

Abstract
c-Jun is a major component of the AP-1 transcription factor and plays a key role in regulation of diverse biological processes including proliferation and apoptosis. Treatment of a wide variety of cells with the microtubule inhibitor vinblastine leads to a robust increase in c-Jun expression, JNK-mediated c-Jun phosphorylation, and activation of AP-1-dependent transcription. However, the role of c-Jun induction in the response of cells to vinblastine remains obscure. In this study we used MCF7 breast cancer cell lines that express the dominant-negative form of c-Jun, TAM-67, as well as cells that overexpress c-Jun, under the control of an inducible promoter. Vinblastine induced c-Jun protein expression, c-Jun phosphorylation, and AP-1 activation in MCF7 cells, and these parameters were strongly inhibited by inducible TAM-67 expression and strongly enhanced by inducible c-Jun expression. Vinblastine-induced cell death was not affected by TAM-67 expression whereas cells were protected by c-Jun overexpression. Further investigation revealed that apoptotic and senescent cells were observed after vinblastine treatment and that both outcomes were strongly inhibited by c-Jun overexpression. Although c-Jun expression inhibited cell death, it did not affect the ability of vinblastine to induce mitotic arrest. These results indicate that c-Jun expression plays a protective role in the cellular response to vinblastine and operates post-mitotic block to inhibit drug-induced apoptosis and senescence.
Keywords: c-Jun, TAM-67, AP-1, vinblastine, apoptosis, senescence
1. Introduction
Microtubule inhibitors, especially vinca alkaloids and paclitaxel and its derivatives, are widely used in cancer chemotherapy. The primary mechanism of action of these drugs is to bind to tubulin or microtubules and disrupt mitotic spindle dynamics leading to M-phase arrest [1]. In general, mitotic arrest leads to cell death by apoptosis. However, if apoptosis is suppressed or mitotic checkpoint signaling perturbed, other outcomes, such as aneuploidy, mitotic catastrophe, or senescence, can result [2–5]. There is a great deal of interest in unraveling the molecular links between mitotic arrest and the eventual outcome, whether it is the immediate or eventual death of the cell, or the survival of an impaired cell. Information in this area is critical as it contributes to our knowledge of basic cellular processes and is essential for a better understanding of the mechanism of action of drugs that interfere with microtubule dynamics.
Our previous studies in KB-3 cells have shown that vinblastine activates the c-Jun NH2-terminal protein kinase (JNK) signal transduction pathway, leading to phosphorylation and activation of c-Jun, and in turn activation of AP-1-dependent transcription [6–8]. Through an auto-amplification loop involving AP-1 sites in the c-Jun promoter, these events result in a robust increase in c-Jun protein expression. Indeed, we have observed that vinblastine causes highly increased c-Jun expression in all cell lines examined to date, suggesting it is a common and perhaps universal response to microtubule inhibition1. While it is established that c-Jun plays diverse roles in numerous cellular functions, including cell proliferation, differentiation, and death, its precise role in cellular responses to microtubule inhibitors is controversial. Theoretically, increased c-Jun expression could represent a protective mechanism in aid of cell survival; a destructive mechanism actively involved in the cell death process; or may simply be a bystander event with no direct role in the response to mitotic block.
Stable expression of a c-Jun dominant-negative, TAM-67, in KB-3 cells caused a modest resistance to vinblastine and a delay in caspase activation induced by the drug [6]. However, these properties were context-dependent, in that the TAM-67-expressing cells were resistant only when cells were exposed to drug for short intervals, and relative to control cells were not resistant upon continuous drug treatment2. More recent studies indicated that the kinetics and extent of apoptosis are similar for both wild-type and c-Jun knockout fibroblasts [9]. In contrast, fibroblasts expressing high levels of human c-Jun were found to be highly resistant to vinblastine [9]. However, cell systems stably expressing dominant-negative forms of a multi-functional transcription factor or with specific genetic lesions do not necessarily represent the best models because of possible cellular adaptation and compensatory mechanisms.
In order to avoid these potential drawbacks we sought to determine the effect of c-Jun inhibition or c-Jun overexpression on vinblastine sensitivity in a more controlled and defined system. For this purpose we utilized MCF7 cells that have been developed to express either the c-Jun dominant-negative TAM-67 or human c-Jun under the control of an inducible promoter [10]. Using the Tet-off system, TAM-67 or c-Jun can be expressed in a manner that is dependent upon the removal of doxycycline from the medium. In this study, we exploited this powerful system to investigate the role of c-Jun and its inhibition on the sensitivity of breast cancer cells to vinblastine. We show that MCF7 cells are highly similar to KB-3 cells in that they exhibit a robust increase in c-Jun expression, c-Jun phosphorylation, and AP-1 activation in response to vinblastine treatment. While inducible TAM-67 expression blocks these events as predicted, there is no change in the sensitivity of the cells to vinblastine. In contrast, inducible overexpression of c-Jun renders the cells markedly resistant to vinblastine, through a mechanism that operates post-mitotic block to prevent drug-induced apoptosis and senescence.
2. Materials and Methods
Materials
Flag-tag antibody was obtained from Sigma Chemical (St. Louis, MO). C-terminal c-Jun antibody (sc-44) and actin antibody (sc-1616) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and N-terminal c-Jun antibody (J319200) was from Transduction Laboratories (San Diego, CA). Phospho-c-Jun (Ser63) antibody (9261S) was from Cell Signaling (Beverly, MA). For immunoblotting primary antibodies were used at 1:1000 dilution and secondary antibodies at 1:5000 dilution.
Cell lines
MCF7 cells were purchased from American Type Culture Collection and were maintained in Earle’s minimal essential medium (MEM) with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin. MCF7 vector cells and MCF7-TAM-67 cells were described previously [10] and cells with inducible expression of c-Jun, termed MCF7-c-Jun cells, were isolated using the same strategy. These cell lines were maintained in improved MEM with 10% FBS, 100 μg/ml geneticin, 100 μg/ml hygromycin, 1 μg/ml of doxycycline, 2 mM L-glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin [10]. c-Jun or TAM-67 expression was induced in MCF7-c-Jun or MCF7-TAM67 cells respectively by washing cells twice in PBS and replating in complete medium without doxycycline.
