation of cells.
Northern Blotting
Isolation of total RNA and Northern blotting was performed as described previously (33). cDNA for NOR1 was used as described recently (34). Blots were cohybridized with cDNA encoding the constitutively expressed housekeeping gene 36B4 to assess equal loading of samples.
Western Blot Analysis
Western blotting was performed as described recently (35). Primary antibodies were obtained from the following suppliers: NOR1 (2ZH7833H; R & D Systems); phospho-p44/42 MAPK (Thr-202/Tyr-204), p44/42 MAPK, phospho-CREB (Ser-133), CREB (Cell Signaling); GAPDH (FL 335; Santa Cruz Biotechnology); cyclin D1 (05-815, Upstate); cyclin D2 (M-20; Santa Cruz Biotechnology). Blots were cohybridized with p44/42 MAPK, CREB, or GAPDH.
Plasmids, Transient Transfections, and Luciferase Assay
The NOR1 promoter constructs and the NBRE(x8)-Luc reporter construct were used as described recently (36, 37). The CRE-mutated NOR1 promoter constructs were kindly provided by Dr. Hiroshi Tokumitsu (Kagawa Medical University, Kagawa, Japan) (38). The dominant-negative CREB (ACREB) construct was provided by Dr. Jane E.-B. Reusch (University of Colorado Health Sciences Center, Aurora, CO) (39). SMC were transfected for 6–8 h with 2 μg of reporter DNA using Lipofectamine 2000 (Invitrogen). Following transfection, cells were serum-deprived in 0.1% FBS for 24 h and subsequently stimulated with PDGF (25 ng/ml) for the indicated time points. Luciferase activity was assayed using a dual luciferase reporter assay (Promega). Transfection efficiency was normalized to Renilla luciferase activities generated by cotransfection of 10 ng/well pGL4.74[hRluc/TK] (Promega).
Chromatin Immunoprecipitation (ChIP) Assays
ChIP assays were performed using an EZ-ChIP assay kit (Upstate) according to the manufacturer’s instructions. Briefly, quiescent SMC were stimulated with PDGF (25 ng/ml) for the indicated time points. Cells were harvested, and soluble chromatin was prepared. Chromatin was immunoprecipitated using an antibody (4 μg) directed against Ser-133-phospho-CREB (catalog number 9192; Cell Signaling). Final DNA extractions were PCR-amplified using the following primer pairs that cover the three CRE consensus sequences between −79 and −46 in the NOR1 promoter: forward, 5′-ACACGAACACACACACCCT-3′; reverse, 5′-TCCATAGAGTGCCTGGAATGCGA-3′.
siRNA Knockdown of CREB Expression
siRNA experiments were performed using the SMARTpool technology (Dharmacon RNA Technologies), which provides a mix of four different proprietary siRNAs specific for rat CREB. SMC were plated at a density of 1.0 × 105 cells on 6-well plates and transfected with 150 nmol/liter of CREB siRNA or scrambled siRNA using Lipofectamine 2000 (Invitrogen). Following transfection, cells were serum-deprived in 0.1% FBS for 12 h and subsequently stimulated with PDGF (25 ng/ml) for 6 h. Total cell lysates were collected and analyzed for CREB and NOR1 protein expression by Western blotting.
Isolation of Murine Aortic Vascular SMC
NOR1-deficient mice have been described previously (22). Heterozygote NOR1 mice were interbred to generate littermate NOR1−/− and NOR1+/+ mice used for the isolation of aortic vascular SMC. Five different preparations of NOR1−/− and wild type SMC were isolated from the aortas of 8–10-week-old male matched littermate mice. The aortas were excised, washed in PBS, and incubated in DMEM containing 1 mg/ml collagenase type 2 (Worthington) for 15 min. The adventitia was removed; the aortas were incised longitudinally, and the endothelial cells were gently scraped off. The vessels were then transferred to culture dishes containing DMEM with 0.5 mg/ml collagenase type 1 (Worthington) and 0.125 mg/ml elastase type III (Sigma). Subsequently, the vessels were minced with scissors, incubated in 37 °C, 5% CO2 humidified incubator, and mechanically dispersed by pipeting vigorously every 10 min for 40–60 min until >90% of the cells were dispersed under the microscope. The cells were centrifuged at 1600 rpm for 5 min then resuspended in 3 ml of DMEM supplemented with 20% FBS, 2% penicillin/streptomycin, and cultured in 60-mm plates. Cultured cells were verified as SMC by immunostaining using anti-α-smooth muscle actin (Sigma) and calponin antibodies (Sigma).
Cell Proliferation Assays
NOR1+/+ and NOR1−/− murine aortic vascular SMC were plated at a density of 0.5 × 105 cells/plate on 60-mm plates and maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Proliferation assays were performed with exponentially growing cells in 10% FBS or following serum deprivation in 0.1% FBS for 24 h followed by stimulation with PDGF (25 ng/ml). After 2, 4, 6, and 8 days, cells were harvested, and cell proliferation was analyzed by cell counting using a hemocytometer. To re-express NOR1 protein in NOR1−/− cells, NOR1-deficient SMC were transfected with 1 μg of pCMV-NOR1 or an expression vector overexpressing green fluorescent protein (pCMV-GFP) as control. Cells were maintained in DMEM supplemented with 20% FBS. Cell proliferation was analyzed after 3 days by cell counting using a hemocytometer. For all experiments, wild type and NOR1-deficient SMC of similar passages (passage 3–7) were employed. Experiments were performed in triplicates using five different preparations of aortic vascular SMC.
Statistical Analysis
Analysis of variance and paired or unpaired t test were performed for statistical analysis as appropriate. p values less than 0.05 were considered to be statistically significant. Results are expressed as mean ± S.E.
RESULTS
NOR1 Is Expressed in Human Atherosclerotic Lesions
The specificity of the employed NOR1 antibody was first confirmed in HEK293 cells, which had been transfected with a eukaryotic expression vector overexpressing NOR1 protein. These experiments demonstrated NOR1 staining only in cells transfected with the NOR1 expression vector and not with the control vector. The staining pattern revealed a typical nuclear localization of NOR1 protein as depicted in the supplemental Fig. 1.
FIGURE 1. NOR1 protein is expressed in SMC of human coronary artery atherosclerotic lesions.
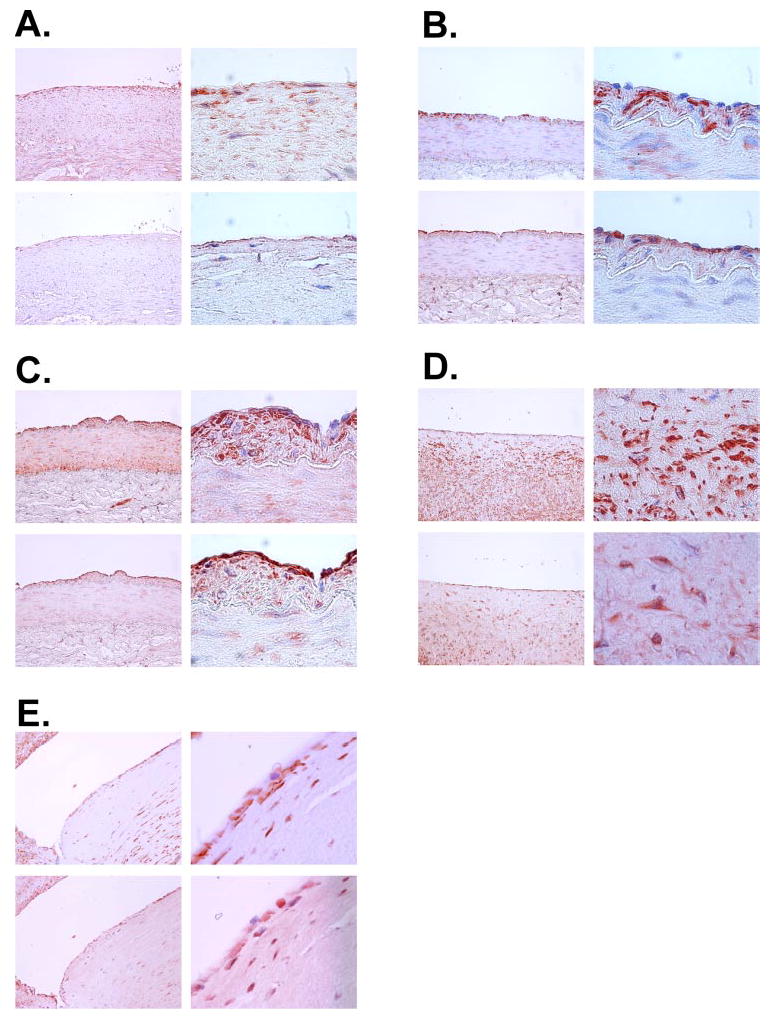
Paraffin-embedded serial sections of normal human coronary arteries (A) and arteries containing atherosclerotic lesions of different stages (B–E) were stained for smooth muscle α-actin (upper panels) and NOR1 (lower panels). Objective magnifications are ×20 (left panels) and ×100 (right panels).
To analyze NOR1 protein expression in normal arteries and arteries containing atherosclerotic lesions of different stages, immunohistochemical analysis was performed using tissues derived from coronary arteries of explanted hearts during cardiac transplantation. Serial sectioning and staining for smooth muscle α-actin and NOR1 revealed that NOR1 expression is almost absent in the media of normal arteries, although some staining is observed in endothelial cells (Fig. 1A). In contrast, in early stages of atherosclerosis (Fig. 1, B and C) and fibroatheromas (Fig. 1D), NOR1 is strongly expressed in neointimal SMC. Higher magnifications indicate that in these cells NOR1 expression is typically observed in the nucleus. In more advanced atherosclerotic lesions (Fig. 1E), NOR1 expression was observed in SMC of the fibrous cap as well as in medial SMC. In addition, these lesions revealed NOR1 expression in the core of the atheroma.
PDGF Induces NOR1 mRNA and Protein Expression through ERK-MAPK-dependent Signaling Pathways
In response to vascular injury, secreted growth factors stimulate SMC proliferation that perpetuates neointimal hyperplasia contributing to the pathogenesis of atherosclerosis (2, 4, 40). PDGF is one of the most potent mitogenic growth factors secreted after vascular injury and induces SMC migration and proliferation (29). In an effort to identify novel transcription factors regulating SMC proliferation, we screened for the expression of nuclear receptors in quiescent SMC stimulated with PDGF. In Northern blotting experiments, NOR1 mRNA was transiently induced after stimulation with PDGF (Fig. 2A). The NOR1 cDNA recognized two transcripts (4.0 and 5.7 kb) because of alternative splicing from the NOR1 gene (37). Consistent with previous reports characterizing NOR1 as an early response gene (20), maximal NOR1 mRNA expression was observed after 2 h. As further depicted in Fig. 2B, Western blotting experiments identified maximal NOR1 protein expression 6 h after stimulation with PDGF.
FIGURE 2. PDGF induces NOR1 expression through ERK/MAPK-dependent signaling pathways.
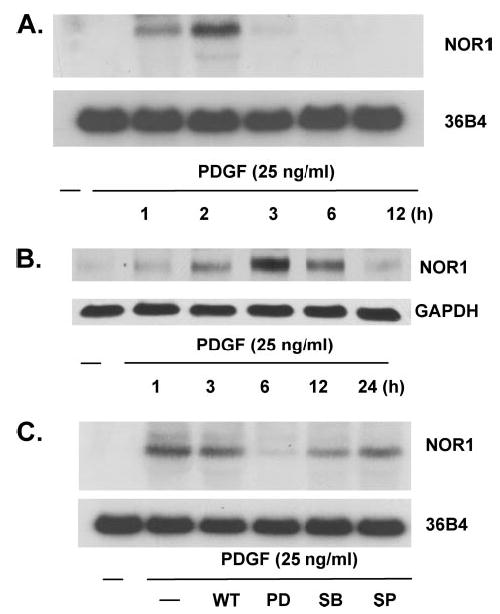
SMC were serum-deprived for 48 h and stimulated with PDGF (25 ng/ml). A, mRNA was isolated at the indicated time points and analyzed for NOR1 expression by Northern blotting. Cohybridization for the housekeeping gene 36B4 was performed as internal control. B, whole cell proteins were isolated at the indicated time points and analyzed for NOR1 protein expression by Western blotting. Cohybridization for GAPDH was performed to assess equal loading. C, quiescent SMC were pretreated with wortmannin (WT, 200 mM), PD98059 (PD, 10 mM), SB203580 (SB, 15 mM), and SP600125 (SP, 25 mM) for 30 min prior to stimulation with PDGF (25 ng/ml). RNA was isolated 2 h after stimulation and analyzed for NOR1 mRNA expression by Northern blotting. The autoradiograms shown are representative of three independently performed experiments using different cell preparations.
Previous work has established a critical role for ERK-MAPK in PDGF signaling, vascular SMC proliferation, and arterial injury (41, 42). We next investigated whether ERK-MAPK signaling cascades mediate PDGF-induced expression of NOR1. Pretreatment of quiescent SMC with the pharmacologic inhibitor of MEK1 PD98059 (10 μM) almost completely abolished PDGF-induced NOR1 expression (Fig. 2C). In contrast, inhibition of p38 MAPK (SB203580, 15 μM), phosphatidylinositol 3-kinase (wortmannin, 200 μM), or c-Jun NH2-terminal kinase (JNK) signaling (SP600125, 25 μM) revealed only a modest effect on PDGF-induced NOR1 mRNA expression. These observations reveal that PDGF-induced NOR1 expression in SMC is primarily mediated through the RAS-MEK-ERK1/2 signaling pathway.
The Proximal NOR1 Promoter Confers the Transcriptional Regulation in Response to PDGF
To investigate the transcriptional mechanisms regulating NOR1 promoter activity, SMC were transiently transfected with a 4.0-kb NOR1 promoter fragment. Stimulation of quiescent SMC with PDGF resulted in 4.75 ± 0.38-fold induction of NOR1 promoter activity (Fig. 3A). A series of 5′-deletion constructs was employed to further identify the minimal responsive promoter region that confers the transcriptional regulation of the NOR1 promoter in response to PDGF. Although PDGF induced NOR1 promoter activity of a −162-NOR1 promoter construct, the −42-NOR1 promoter construct was not inducible (Fig. 3B). These findings indicate that PDGF-induced NOR1 promoter activity is dependent on transcription factor-binding sites located between −162 to −42 relative to the transcription initiation site.
FIGURE 3. The proximal NOR1 promoter confers the transcriptional regulation in response to PDGF.