Cell extraction and immunoblotting
Cells were grown in 60 mm dishes. Whole-cell extracts were prepared by suspending cells (3 x 106) in 0.25 ml of lysis buffer (25 mM HEPES (pH 7.5), 0.3 M NaCl, 0.2% SDS, 0.5% sodium deoxycholate, 0.2 mM EDTA, 0.5 mM DTT, 20 mM ß-glycerophosphate, 1 mM Na3VO4, 0.1% Triton X-100, 20 μg/ml aprotinin, 50 μg/ml leupeptin, 10 μM pepstatin, 0.1 μM okadaic acid, and 1 mM phenylmethylsulfonyl fluoride). After 15 min on ice, extracts were sonicated (three times for 10 s each), insoluble material was removed by centrifugation (15 min at 12,000 × g), and the protein concentration in the supernatant was determined using the Bio-Rad protein assay. Immunoblotting was performed as described previously [7], using 30–80 μg protein/lane.
AP-1 transcriptional activity
Cells (0.2× 106/well) were plated in a 12-well plate for 24 h and transfected with 1 μg of TRE-Luc construct (firefly luciferase under control of two copies of TPA response element), together with 0.1 μg of pRL-TK-Luc construct (Renilla luciferase under control of TK promoter) to normalize transfection efficiency. The DNA was mixed with 4 μL of Lipofectamine (Invitrogen) and 7 uL of Lipofectamine Plus and 0.1 mL of serum-free media, transfection was for 4–5 h, followed by incubation in normal media. After 24 h, cells were treated with vinblastine or vehicle (0.1% DMSO) for 18 h. Cells were harvested for determination of firefly and Renilla luciferase activities by the Dual-Luciferase® Reporter Assay System (Promega). Triplicate assays were performed for each condition. The results were expressed as average relative firefly luciferase activity, normalized to Renilla luciferase activity.
Flow cytometric cell cycle analysis
DNA content was examined by propidium iodine staining and flow cytometry as described in detail previously [6]. Data were analyzed using the ModFit DNA analysis program.
Cell viability, apoptosis and senescence assays
Cell viability was determined by trpan blue exclusion. Cells (0.2×106/well) were plated in a 12-well plate, treated with vinblastine or vehicle (0.1% DMSO) for 3 days and collected by trypsinization. Cell viability was measured by counting the number of trypan blue-excluding intact cells. Apoptosis was measured using the DNA fragmentation cell death detection kit (Roche Applied Science) as described in detail previously [11]. Cellular senescence was determined using a kit from Cell Signaling Technology. The kit histochemically detects β-galactosidase activity in cultured cells at pH 6, a known characteristic of senescent cells. β-galactosidase activity in cultured cells at pH 6 is found only in senescent cells not in immortal, quiescent or presenescent cells. Cells at exponential growth (80% confluence) were treated with vinblastine or vehicle (0.1% DMSO) for 2 days and adherent and non-adherent cells were pooled. Cells were washed with cold PBS twice and fixed in 4% formaldehyde solution for 15 minutes at room temperature. Staining buffer provided in the kit was prepared fresh just before use and cells were incubated for 12 h at 37°C. Development of blue color was monitored under a microscope (200 × magnification) and representative fields selected for photographic recording. At least three separate fields of view were selected to determine the number of blue staining cells versus total number of cells under each condition.
3. Results
Vinblastine induces c-Jun expression and phosphorylation in MCF7 cells
MCF7 cells were treated with vinblastine and extracts prepared and analyzed for c-Jun expression and JNK-mediated phosphorylation by immunoblotting (Fig. 1). c-Jun expression was barely detectable in control cells but strongly induced after vinblastine treatment, with expression sustained over a prolonged period of 6 to 30 h. c-Jun amino-terminal phosphorylation, evaluated with a phospho-specific (Ser63) c-Jun antibody, followed a similar time-course. These results are very similar to our previous findings in KB-3 cells [6], and together with similar data from several other cell lines, suggest that c-Jun induction/phosphorylation is a common and perhaps universal response to vinblastine.
Fig. 1.
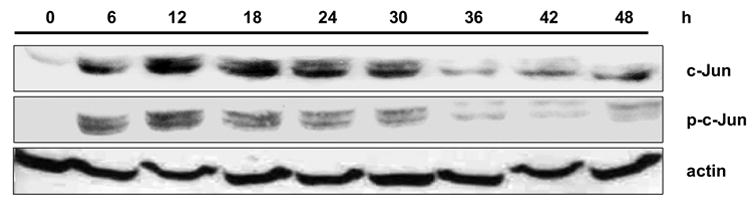
Vinblastine induces expression and phosphorylation of c-Jun in MCF7 cells. Cells were treated with 0.3 μM vinblastine for the times indicated, and cell extracts prepared and subjected to immunoblotting for c-Jun, phospho-c-Jun, and actin, as indicated.
Inducible expression of c-Jun and TAM-67
In order to determine the role of c-Jun induction in the cellular response to microtubule inhibition, we utilized a system we developed for the inducible expression of either c-Jun or of a c-Jun dominant-negative, TAM-67, which encodes c-Jun with truncation of the N-terminal transactivation domain [12]. MCF7-c-Jun cells express flag-tagged c-Jun and MCF7-TAM-67 cells express flag-tagged TAM-67 under the control of an inducible promoter [10]. These cell lines express the desired protein upon removal of doxycycline from the medium. A representative clone of MCF7-c-Jun cells was first examined (Fig. 2A). In the presence of doxycycline, c-Jun was undetectable (endogenous c-Jun could be visualized with longer exposures of the blot to film). After the removal of doxycycline from the medium, there was a time-dependent increase in the expression of c-Jun which was detectable with either c-Jun-specific antibody or an antibody to the flag tag. A representative clone of MCF7-TAM-67 cells was examined next (Fig. 2B). Time-dependent expression of TAM-67, detected with either an antibody to the C-terminus of c-Jun or to the flag tag, was observed upon removal of doxycycline from the medium. Neither c-Jun overexpression nor TAM-67 expression was observed in vector-expressing MCF7 cells, as expected (data not shown). Indeed, in all experiments described here, vector-expressing MCF7 cells were indistinguishable from parental MCF7 cells.
Fig. 2.