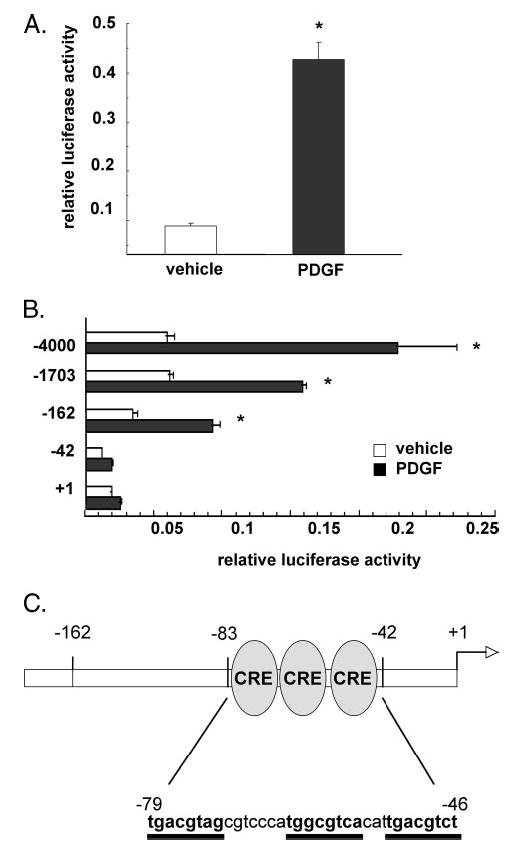
A, SMC were transiently transfected with a 4.0-kb NOR1 promoter construct, serum-deprived, and stimulated with vehicle (white bars) or PDGF (25 ng/ml, black bars) for 6 h. B, SMC were transfected with the indicated 5′-deletion series of the 4.0-kb NOR1 promoter. Serum-deprived cells were stimulated with either vehicle (white bars) or PDGF (25 ng/ml, black bars) for 6 h. Following stimulation, cells were harvested, and luciferase activities were analyzed. Transfection efficiency was adjusted by normalizing firefly luciferase activities to Renilla luciferase activities generated by cotransfection with 10 ng of pRL-CMV. Data are presented as mean ± S.E. from three independently performed experiments (*, p < 0.05 versus vehicle). C, schematic illustration of the three cAMP-responsive elements located in the NOR1 promoter at −79, −64, and −53 from the transcription initiation site.
PDGF-induced NOR1 Promoter Activity Depends on CRE Sites Located between −79 and −46
NOR1 mRNA is rapidly induced in response to PDGF, and previous studies have characterized NOR1 as an early response gene (20). Therefore, it is likely that PDGF modifies transcription factors acting on the NOR1 promoter primarily by rapid post-translational modification, such as phosphorylation, rather than inducing their de novo expression. Interestingly, the proximal NOR1 promoter contains three CRE-binding sites at −79, −64, and −53 (37) (Fig. 3C). To further determine the functional relevance of these CRE sites for the induction of NOR1 promoter activity by PDGF, we next employed a 1.7-kb NOR1 promoter construct bearing site-directed mutations of these CRE sites. As shown in Fig. 4A, mutation of each of these CRE sites abrogated the induction of NOR1 promoter activity induced by PDGF. To further address the role of CREB for the transcriptional induction of NOR1, we next performed transactivation assays using the 1.7-kb NOR1 promoter construct cotransfected with a eukaryotic expression vector overexpressing a dominant-negative CREB mutant. In SMC overexpressing dominant-negative CREB, the effect of PDGF to induce NOR1 promoter activity was completely lost (Fig. 4B).
FIGURE 4. PDGF-induced NOR1 expression is mediated through CREB-dependent transactivation of the NOR1 promoter.
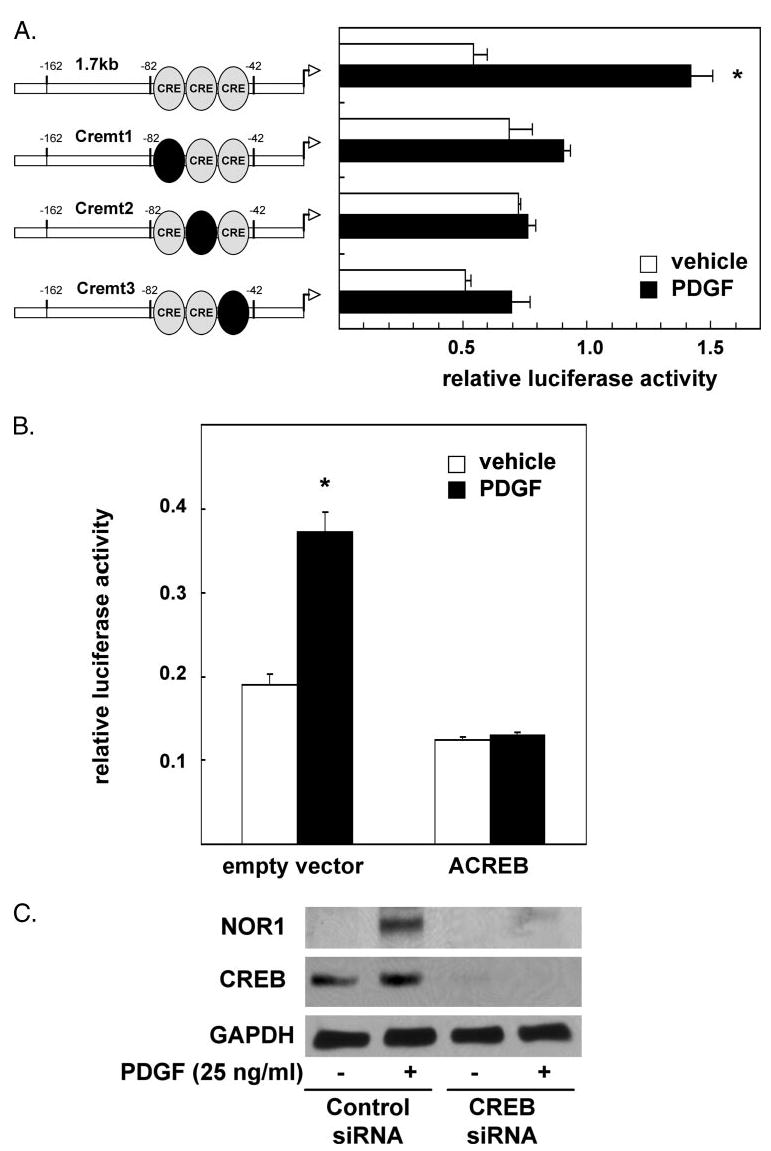
A, SMC were transiently transfected with 1.7-kb NOR1 promoter constructs bearing mutations of the CRE sites at −79 (Cremt1), −64 (Cremt2), and −53 (Cremt3). B, SMC were cotransfected with the 1.7-kb NOR1 promoter construct and an empty control vector or an expression vector overexpressing a dominant-negative CREB mutant (ACREB). Following transfection, cells were serum-deprived and stimulated with vehicle (white bars) or PDGF (25 ng/ml, black bars) for 6 h. Luciferase activities were analyzed as described in the legend to Fig. 3. Data are presented as mean ± S.E. from three independently performed experiments (*, p < 0.05 versus vehicle). C, SMC were transfected either with scrambled siRNA or CREB siRNA, serum-deprived, and stimulated with PDGF (25 ng/ml) for 6 h. Whole cell lysates were analyzed for NOR1 and total CREB protein expression by Western blotting. Cohybridization for GAPDH was performed to assess equal loading.
Next we used siRNA experiments to further confirm a key role of CREB for the induction of NOR1 expression by PDGF. As depicted in Fig. 4C, transfection of SMC with CREB siRNA resulted in an almost complete inhibition of CREB expression. This loss of CREB protein expression was associated with a substantial decrease in PDGF-induced NOR1 expression. In concert, these experiments suggest that the induction of NOR1 expression by PDGF is mediated through a CREB-dependent transactivation of the NOR1 promoter.