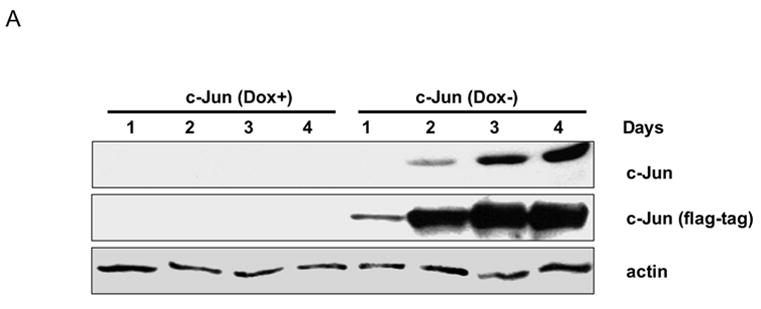
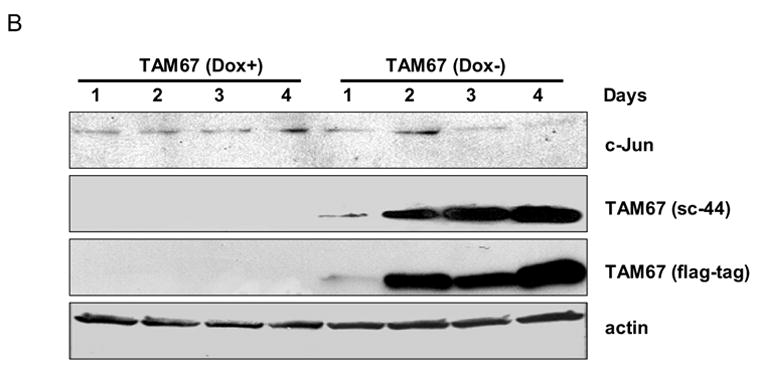
Inducible expression of c-Jun or TAM-67. A. MCF7-c-Jun cells were maintained in the presence (Dox+) or absence (Dox-) of doxycycline for the times indicated, and cell extracts prepared and subjected to immunoblotting with anti-c-Jun, anti-flag-tag, and anti-actin antibodies, as indicated. B. MCF7-TAM-67 cells were maintained in the presence (Dox+) or absence (Dox-) of doxycycline for the times indicated, and cell extracts prepared and subjected to immunoblotting for detection of endogenous c-Jun or TAM-67, the latter using either an antibody (sc-44) to the COOH terminus of c-Jun, or using anti-flag-tag antibody, as indicated. Actin served as a loading control.
Effects of vinblastine on c-Jun and phospho-c-Jun in MCF7-c-Jun and MCF7-TAM-67 cells
MCF7-c-Jun and MCF7-TAM-67 cells were next treated with vinblastine, and cell extracts examined for c-Jun and phospho-c-Jun expression. In the presence of doxycycline, c-Jun expression and phosphorylation in vinblastine-treated MCF7-c-Jun cells was first observed at 6 h, similar to that in control MCF7 cells (Fig. 3, top panel). However, in contrast to control cells, these elevated levels were maintained for longer periods, up to 48 h. MCF7-c-Jun cells were then cultured in the absence of doxycycline for 3 d to induce c-Jun over-expression, and then treated with vinblastine. The effects of doxycycline removal and vinblastine treatment were clearly additive, with very high levels of c-Jun and c-Jun phosphorylation achieved under these conditions (Fig. 3, lower panel). Note that the c-Jun and phospho-c-Jun blots in the upper panel of Fig. 3 were prepared under identical conditions and with identical ECL exposures as those in the lower panel, such that the image densities are directly comparable.
Fig. 3.
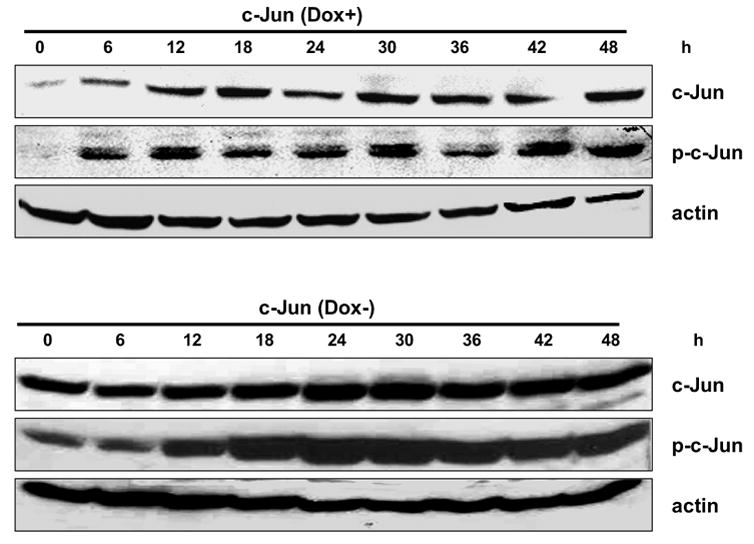
Effect of vinblastine on c-Jun expression and phosphorylation in MCF7-c-Jun cells. MCF7-c-Jun cells were maintained either in the presence (top panel) or absence (lower panel) of doxycycline for 3 d and then treated with 0.3 μM vinblastine for the times indicated. Cell extracts were prepared and subjected to immunoblotting for c-Jun, phospho-c-Jun, or actin, as indicated.
MCF7-TAM-67 cells were examined next. In the presence of doxycycline and thus absence of TAM-67 expression, c-Jun expression and phosphorylation in vinblastine-treated MCF7-TAM-67 cells were similar in extent as control MCF7 cells, though the elevated levels were maintained for a longer period (Fig. 4, top panel). When MCF7-TAM-67 cells were cultured in the absence of doxycycline for 3 d to induce TAM-67 expression, and then treated with vinblastine, there was a marked decrease in phospho-c-Jun levels (Fig. 4, lower panel). This is similar to observations made previously in KB-3 cells stably transfected with TAM-67 [6]. The most likely interpretation is that TAM-67 dimerizes with endogenous c-Jun and, because TAM-67 lacks the amino-terminal domain and JNK docking site, JNK-mediated phosphorylation of the normal c-Jun partner is prevented. This is consistent with the proposed mechanism of JNK-mediated phosphorylation of sites in the amino-terminus of c-Jun homo/heterodimers [13]. TAM-67 expression also caused a reduction in vinblastine-induced c-Jun expression (Fig. 4, lower panel).
Fig. 4.
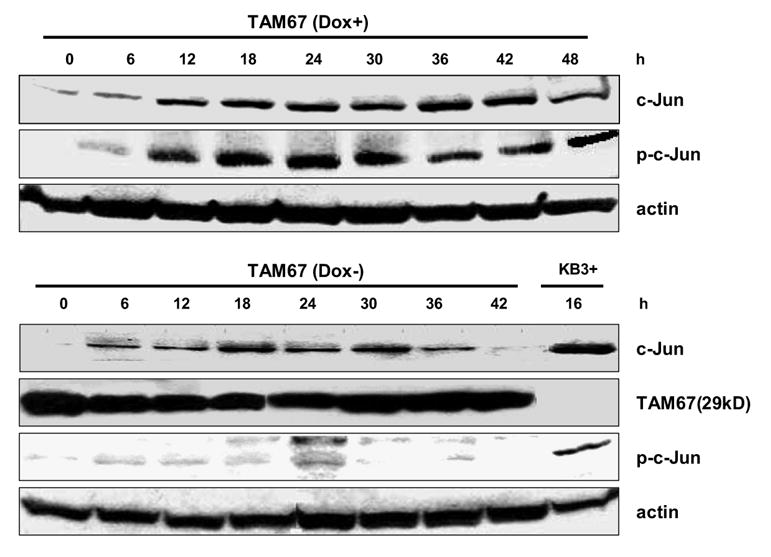
Effect of vinblastine on c-Jun expression and phosphorylation in MCF7-TAM-67 cells. MCF7-TAM-67 cells were maintained either in the presence (top panel) or absence (lower panel) of doxycycline for 3 d and then treated with 0.3 μM vinblastine for the times indicated. Cell extracts were prepared and subjected to immunoblotting for detection of c-Jun, phospho-c-Jun, TAM-67, or actin, as indicated. The right-hand lane in the lower panel (KB3+) represents an extract from vinblastine-treated KB-3 cells as a positive control for c-Jun and phospho-c-Jun.