PDGF Induces CREB Phosphorylation at Ser-133 and Subsequent Binding to the NOR1 Promoter
CREB phosphorylation at Ser-133 is a pivotal step for the induction of its transcriptional activity (43). To determine further whether CREB phosphorylation at Ser-133 by PDGF precedes binding and transactivation of the NOR1 promoter, we stimulated quiescent SMC with PDGF and analyzed CREB phosphorylation at Ser-133. The employed antibody also detects the phosphorylated form of the slightly smaller CREB-related protein, ATF-1. As shown in Fig. 5A, PDGF induced a rapid phosphorylation of CREB after 15 min in the absence of changes in total CREB protein expression. Consistent with PDGF-induced NOR1 mRNA expression being mediated through ERK-MAPK signaling and dependent on CREB expression, PDGF-induced CREB phosphorylation was also inhibited by PD98059 (Fig. 5B).
FIGURE 5. PDGF stimulates Ser-133 phosphorylation of CREB and binding to the NOR1 promoter through the ERK-MAPK signaling pathway.
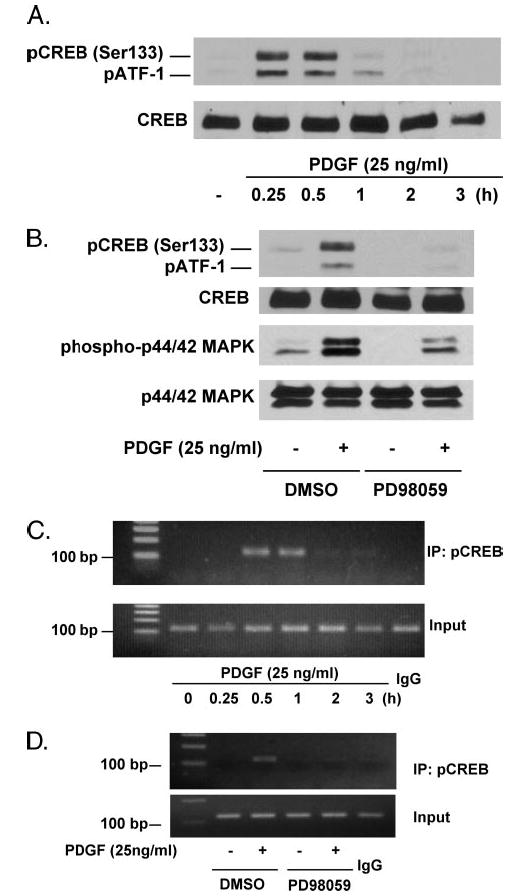
A, serum-deprived SMC were stimulated with PDGF (25 ng/ml) and analyzed for Ser-133-phosphorylated CREB by Western blotting. Cohybridization for total CREB was employed as control. B, serum-deprived SMC were preincubated with vehicle or PD98059 (10 μmol/liter) for 30 min and stimulated with PDGF (25 ng/ml) for 15 min. Whole cell lysates were analyzed for Ser-133-phosphorylated CREB and phosphorylated p44/42 MAPK. Cohybridization for total CREB and total p44/42 MAPK was employed as control. C, quiescent SMC were stimulated with PDGF (25 ng/ml) for the indicated time points. Following chromatin immunoprecipitation (IP) using a Ser-133-phospho-CREB antibody (4 μg) or control IgG, PCR analysis was performed using primer pairs that cover the CRE sites between −79 and −46 of the NOR1 promoter. Total extract (Input) was used as positive PCR control. D, serum-deprived SMC were preincubated with vehicle or PD98059 (10 μmol/liter) for 30 min prior to stimulation with PDGF (25 ng/ml) for 30 min. Chromatin immunoprecipitation of Ser-133-phosphorylated CREB to the endogenous NOR1 promoter was performed as described in C. All autoradiograms shown are representative of three independently performed experiments. DMSO indicates Me2SO.
We next analyzed whether PDGF stimulates binding of Ser-133-phosphorylated CREB to the CRE sites of the endogenous NOR1 promoter located between −79 and −46 from the transcription initiation site. Chromatin immunoprecipitation with an antibody specific for Ser-133-phosphorylated CREB and subsequent PCR amplification, using primer pairs that cover the CRE sites between −79 and −46, demonstrated that PDGF induced rapid binding of Ser-133-phosphorylated CREB to these CRE sites after 30 min (Fig. 5C). This binding of Ser-133-phosphorylated CREB to the NOR1 promoter was inhibited by PD98059 and, therefore, was dependent on ERK-MAPK signaling (Fig. 5D). In concert, these findings indicate that PDGF induces NOR1 transcription through Ser-133 phosphorylation of CREB and subsequent transactivation of the proximal NOR1 promoter.
NOR1 Is Functional in SMC and Transactivates NBRE Response Elements
NOR1 is thought to exert its biological function as a transcription factor by binding to specific NBRE consensus sequences within the promoter region of target genes (13–16). To determine further whether growth factor stimulation of SMC results in an induction of NBRE transactivity, SMC were transfected with a reporter construct driven by multiple NBRE sites. As demonstrated in Fig. 6, PDGF stimulation significantly increased NBRE reporter activity compared with unstimulated cells (2.05 ± 0.4-fold; p < 0.05). Therefore, PDGF not only stimulates NOR1 expression in SMC but also induces NOR1 transcriptional activity.
FIGURE 6. PDGF induces NBRE transactivation.
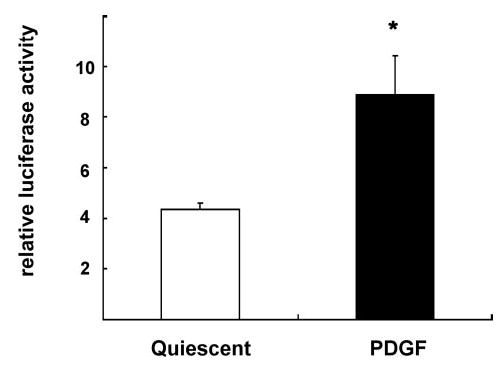
SMC were transiently transfected with a reporter construct containing eight copies of the NBRE consensus sequence upstream of a minimal prolactin promoter driving expression of the firefly luciferase gene. Following transfection, cells were serum-deprived and stimulated with PDGF (25 ng/ml) for 12 h. Data are expressed as normalized luciferase activity and presented as mean ± S.E. (*, p < 0.05 versus quiescent cells).
SMC Proliferation Is Attenuated in NOR1-deficient Cells
To determine the functional relevance of increased NOR1 expression in response to mitogenic stimulation in SMC, we employed primary aortic SMC isolated from NOR1-deficient and littermate wild type mice. SMC from both genotypes were plated in equal densities in DMEM supplemented with FBS and analyzed for cell proliferation after 2, 4, 6, and 8 days. As depicted in Fig. 7A, SMC isolated from wild type mice had a selective growth advantage relative to NOR1-deficient cells and showed substantially higher proliferation rates at 4, 6, and 8 days. The average population doubling times calculated for the first 6 days were 16.3 and 34.3 h for wild type and NOR1-deficient SMC, respectively (p < 0.005), indicating that wild type SMC double their population twice as fast as NOR1-deficient SMC. Similarly, PDGF-induced SMC proliferation of serum-deprived NOR1-deficient SMC was abolished compared with wild type cells (Fig. 7B).
FIGURE 7. NOR1 expression is required for SMC proliferation.
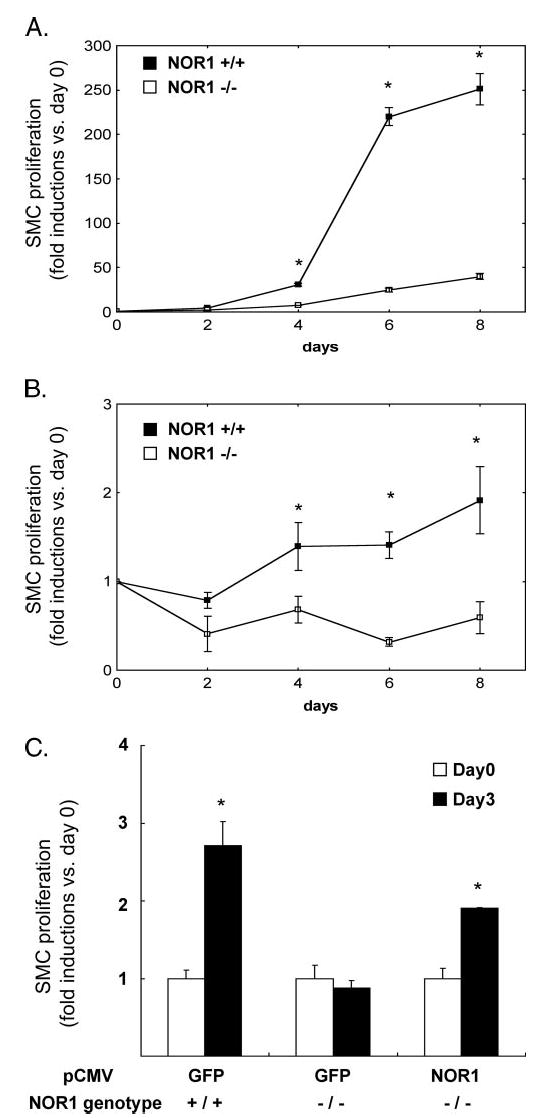
Mouse aortic SMC were isolated from littermate wild type mice and NOR1-deficient mice. Equal numbers of cells (0.5 × 105 cells/plate) were plated on 60-mm plates. A, SMC were maintained in DMEM supplemented with 10% FBS. After 2, 4, 6, and 8 days, the cells were harvested, and cell proliferation was analyzed by cell counting using a hemocytometer. Cell proliferation was expressed as fold induction compared with day 0 and presented as mean ± S.E. (*, p < 0.05 versus NOR1−/−). B, serum-deprived SMC were stimulated with PDGF (25 ng/ml) for the indicated time points, and cell proliferation was analyzed by cell counting. Cell proliferation was expressed as fold induction compared with day 0 and presented as mean ± S.E. (*, p < 0.05 versus NOR1−/−). C, NOR1 wild type or NOR1-deficient SMC were transfected with eukaryotic expression vectors overexpressing either GFP as control or NOR1. Following transfection, cells were maintained in DMEM supplemented with 20% FBS, and cell proliferation was analyzed after 3 days. Proliferation was expressed as fold induction compared with day 0 and presented as mean ± S.E. (*, p < 0.05 versus day 0).
We next analyzed whether re-expression of NOR1 in NOR1-deficient SMC restores cell proliferation. In these experiments, NOR1-deficient SMC were transfected with expression vectors over-expressing either NOR1 or GFP as control, and their proliferation rates were compared with wild type cells transfected with GFP. Wild type cells revealed a 2.7-fold increase in cell number after 3 days, which was completely absent in NOR1-deficient cells. In contrast, in NOR1-deficient SMC transfected with the NOR1 expression vector, cell proliferation was partially restored and increased by 2.0-fold in Fig. 7C. In concert, these data demonstrate that NOR1 deficiency is associated with decreased SMC proliferation providing strong evidence for a key role of NOR1 in regulating SMC proliferation.
NOR1 Promotes SMC Proliferation by Regulating Cyclin D Expression
Very few NR4A target genes have been identified, and NOR1 target genes in SMC currently remain to be identified. Cyclin D1 is required for SMC proliferation (44), and recent studies identified cyclin D2 as a target gene for the related NR4A receptor Nur77 in monocytes (45). Based on this evidence, we analyzed expression levels of cyclin D1 and D2 in wild type and NOR1-deficient SMC. As shown in Fig. 8A, PDGF induced NOR1 expression only in wild type SMC, and no band was observed in NOR1−/− cells. PDGF-induced cyclin D1 expression was almost completely abolished in NOR1-deficient cells (Fig. 8B). In addition, PDGF-induced cyclin D2 expression was decreased. However, basal expression levels of both cyclins appeared to be similar in quiescent wild type and NOR1-deficient SMC. These results suggest that cyclin D1 and D2 function as NOR1 target genes and provide a likely molecular mechanism by which NOR1 mediates mitogen-induced SMC proliferation.
FIGURE 8. NOR1 stimulates SMC proliferation by regulating cyclin D1 and D2 expression.
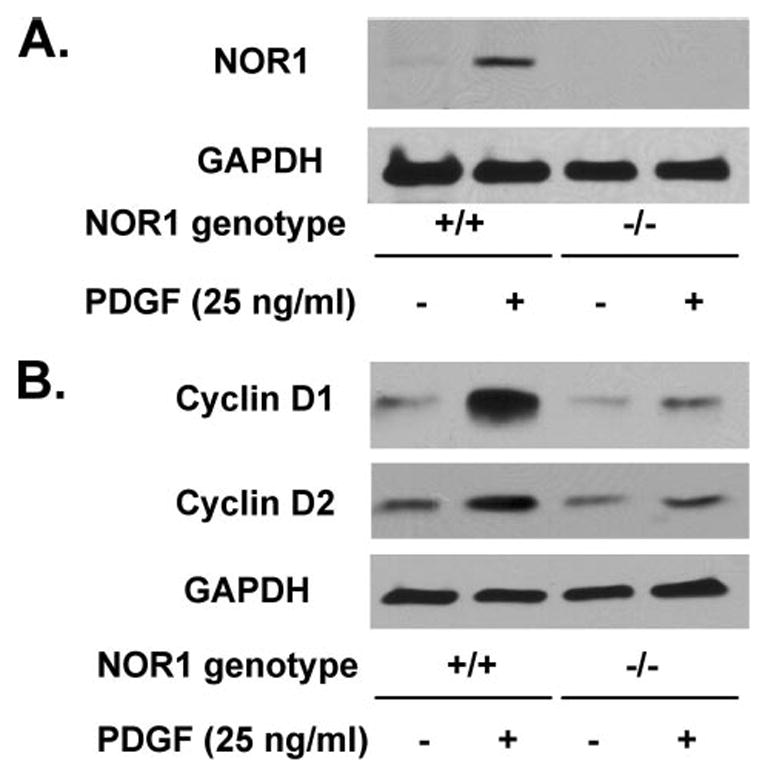
A, serum-deprived NOR1 wild type and NOR1−/− SMC were stimulated with PDGF (25 ng/ml) for 6 h, and whole cell lysates were analyzed for NOR1 protein expression by Western blotting. B, serum-deprived NOR1 wild type and NOR1−/− SMC were stimulated with PDGF (25 ng/ml) for 24 h and analyzed for cyclin D1 and D2 expression by Western blotting. Cohybridization for GAPDH was employed as control to assess for equal loading.