Effect of vinblastine on AP-1 transcriptional activity in MCF7-c-Jun and MCF7-TAM-67 cell lines
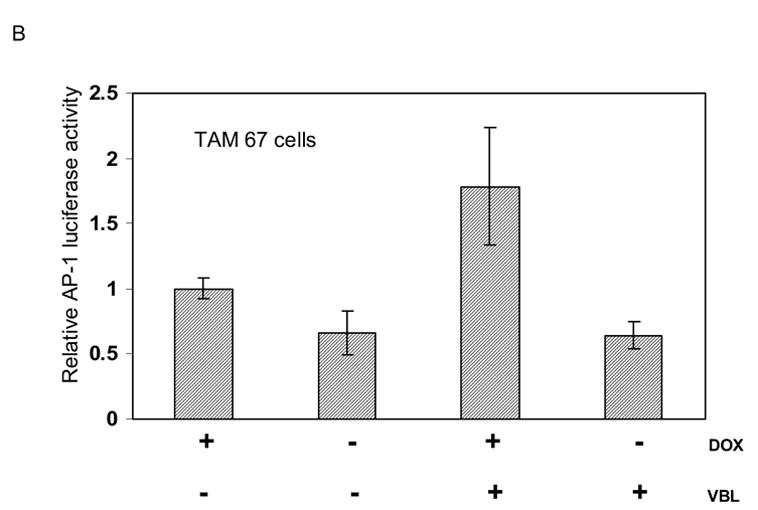
We next determined the effect of c-Jun overexpression and vinblastine treatment, alone and together, on AP-1 transcriptional activity. In MCF7-c-Jun cells, doxycycline removal induced c-Jun overexpression which in turn induced an increase in AP-1 activity (Fig. 5A). Vinblastine also stimulated AP-1 activity although to a lesser extent (Fig. 5A). The approximately 3-fold increase with vinblastine treatment is similar to that observed previously in vinblastine-treated KB-3 cells [6]. When c-Jun overexpressing cells were treated with vinblastine, effects on AP-1 activity were additive. This is consistent with the high levels of phosphorylated c-Jun seen under this condition (Fig. 3, lower panel). In MCF7-TAM-67 cells, TAM-67 expression inhibited both basal and vinblastine-induced AP-1 activity consistent with its action as a c-Jun antagonist (Fig. 5B).
Fig. 5.
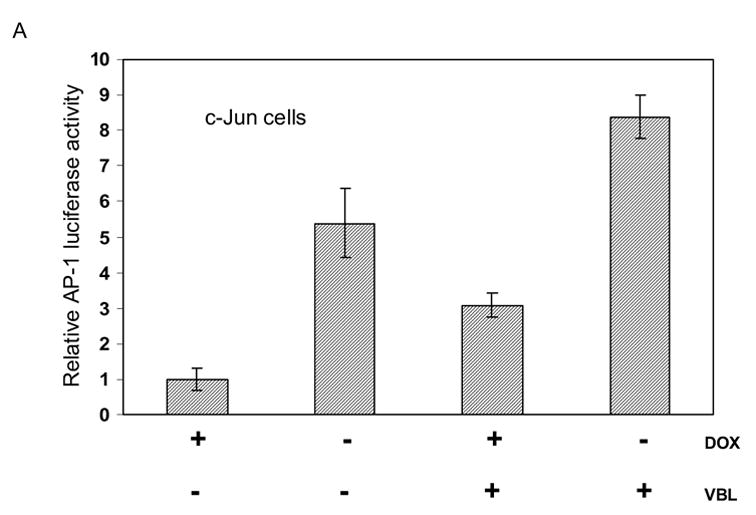
A. Effects of c-Jun expression and vinblastine treatment on AP-1 transcriptional activity. MCF7-c-Jun cells were either untreated or treated with 0.3 μM vinblastine (VBL) for 18 h, either in the presence of doxycycline (+DOX) to repress c-Jun expression, or in the absence of doxycycline (−DOX) for 3 d to induce c-Jun expression. AP-1-dependent transcriptional activity was determined using a luciferase reporter system as described in Methods and Materials. Results shown represent mean ± S.D. (n = 6).
B. Effects of TAM-67 expression and vinblastine treatment on AP-1 transcriptional activity. MCF7-TAM-67 cells were either untreated or treated with 0.3 μM vinblastine (VBL) for 18 h, either in the presence of doxycycline (+DOX) to repress TAM-67 expression, or in the absence of doxycycline (−DOX) for 3 d to induce TAM-67 expression. AP-1 dependent transcription was determined using a luciferase reporter system as described in Methods and Materials. Results shown represent mean ± S.D. (n = 6).
Effect of c-Jun and TAM-67 expression on vinblastine-induced cell death
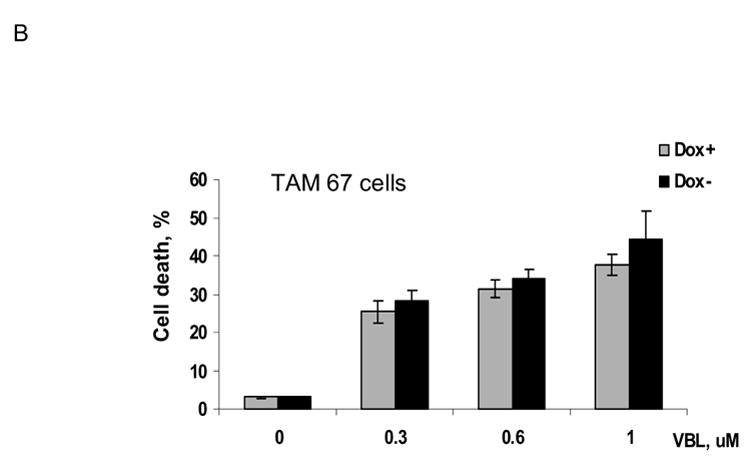
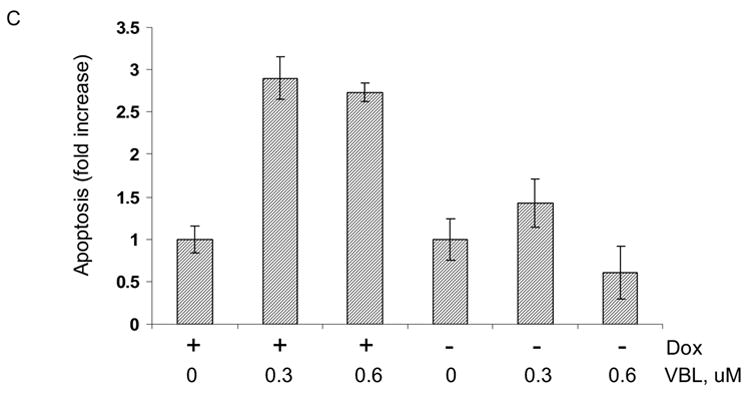
MCF7-c-Jun cells were either maintained in the presence of doxycycline, or its absence for 3 d to induce c-Jun expression, and were then untreated or treated with vinblastine at several concentrations for 3 d. Cell death was assessed by trypan blue exclusion (Fig. 6A). In the presence of doxycycline, increasing concentrations of vinblastine led to an increase in the proportion of dead cells. However, in the absence of doxycycline, under condition of high c-Jun expression, cells were protected against death. At each concentration of vinblastine there was a highly statistically significant decrease in the extent of cell death upon c-Jun expression (P values of <0.02 at 0.3 μM, <0.0006 at 0.6 μM, and <0.001 at 1 μM vinblastine, using two-tailed t test). In contrast, the extent of cell death was not significantly altered upon expression of TAM-67 in MCF7-TAM-67 cells (Fig. 6B). To further investigate the effect of c-Jun overexpression on cell death, apoptosis assays were conduced with untreated and vinblastine-treated MCF7-c-Jun cells maintained in the presence or absence of doxycycline (Fig. 6C). Vinblastine-induced apoptosis was inhibited upon overexpression of c-Jun, consistent with the data of Fig. 6A, again indicating a protective effect of c-Jun expression.
Fig. 6.
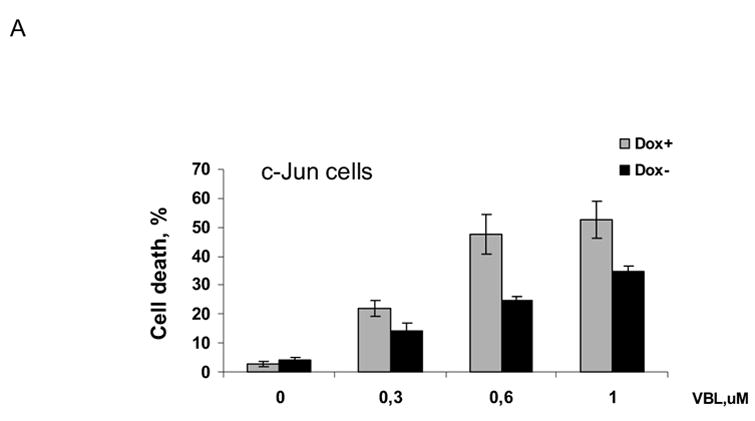
Effects of c-Jun or TAM-67 expression on vinblastine-induced cell death. A. MCF7-c-Jun cells were maintained either in the presence of doxycyline or in the absence of doxycycline for 3 d and then untreated or treated with the indicated concentration of vinblastine (VBL) for 3 d. Cell death was determined by trypan blue exclusion as described in Materials and Methods. Results represent mean ± S.D. (n = 6). The extent of cell death was highly significantly different at each vinblastine concentration in cells overexpressing c-Jun versus those not overexpressing c-Jun (* P <0.02, ** P <0.0006, and ***P <0.001, by two-tailed t test).
B. MCF7-TAM-67 cells were maintained either in the presence of doxycycline or in the absence of doxycycline for 3 d and then untreated or treated with the indicated concentration of vinblastine (VBL) for 3 d. Cell death was determined using trypan blue exclusion as described in Materials and Methods. Results represent mean ± S.D. (n = 6).
C. Effect of c-Jun expression on vinblastine-induced apoptosis. MCF7-c-Jun cells were maintained either in the presence of doxycycline or absence of doxycycline for 3 d and then untreated or treated with the indicated concentration of vinblastine (VBL) for 2 d. The extent of apoptosis, indicated as a fold-increase over the value in untreated cells, was determined using a Cell Death ELISA kit, as described in Materials and Methods. Results represent mean ± S.D. (n = 6).
Post-mitotic effect of c-Jun expression on cell death
Cell death in response to microtubule inhibitors such as vinblastine proceeds via mitotic arrest and subsequent cell death through apoptosis or other mode [1]. The inhibition of cell death by c-Jun overexpression could be due to inhibition of mitotic arrest, or inhibition at a step subsequent to mitotic arrest. To determine at what step c-Jun exerted its effect, cells were maintained in the presence or absence of doxycycline and then treated with 0.3 μM vinblastine from 12 to 72 h. At regular intervals cells were collected and subjected to propidium iodide staining and flow cytometry to determine the percentage of cells at different stages of the cell cycle. Cells with sub-G1 DNA content were considered apoptotic. As shown in Fig. 7, vinblastine treatment of MCF7-c-Jun cells maintained in the presence of doxycycline resulted in a time-dependent increase in the proportion of cells mitotically arrested, i.e. in G2-M phase, at the expense of cells in other phases of the cell cycle. After 3 days of vinblastine treatment, most of the cells were either mitotic or apoptotic. In MCF7-c-Jun cells maintained in the absence of doxycycline to induce c-Jun overexpression, a similar trend was observed, in that there was an increase in the proportion of mitotically arrested cells with increasing times of vinblastine treatment (Fig. 7). Thus, c-Jun expression did not alter the ability of vinblastine to induce mitotic arrest, and the protective effect appears to be post-mitotic block. In addition, after 72 h vinblastine treatment, the proportion of sub-G1 apoptotic cells was 5 % in the population overexpressing c-Jun compared to 15 % in the population not overexpressing c-Jun (average of two determinations). These results confirm those of Fig. 6C and highlight the protective effect of c-Jun expression on the extent of apoptosis.
Fig. 7.