DISCUSSION
NOR1 is the most recently cloned member of the NR4A subfamily of the nuclear receptor superfamily (19). Recent evidence has implicated members of this subfamily as important transcriptional regulators of key cellular functions, including inflammation, proliferation, differentiation, and apoptosis (11, 12, 45). However, the transcriptional regulation of NOR1 in SMC, its functional role in cardiovascular disease, and its target genes remain to be investigated. In this study we demonstrate NOR1 protein expression in neointimal SMC of human atherosclerotic lesions. PDGF, which plays a prominent role for the proliferation of neointimal SMC following vascular injury (46), induces NOR1 expression through a CREB-dependent transactivation of the NOR1 promoter. Finally, our results using SMC isolated from NOR1-deficient mice demonstrate that NOR1 expression is required for SMC proliferation by regulating cyclin D1 and D2 expression. These observations outline a previously unrecognized mechanism regulating SMC proliferation and identify NOR1 as a novel regulator of mitogen-induced SMC proliferation.
The media of normal arteries consist of quiescent SMC proliferating at low frequency (47). In response to injury, these SMC resume a proliferative phenotype that constitutes a critical event for the development of the neointima of an atherosclerotic plaque (2, 4). Concomitant with the phenotypic shift from quiescent SMC resident in the uninjured vessel wall to proliferating SMC present in the neointima of an atherosclerotic lesion, we observed a substantial up-regulation of NOR1 protein expression in neointimal SMC. Consistent with these observations, Martinez-Gonzalez et al. (25) have recently detected NOR1 mRNA by reverse transcription-PCR in atherosclerotic lesions, although Bonta et al. (26) identified NOR1 expression primarily in macrophage-rich cores of atherosclerotic plaques. In particular, the high level expression of NOR1 in proliferating neointimal SMC indicates that this receptor may be functionally involved in the regulation of neointimal SMC proliferation. This concept is further supported by our in vitro data demonstrating low expression of NOR1 in quiescent SMC, which is highly induced upon stimulation with the mitogenic growth factor PDGF.
In contrast to the large number of nuclear receptors that require ligand-dependent receptor activation, including PPAR and LXR, members of the NR4A subfamily, like NOR1, lack a classical ligand binding domain and function as constitutively active transcription factors (10, 17). Based on this unique feature and the rapid induction of NR4A receptors as immediate early genes, it is likely that the transcriptional activity of NOR1 to modulate target gene expression is primarily regulated through mechanisms involving NOR1 expression (20). To further characterize the molecular mechanisms governing NOR1 expression, we initially analyzed the signaling pathways controlling mitogen-induced NOR1 expression in SMC. Interestingly, pharmacologic inhibition of the ERK-MAPK signaling pathway with the MEK inhibitor PD98059 prevented PDGF-induced NOR1 expression in SMC as well as CREB Ser-133 phosphorylation and its subsequent binding to the NOR1 promoter. The Ras → Raf → MEK → MAPK pathway is strongly activated during neointimal SMC proliferation in atherosclerotic lesions (49). In addition, ERK-MAPK signaling is induced by stimulation of quiescent SMC with PDGF and has been characterized as a key signaling pathway involved in SMC proliferation (50, 51). These studies are in further support of our observation that NOR1 expression in SMC is regulated via ERK-MAPK-dependent signaling cascades.
The NOR1 promoter contains a number of putative binding sites for transcription factors, which are either induced or activated via the PDGF → ERK-MAPK cascade. Previous studies have characterized NOR1 as an early response gene, and induction of NOR1 mRNA expression does not require de novo protein synthesis in cancer cells (20) and osteoblasts (52). Therefore, it is likely that PDGF modifies transcription factors acting on the NOR1 promoter primarily by rapid post-translational modification, such as phosphorylation, rather than inducing their de novo expression. To further characterize the transcriptional regulation of the NOR1 promoter in response to PDGF, we first employed a series of 5′-deletion constructs. The minimal responsive NOR1 promoter, which conferred the transcriptional induction in response to PDGF, was dependent on transcription factor-binding sites located between −162 and −42. We have recently identified that this proximal NOR1 promoter contains three CRE sites at −79, −64, and −53, which are positionally conserved across species, suggesting that this sequence has important regulatory functions (37). Our results demonstrate that mutation of any of the three CRE sites significantly impaired the promoter activity in response to PDGF, indicating that each of the CRE sites in the NOR1 promoter are required for the full induction of NOR1 gene transcription. Although these findings are consistent with recent studies (38, 53), the precise molecular role of each of the CREB-binding sites in the promoter is currently incompletely understood and warrants further studies. However, to further confirm that CREB is involved in the regulation of the NOR1 promoter by PDGF, we performed site-directed mutagenesis and transactivation studies using eukaryotic expression vectors. These experiments revealed that mutagenesis or overexpression of a dominant-negative CREB mutant abolished PDGF-induced NOR1 promoter activity. Finally, siRNA-mediated depletion of CREB inhibited PDGF-induced NOR1 expression, and based on these observations CREB appears to be an important transcriptional activator of the NOR1 promoter in SMC.
CREB phosphorylation at Ser-133 resulting in recruitment of the coactivators CREB-binding protein and p300 has been recognized to be crucial for transcriptional activation of CREB (43). Consistent with this evidence, we observed rapid PDGF-induced Ser-133 phosphorylation of CREB in SMC. In addition, by using ChIP assays we provide the first evidence for a direct binding of Ser-133-phosphorylated CREB to the consensus sites within the endogenous NOR1 promoter. CREB Ser-133 phosphorylation has been demonstrated to be induced by PDGF in SMC (54), and PDGF-induced CREB phosphorylation is mediated through ERK-MAPK signaling (55). Additionally, recent in vivo studies by Tokunou et al. (56) observed that proliferating neointimal SMC express high levels of phosphorylated CREB, thereby providing the basis for induced NOR1 expression during neointimal SMC proliferation.
Activation of several nuclear receptors has been implicated in the regulation of SMC proliferation (57, 58). Our previous work has suggested that PPARγ ligands inhibit cell cycle progression in SMC through a receptor-dependent mechanism (33, 59, 60). In addition, we have identified the expression of LXRα and -β in SMC and demonstrated that LXR agonists inhibit SMC proliferation and prevent the development of neointima formation (9). In contrast to the transcriptional suppression of SMC proliferation by PPARγ and LXRα/β, our current experiments outline opposite effects on SMC proliferation for NOR1. Using NOR1-deficient SMC, we demonstrate that NOR1 expression is required for SMC proliferation in response to mitogenic stimulation. These findings are consistent with a recent study demonstrating decreased proliferation of cells treated with NOR1 antisense oligonucleotides (25). However, to date specific NOR1 target genes in SMC have not been identified. Interestingly, Pei et al. (45) have recently characterized cyclin D2 as direct target gene for NOR1 in monocytes. Based on the evidence that cyclin D1 controls SMC proliferation by regulating G1 → S phase progression of the cell cycle (44), we analyzed PDGF-induced cyclin D1 and D2 expression in NOR1-deficient SMC. Mitogen-induced expression levels of both cyclin D1 and D2 were substantially decreased in NOR1-deficient cells compared with wild type cells. These observations identify cyclin D1 and D2 as NOR1 target genes in SMC and provide a likely molecular mechanism by which NOR1 promotes SMC proliferation. However, additional studies are warranted to further determine the molecular mechanisms by which NOR1 regulates cyclin D1 and D2 expression in SMC.