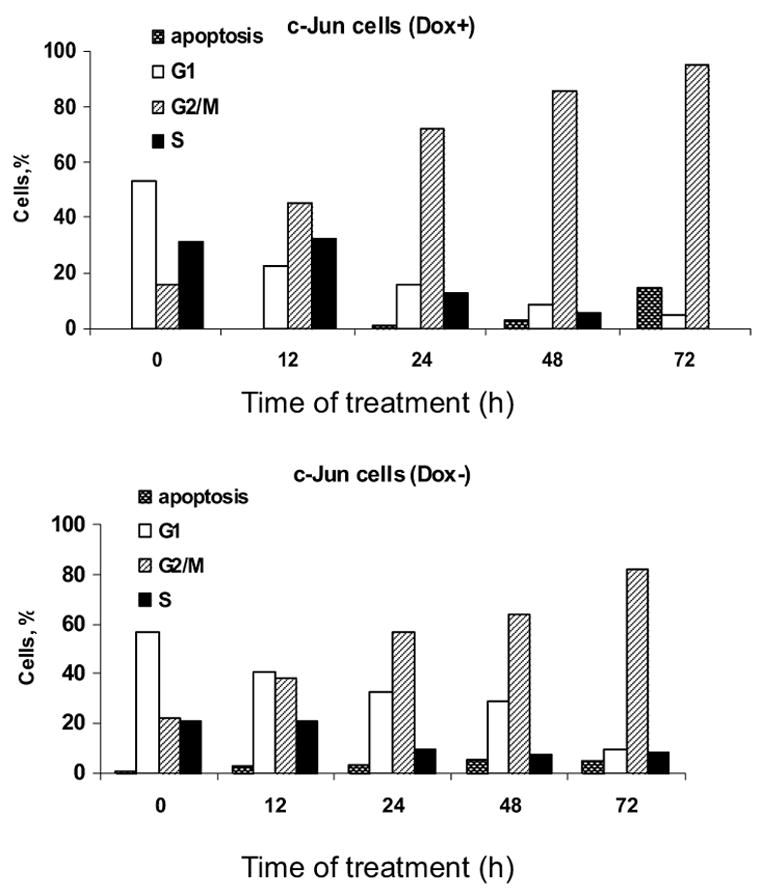
Vinblastine induces mitotic arrest in the absence or presence of c-Jun overexpression. MCF7-c-Jun cells were maintained in the presence of doxycycline (top panel) or absence of doxycycline (bottom panel) for 3 d and then treated with 0.3 μM vinblastine for the times indicated. DNA content was determined by propidium iodide staining and flow cytometry as described in Materials and Methods. The proportion of cells in different phases of the cell cycle, as well as cells with sub-G1 DNA content, are shown. The results are representative of two independent experiments.
Inhibition of vinblastine-induced senescence by c-Jun expression
Analysis of vinblastine-treated cells revealed that only a relatively low proportion of the total population of cells exhibited sub-G1 DNA content indicative of an apoptotic cell death (Fig. 7). In addition, recent evidence has suggested that another outcome after mitotic arrest is senescence [5], defined as an irreversible cell cycle arrest. We therefore evaluated whether vinblastine treated cells underwent senescence as an alternate or secondary outcome. For this purpose we used a specific assay which stains for the presence of β-galactosidase activity, a marker of senescent cells, as described in Materials and Methods. In control MCF7-vector cells and control MCF7-c-Jun cells maintained with doxycycline, a relatively low percentage of cells stained positive for β-galactosidase activity (Fig. 8A top panel, Fig. 8B). After vinblastine treatment of MCF7-vector cells or MCF7-c-Jun cells maintained in the presence of doxycycline, a much higher percentage of cells, close to 20% of the total, stained positive for β-galactosidase activity (Fig. 8A bottom panel, Fig. 8B). However, for MCF7-c-Jun cells maintained in the absence of doxycycline to induce c-Jun expression, the percentage of cells staining positive for β-galactosidase activity was much lower and was not significantly increased after vinblastine treatment (Fig. 8A, Fig. 8B).
Fig. 8.
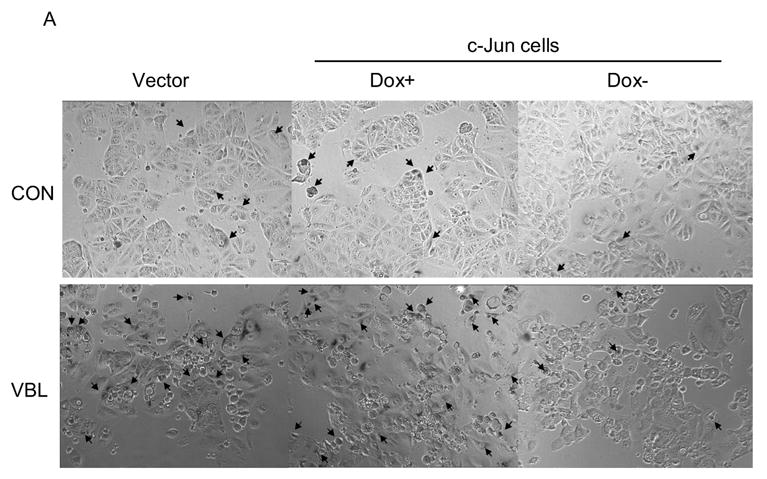
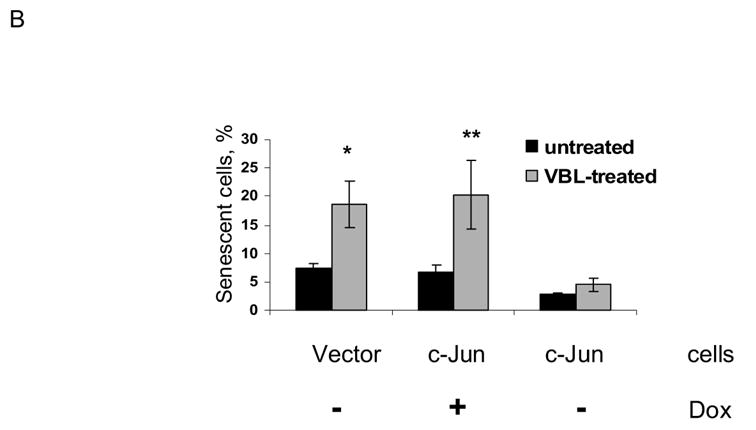
Vinblastine induces senescence in MCF7 cells which is inhibited by c-Jun expression. MCF7-vector cells in doxycycline-free medium (vector), or MCF7-c-Jun cells maintained either in the presence or absence of doxycycline (DOX) for 3 d, were untreated (CON) or treated with 0.3 μM vinblastine (VBL) for 2 d and stained for β-galactosidase activity, as described in Materials and Methods. A. Representative micrographs. Cells with blue staining are indicated by arrows. B. Quantitation of results. Percent senescent cells are shown for the different conditions (mean ± S.D., n = 6). (* P < 0.016 and ** P < 0.018 by two-tailed t test).
4. Discussion
c-Jun was originally identified as one of the immediate-early mitogen regulated genes [14]. A role in cell cycle regulation has been clearly established. One mechanism appears to be due to the ability of c-Jun to transcriptionally down-regulate p53 and in turn the p53-regulated gene p21, thus removing a block on cell cycle progression [15]. Another mechanism involves the induction of cyclin D1 by AP-1 [16]. c-Jun and its upstream regulator JNK have also been implicated in apoptosis [17]. c-Jun is strongly induced by UV irradiation, a genotoxic agent that causes cell cycle arrest and apoptosis [18]. The role of c-Jun in the UV response appears to parallel the role of c-Jun in cell proliferation, in that a major function in the UV response is in negative regulation of p53, allowing UV-damaged cells to re-enter the cell cycle [18]. Cell cycling is an important and perhaps underappreciated component of many apoptotic pathways, in that cell damage is most readily recognized after the passage of a cell through a cell cycle checkpoint. Thus a cycling damaged cell can be appropriately directed to an apoptotic outcome after failing a checkpoint. Such considerations reconcile in part the apparently contradictory participation of c-Jun in cell proliferation as well as cell death.
In addition to being strongly induced in response to mitogenic stimulation or UV irradiation, previous studies in our laboratory have shown that microtubule inhibitors such as vinblastine also strongly induce c-Jun. We have observed this in several different cell lines including KB-3 cells [6], HCT-116 cells1,CHO cells2, and MCF-7 cells (this study). The induction of c-Jun by vinblastine also occurs in p53 null cells1 consistent with c-Jun acting upstream of p53. c-Jun induction by vinblastine in KB-3 cells occurs via JNK-mediated phosphorylation of pre-existing c-Jun protein, which in turn leads to the induction of c-jun mRNA through AP-1 sites in the c-jun promoter, and production of more c-Jun protein, generating a strong auto-amplification loop3. The common occurrence of c-Jun induction and AP-1 activation in response to vinblastine and other related agents suggests an important role in the cellular response to mitotic arrest. We have used several approaches to determine the role of c-Jun in this context including cells stably expressing TAM-67 [6] and the use of c-Jun null fibroblasts [9]. However, these systems do not necessarily represent the best models because of possible cellular adaptation and compensatory mechanisms. In this paper we have used a more controlled cell system that enables the inducible expression of either dominant-negative c-Jun or c-Jun itself.
Our results have shown that inducible expression of TAM-67 effectively blocks vinblastine-induced c-Jun phosphorylation and AP-1 activation but does not significantly affect cell death. Thus it appears that c-Jun/AP-1 is not playing a destructive role directly involved in the cell death process. Similarly, KB-3 cells, a HeLa subline, stably expressing TAM-67 are equally as sensitive as control cells when exposed continuously to vinblastine2, and only show relative resistance when exposed to the drug for short durations [6]. Interestingly, earlier studies in HeLa cells showed different outcomes after pulse and continuous exposure to another microtubule inhibitor, colcemid [19]. In that study, continuous drug exposure led to mitotic block and apoptosis occurring at a period equivalent to one cell cycle later. However, if exposed to drug briefly or to lower concentrations, cells with multipolar mitoses and hypodiploid daughter cells were observed and apoptosis occurred in interphase after mitotic exit. Thus HeLa cells exhibit different fates depending on the conditions of drug exposure. The differential sensitivity of KB-3 cells stably expressing TAM-67 to pulse or continuous vinblastine treatment can be explained if it is postulated that c-Jun plays a more prominent role in the fate of cells after milder conditions of drug exposure.
We found that while expression of TAM-67 did not affect MCF7 cell sensitivity to vinblastine, overexpression of c-Jun had a strongly protective effect. Similarly, we found previously that mouse fibroblasts overexpressing human c-Jun were significantly more resistant to vinblastine relative to control cells [9]. The findings with MCF7 cells reported here were based on several different assays. Trypan blue staining was used as a general method for evaluation of the number of viable and non-viable cells in the population. This is particularly valuable as it is well established that cells can undergo different fates in response to microtubule inhibitors [19–23]. Our results showed that at several different concentrations of vinblastine there was a highly significant decrease in the number of non-viable cells when c-Jun was overexpressed (Fig. 6A). We used a specific apoptosis assay to determine whether apoptosis was one of the outcomes of vinblastine treatment of MCF7 cells and found this to be the case. Consistent with the data derived from trypan blue staining, c-Jun overexpression reduced the extent of apoptotic cell death (Fig. 6C). When cells with sub-G1 DNA content were monitored by flow cytometry, only a relatively small proportion of the total cell population exhibited fragmented DNA even after 3 days of drug treatment (Fig. 7). A much higher proportion of the cells were trypan blue inclusive under similar conditions. These results suggest that vinblastine may induce more than one mode of cell death in MCF-7 cells. This would not be unexpected as earlier studies have revealed that cells treated with a microtubule inhibitor can undergo cell death via one of many different pathways. In some circumstances, apoptosis can be triggered soon after mitotic arrest, whereas in other cases apoptosis ensues only after a long delay [19–23]. Furthermore, it is now recognized that cells may die after mitotic arrest through means other than apoptosis. For example, another fate is mitotic catastrophe, which has been defined as cell death occurring during or after a failed mitosis and one that shares some of the features of apoptosis [3].
Previous studies have demonstrated damage-induced senescence of tumor cells which is most prevalent following DNA damage, and that senescence is also induced by microtubule inhibitors although to a lesser extent [24]. We observed that a significant fraction of MCF7 cells became senescent following vinblastine treatment. We do not know whether apoptosis and senescence represent two independent outcomes for individual cells in the population or whether there exists a relationship between these fates. For example, the vinblastine-induced senescent-like phenotype may be transient and followed by apoptosis. Nonetheless, it is significant that c-Jun inhibits both outcomes. If these processes are not related then it would need to be postulated that c-Jun functions independently in each pathway. On the other hand, because c-Jun inhibits both outcomes, it may be more likely that they are inter-related. More detailed studies on the fate of individual cells are in progress to answer this question. Mechanistically, the inhibition of cell death by c-Jun overexpression presumably reflects the ability of c-Jun to alter the expression level of mediators of the death pathways that function post-mitotic block. Because there are many cell death pathways and each are molecularly complex, defining the mechanism of c-Jun protection is not a trivial task. Current work is focused on determining the effect of c-Jun expression on gene and protein expression profiles, a strategy that has been used successfully previously to identify c-Jun-responsive genes [25]. Further work in this area will be especially important as a major implication of the findings reported here is that the sensitivity of tumor cells to microtubule inhibiting drugs may be influenced by endogenous c-Jun levels.