In summary, data provided in this study demonstrate that NOR1 expression may not merely increase in response to mitogenic stimulation but may play a direct causal role for SMC proliferation by inducing the expression of proliferative target genes such as cyclin D1 and D2. However, future studies will determine the role of NOR1 in vascular diseases in vivo using murine models. Our observations in concert with recent studies characterizing NR4A receptors as important regulators of macrophage inflammation (45) and foam cell formation (26) suggest that this family of transcription factors may play a pivotal role in the development of vascular diseases. In addition, NR4A receptors have recently been identified to regulate glucose homeostasis (48), and therefore, these receptors may provide an important link between diabetes and cardiovascular disease.
Supplementary Material
The abbreviations used are
- SMC
smooth muscle cells
- PPAR
peroxisome proliferator-activated receptor
- LXR
liver X receptor
- NOR1
neuron-derived orphan receptor-1
- PDGF
platelet-derived growth factor
- ERK
extracellular signal-regulated kinase
- CREB
cAMP-response element-binding protein
- NBRE
NGFI-B response elements
- ChIP
chromatin immunoprecipitation
- siRNA
short interfering RNA
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- CRE
cAMP-response element
- MAPK
mitogen-activated protein kinase
- MEK
MAPK/ERK kinase
Footnotes
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
Supplemental Material can be found at: http://www.jbc.org/cgi/content/full/M603436200/DC1
References
- 1.Libby P. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Dzau VJ, Braun-Dullaeus RC, Sedding DG. Nat Med. 2002;8:1249–1256. doi: 10.1038/nm1102-1249. [DOI] [PubMed] [Google Scholar]
- 3.Costa MA, Simon DI. Circulation. 2005;111:2257–2273. doi: 10.1161/01.CIR.0000163587.36485.A7. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz SM, deBlois D, O’Brien ERM. Circ Res. 1995;77:445–465. doi: 10.1161/01.res.77.3.445. [DOI] [PubMed] [Google Scholar]
- 5.Hsueh WA, Bruemmer D. Hypertension. 2004;43:297–305. doi: 10.1161/01.HYP.0000113626.76571.5b. [DOI] [PubMed] [Google Scholar]
- 6.Laffitte BA, Tontonoz P. Curr Atheroscler Rep. 2002;4:213–221. doi: 10.1007/s11883-002-0022-6. [DOI] [PubMed] [Google Scholar]
- 7.Law RE, Meehan WP, Xi XP, Graf K, Wuthrich DA, Coats W, Faxon D, Hsueh WA. J Clin Investig. 1996;98:1897–1905. doi: 10.1172/JCI118991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Law RE, Goetze S, Xi XP, Jackson S, Kawano Y, Demer L, Fishbein MC, Meehan WP, Hsueh WA. Circulation. 2000;101:1311–1318. doi: 10.1161/01.cir.101.11.1311. [DOI] [PubMed] [Google Scholar]
- 9.Blaschke F, Leppanen O, Takata Y, Caglayan E, Liu J, Fishbein MC, Kappert K, Nakayama KI, Collins AR, Fleck E, Hsueh WA, Law RE, Bruemmer D. Circ Res. 2004;95:110–123. doi: 10.1161/01.RES.0000150368.56660.4f. [DOI] [PubMed] [Google Scholar]
- 10.Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Science. 2001;294:1866–1870. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- 11.Martinez-Gonzalez J, Badimon L. Cardiovasc Res. 2005;65:609–618. doi: 10.1016/j.cardiores.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Maruyama K, Tsukada T, Ohkura N, Bandoh S, Hosono T, Yamaguchi K. Int J Oncol. 1998;12:1237–1243. doi: 10.3892/ijo.12.6.1237. [DOI] [PubMed] [Google Scholar]
- 13.Wilson TE, Fahrner TJ, Johnston M, Milbrandt J. Science. 1991;252:1296–1300. doi: 10.1126/science.1925541. [DOI] [PubMed] [Google Scholar]
- 14.Castro DS, Hermanson E, Joseph B, Wallen A, Aarnisalo P, Heller A, Perlmann T. J Biol Chem. 2001;276:43277–43284. doi: 10.1074/jbc.M107013200. [DOI] [PubMed] [Google Scholar]
- 15.Forman BM, Umesono K, Chen J, Evans RM. Cell. 1995;81:541–550. doi: 10.1016/0092-8674(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 16.Perlmann T, Jansson L. Genes Dev. 1995;9:769–782. doi: 10.1101/gad.9.7.769. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z, Benoit G, Liu J, Prasad S, Aarnisalo P, Liu X, Xu H, Walker NP, Perlmann T. Nature. 2003;423:555–560. doi: 10.1038/nature01645. [DOI] [PubMed] [Google Scholar]
- 18.Codina A, Benoit G, Gooch JT, Neuhaus D, Perlmann T, Schwabe JW. J Biol Chem. 2004;279:53338–53345. doi: 10.1074/jbc.M409096200. [DOI] [PubMed] [Google Scholar]
- 19.Ohkura N, Hijikuro M, Yamamoto A, Miki K. Biochem Biophys Res Commun. 1994;205:1959–1965. doi: 10.1006/bbrc.1994.2900. [DOI] [PubMed] [Google Scholar]
- 20.Maruyama K, Tsukada T, Bandoh S, Sasaki K, Ohkura N, Yamaguchi K. Cancer Lett. 1995;96:117–122. doi: 10.1016/0304-3835(95)03921-i. [DOI] [PubMed] [Google Scholar]
- 21.Zetterstrom R, Solomin L, Mitsiadis T, Olson L, Perlmann T. Mol Endocrinol. 1996;10:1656–1666. doi: 10.1210/mend.10.12.8961274. [DOI] [PubMed] [Google Scholar]
- 22.Ponnio T, Burton Q, Pereira FA, Wu DK, Conneely OM. Mol Cell Biol. 2002;22:935–945. doi: 10.1128/MCB.22.3.935-945.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ponnio T, Conneely OM. Mol Cell Biol. 2004;24:9070–9078. doi: 10.1128/MCB.24.20.9070-9078.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng LE, Chan FK, Cado D, Winoto A. EMBO J. 1997;16:1865–1875. doi: 10.1093/emboj/16.8.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez-Gonzalez J, Rius J, Castello A, Cases-Langhoff C, Badimon L. Circ Res. 2003;92:96–103. doi: 10.1161/01.es.0000050921.53008.47. [DOI] [PubMed] [Google Scholar]
- 26.Bonta PI, van Tiel CM, Vos M, van Thienen JV, Ferreira V, Arkenbout EK, Seppen J, Spek CA, van der Poll T, Pannekoek H, de Vries CJM. Arterioscler Thromb Vasc Biol. 2006;26:2288–2294. doi: 10.1161/01.ATV.0000238346.84458.5d. [DOI] [PubMed] [Google Scholar]