Acknowledgments
This work was supported by National Institutes of Health Grant CA-75577 (to TCC). We thank Christopher Lyle for assistance with the figures.
Abbreviations
- AP-1
activator or activating protein 1
- JNK
c-Jun NH2-terminal protein kinase
- FBS
fetal bovine serum
- MEM
minimal essential medium
- DMSO
dimethyl sulphoxide
- DTT
dithiothreitol
Footnotes
CS Lyle and TC Chambers, unpublished observations;
M Fan and TC Chambers, unpublished observations;
SN Kolomeichuk, A Bene, CS Lyle, M Rajasekaran, M Upreti, RA Dennis and TC Chambers, manuscript in preparation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 2.Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, Zhivotovsky B, Blagosklonny MV, Malorni W, Knight RA, Piacentini M, Nagata S, Melino G. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005;12:1463–1467. doi: 10.1038/sj.cdd.4401724. [DOI] [PubMed] [Google Scholar]
- 3.Castedo M, Perfettini JL, Roumier T, Valent A, Raslova H, Yakushijin K, Horne D, Feunteun J, Lenoir G, Medema R, Vainchenker W, Kroemer G. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene. 2004;23:4362–4370. doi: 10.1038/sj.onc.1207572. [DOI] [PubMed] [Google Scholar]
- 4.Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer. 2005;5:773–785. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- 5.Shay JW, Roninson IB. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene. 2004;23:2919–2933. doi: 10.1038/sj.onc.1207518. [DOI] [PubMed] [Google Scholar]
- 6.Fan M, Goodwin ME, Birrer MJ, Chambers TC. The c-Jun NH(2)-terminal protein kinase/AP-1 pathway is required for efficient apoptosis induced by vinblastine. Cancer Res. 2001;61:4450–4458. [PubMed] [Google Scholar]
- 7.Berry A, Goodwin M, Moran CL, Chambers TC. AP-1 activation and altered AP-1 composition in association with increased phosphorylation and expression of specific Jun and Fos family proteins induced by vinblastine in KB-3 cells. Biochem Pharmacol. 2001;62:581–591. doi: 10.1016/s0006-2952(01)00694-3. [DOI] [PubMed] [Google Scholar]
- 8.Brantley-Finley C, Lyle CS, Du L, Goodwin ME, Hall T, Szwedo D, Kaushal GP, Chambers TC. The JNK, ERK and p53 pathways play distinct roles in apoptosis mediated by the antitumor agents vinblastine, doxorubicin, and etoposide. Biochem Pharmacol. 2003;66:459–469. doi: 10.1016/s0006-2952(03)00255-7. [DOI] [PubMed] [Google Scholar]
- 9.Obey TB, Lyle CS, Chambers TC. Role of c-Jun in cellular sensitivity to the microtubule inhibitor vinblastine. Biochem Biophys Res Commun. 2005;335:1179–1184. doi: 10.1016/j.bbrc.2005.07.194. [DOI] [PubMed] [Google Scholar]
- 10.Ludes-Meyers JH, Liu Y, Munoz-Medellin D, Hilsenbeck SG, Brown PH. AP-1 blockade inhibits the growth of normal and malignant breast cells. Oncogene. 2001;20:2771–2780. doi: 10.1038/sj.onc.1204377. [DOI] [PubMed] [Google Scholar]
- 11.Upreti M, Lyle CS, Skaug B, Du L, Chambers TC. Vinblastine-induced apoptosis is mediated by discrete alterations in subcellular location, oligomeric structure, and activation status of specific Bcl-2 family members. J Biol Chem. 2006;281:15941–15950. doi: 10.1074/jbc.M512586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown PH, Chen TK, Birrer MJ. Mechanism of action of a dominant-negative mutant of c-Jun. Oncogene. 1994;9:791–799. [PubMed] [Google Scholar]
- 13.Kallunki T, Deng T, Hibi M, Karin M. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell. 1996;87:929–939. doi: 10.1016/s0092-8674(00)81999-6. [DOI] [PubMed] [Google Scholar]
- 14.Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 15.Schreiber M, Kolbus A, Piu F, Szabowski A, Mohle-Steinlein U, Tian J, Karin M, Angel P, Wagner EF. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999;13:607–619. doi: 10.1101/gad.13.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 17.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 18.Shaulian E, Schreiber M, Piu F, Beeche M, Wagner EF, Karin M. The mammalian UV response: c-Jun induction is required for exit from p53-imposed growth arrest. Cell. 2000;103:897–907. doi: 10.1016/s0092-8674(00)00193-8. [DOI] [PubMed] [Google Scholar]
- 19.Sherwood SW, Sheridan JP, Schimke RT. Induction of apoptosis by the anti-tubulin drug colcemid: relationship of mitotic checkpoint control to the induction of apoptosis in HeLa S3 cells. Exp Cell Res. 1994;215:373–379. doi: 10.1006/excr.1994.1354. [DOI] [PubMed] [Google Scholar]
- 20.Woods CM, Zhu J, McQueney PA, Bollag D, Lazarides E. Taxol-induced mitotic block triggers rapid onset of a p53-independent apoptotic pathway. Mol Med. 1995;1:506–526. [PMC free article] [PubMed] [Google Scholar]
- 21.Sorger PK, Dobles M, Tournebize R, Hyman AA. Coupling cell division and cell death to microtubule dynamics. Curr Opin Cell Biol. 1997;9:807–814. doi: 10.1016/s0955-0674(97)80081-6. [DOI] [PubMed] [Google Scholar]
- 22.Casenghi M, Mangiacasale R, Tuynder M, Caillet-Fauquet P, Elhajouji A, Lavia P, Mousset S, Kirsch-Volders M, Cundari E. p53-independent apoptosis and p53-dependent block of DNA rereplication following mitotic spindle inhibition in human cells. Exp Cell Res. 1999;250:339–350. doi: 10.1006/excr.1999.4554. [DOI] [PubMed] [Google Scholar]
- 23.Lanni JS, Jacks T. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol Cell Biol. 1998;18:1055–1064. doi: 10.1128/mcb.18.2.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, Kandel ES, Lausch E, Cristov K, Roninson IB. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999;59:3761–3767. [PubMed] [Google Scholar]
- 25.Kinoshita I, Leaner V, Katabami M, Manzano RG, Dent P, Sabichi A, Birrer MJ. Identification of cJun-responsive genes in Rat-1a cells using multiple techniques: increased expression of stathmin is necessary for cJun-mediated anchorage-independent growth. Oncogene. 2003;22:2710–2722. doi: 10.1038/sj.onc.1206371. [DOI] [PubMed] [Google Scholar]