- 27.Pei L, Castrillo A, Chen M, Hoffmann A, Tontonoz P. J Biol Chem. 2005;280:29256–29262. doi: 10.1074/jbc.M502606200. [DOI] [PubMed] [Google Scholar]
- 28.Liu D, Jia H, Holmes DIR, Stannard A, Zachary I. Arterioscler Thromb Vasc Biol. 2003;23:2002–2007. doi: 10.1161/01.ATV.0000098644.03153.6F. [DOI] [PubMed] [Google Scholar]
- 29.Bornfeldt KE, Raines EW, Graves LM, Skinner MP, Krebs EG, Ross R. Ann N Y Acad Sci. 1995;766:416–430. doi: 10.1111/j.1749-6632.1995.tb26691.x. [DOI] [PubMed] [Google Scholar]
- 30.Ohkubo T, Ohkura N, Maruyama K, Sasaki K, Nagasaki K, Hanzawa H, Tsukada T, Yamaguchi K. Mol Cell Endocrinol. 2000;162:151–156. doi: 10.1016/s0303-7207(00)00222-7. [DOI] [PubMed] [Google Scholar]
- 31.Bruemmer D, Collins AR, Noh G, Wang W, Territo M, Arias-Magallona S, Fishbein MC, Blaschke F, Kintscher U, Graf K, Law RE, Hsueh WA. J Clin Investig. 2003;112:1318–1331. doi: 10.1172/JCI18141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bruemmer D, Yin F, Liu J, Kiyono T, Fleck E, Van Herle AJ, Law RE. Exp Cell Res. 2003;290:28–37. doi: 10.1016/s0014-4827(03)00311-2. [DOI] [PubMed] [Google Scholar]
- 33.Bruemmer D, Yin F, Liu J, Berger JP, Sakai T, Blaschke F, Fleck E, Van Herle AJ, Forman BM, Law RE. Circ Res. 2003;93:38–47. doi: 10.1161/01.RES.0000088344.15288.E6. [DOI] [PubMed] [Google Scholar]
- 34.Ohkura N, Ito M, Tsukada T, Sasaki K, Yamaguchi K, Miki K. Biochim Biophys Acta. 1996;1308:205–214. doi: 10.1016/0167-4781(96)00101-7. [DOI] [PubMed] [Google Scholar]
- 35.Ogawa D, Stone JF, Takata Y, Blaschke F, Chu VH, Towler DA, Law RE, Hsueh WA, Bruemmer D. Circ Res. 2005;96:59–67. doi: 10.1161/01.RES.0000163630.86796.17. [DOI] [PubMed] [Google Scholar]
- 36.Ohkura N, Hosono T, Maruyama K, Tsukada T, Yamaguchi K. Biochim Biophys Acta. 1999;1444:69–79. doi: 10.1016/s0167-4781(98)00247-4. [DOI] [PubMed] [Google Scholar]
- 37.Ohkura N, Ito M, Tsukada T, Sasaki K, Yamaguchi K, Miki K. Gene (Amst) 1998;211:79–85. doi: 10.1016/s0378-1119(98)00095-x. [DOI] [PubMed] [Google Scholar]
- 38.Inuzuka H, Tokumitsu H, Ohkura N, Kobayashi R. FEBS Lett. 2002;522:88–92. doi: 10.1016/s0014-5793(02)02890-9. [DOI] [PubMed] [Google Scholar]
- 39.Watson PA, Vinson C, Nesterova A, Reusch JEB. Endocrinology. 2002;143:2922–2929. doi: 10.1210/endo.143.8.8959. [DOI] [PubMed] [Google Scholar]
- 40.Ross R, Glomset JA. Science. 1973;180:1332–1339. doi: 10.1126/science.180.4093.1332. [DOI] [PubMed] [Google Scholar]
- 41.Mii S, Khalil RA, Morgan KG, Ware JA, Kent KC. Am J Physiol. 1996;270:H142–H150. doi: 10.1152/ajpheart.1996.270.1.H142. [DOI] [PubMed] [Google Scholar]
- 42.Gennaro G, Menard C, Michaud SE, Deblois D, Rivard A. Circulation. 2004;110:3367–3371. doi: 10.1161/01.CIR.0000147773.86866.CD. [DOI] [PubMed] [Google Scholar]
- 43.Mayr B, Montminy M. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 44.Xiong W, Pestell RG, Watanabe G, Li J, Rosner MR, Hershenson MB. Am J Physiol. 1997;272:L1205–L1210. doi: 10.1152/ajplung.1997.272.6.L1205. [DOI] [PubMed] [Google Scholar]
- 45.Pei L, Castrillo A, Tontonoz P. Mol Endocrinol. 2006;20:786–794. doi: 10.1210/me.2005-0331. [DOI] [PubMed] [Google Scholar]
- 46.Raines EW. Cytokine Growth Factor Rev. 2004;15:237–254. doi: 10.1016/j.cytogfr.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 47.Gordon D, Reidy MA, Benditt EP, Schwartz SM. Proc Natl Acad Sci U S A. 1990;87:4600–4604. doi: 10.1073/pnas.87.12.4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pei L, Waki H, Vaitheesvaran B, Wilpitz DC, Kurland IJ, Tontonoz P. Nat Med. 2006;12:1048–1055. doi: 10.1038/nm1471. [DOI] [PubMed] [Google Scholar]
- 49.Hu Y, Dietrich H, Metzler B, Wick G, Xu Q. Arterioscler Thromb Vasc Biol. 2000;20:18–26. doi: 10.1161/01.atv.20.1.18. [DOI] [PubMed] [Google Scholar]
- 50.Millette E, Rauch BH, Defawe O, Kenagy RD, Daum G, Clowes AW. Circ Res. 2005;96:172–179. doi: 10.1161/01.RES.0000154595.87608.db. [DOI] [PubMed] [Google Scholar]
- 51.Graf K, Xi XP, Yang D, Fleck E, Hsueh WA, Law RE. Hypertension. 1997;29:334–339. doi: 10.1161/01.hyp.29.1.334. [DOI] [PubMed] [Google Scholar]
- 52.Pirih FQ, Nervina JM, Pham L, Aghaloo T, Tetradis S. Biochem Biophys Res Commun. 2003;306:144–150. doi: 10.1016/s0006-291x(03)00931-8. [DOI] [PubMed] [Google Scholar]
- 53.Rius J, Martinez-Gonzalez J, Crespo J, Badimon L. Arterioscler Thromb Vasc Biol. 2004;24:697–702. doi: 10.1161/01.ATV.0000121570.00515.dc. [DOI] [PubMed] [Google Scholar]
- 54.Klemm DJ, Watson PA, Frid MG, Dempsey EC, Schaack J, Colton LA, Nesterova A, Stenmark KR, Reusch JEB. J Biol Chem. 2001;276:46132–46141. doi: 10.1074/jbc.M104769200. [DOI] [PubMed] [Google Scholar]
- 55.Pugazhenthi S, Boras T, O’Connor D, Meintzer MK, Heidenreich KA, Reusch JEB. J Biol Chem. 1999;274:2829–2837. doi: 10.1074/jbc.274.5.2829. [DOI] [PubMed] [Google Scholar]
- 56.Tokunou T, Shibata R, Kai H, Ichiki T, Morisaki T, Fukuyama K, Ono H, Iino N, Masuda S, Shimokawa H, Egashira K, Imaizumi T, Takeshita A. Circulation. 2003;108:1246–1252. doi: 10.1161/01.CIR.0000085164.13439.89. [DOI] [PubMed] [Google Scholar]
- 57.Bruemmer D, Law RE. Am J Med 115, Suppl. 2003;8A:87–92. doi: 10.1016/j.amjmed.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 58.Bruemmer D, Blaschke F, Law RE. Int J Obes Relat Metab Disord 29, Suppl. 2005;1:26–30. doi: 10.1038/sj.ijo.0802910. [DOI] [PubMed] [Google Scholar]
- 59.Bruemmer D, Berger JP, Liu J, Kintscher U, Wakino S, Fleck E, Moller DE, Law RE. Eur J Pharmacol. 2003;466:225–234. doi: 10.1016/s0014-2999(03)01556-5. [DOI] [PubMed] [Google Scholar]
- 60.Bruemmer D, Yin F, Liu J, Berger JP, Kiyono T, Chen J, Fleck E, Van Herle AJ, Forman BM, Law RE. Mol Endocrinol. 2003;17:1005–1018. doi: 10.1210/me.2002-0410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.