

Abstract
Background and purpose
The current work is based on our previous finding that in neuronal cells nmol/L concentrations of α-tocotrienol (TCT), but not α-tocopherol (TCP), blocked glutamate-induced death by suppressing early activation of c-Src kinase (J Biol Chem 275:13049) and 12-lipoxygenase (12-Lox;J Biol Chem 278:43508).
Methods
Single neuron microinjection technique was employed to compare the neuroprotective effects of TCT with that of the more widely known TCP. Stroke-dependent brain tissue damage was studied in 12-Lox deficient mice and spontaneously hypertensive rats orally supplemented with TCT.
Results
Sub-attomole quantity of TCT, but not TCP, protected neurons from glutamate challenge. Pharmacological as well as genetic approaches revealed that 12-Lox is rapidly tyrosine phosphorylated in the glutamate-challenged neuron and that this phosphorylation is catalyzed by c-Src. 12-Lox deficient mice were more resistant to stroke-induced brain injury than their wild-type controls. Oral supplementation of TCT to spontaneously hypertensive rats led to increased TCT levels in the brain. TCT-supplemented rats showed more protection against stroke-induced injury compared to matched controls. Such protection was associated with lower c-Src activation and 12-Lox phosphorylation at the stroke site.
Conclusion
The natural vitamin E, α-tocotrienol, acts on key molecular checkpoints to protect against glutamate- and stroke-induced neurodegeneration.
Keywords: nutrition, vitamin, pathophysiology
Introduction
Vitamin E is a generic term for tocopherols and tocotrienols 1. Compared to tocopherols, tocotrienols have been poorly studied 2-4. The current work is based on our striking evidence that in neuronal cells nmol/L concentrations of α-tocotrienol, but not α-tocopherol, blocked glutamate-induced death 5–7. These studies from our laboratory presented first evidence showing that at amounts 4–10 fold lower than the levels of α tocotrienol detected in plasma of human supplemented with the vitamin E molecule 8, α-tocotrienol has potent signal transduction regulatory properties that account for its neuroprotective function. Of importance, this striking property was exhibited by a nutrient known to be safe for human consumption. We have reported that glutamate-induced c-Src activation is a key executioner of glutamate-induced neuronal death and that such activation is sensitive to nanomolar concentrations of α-tocotrienol 5. The significance of our report was further enhanced by a later publication demonstrating that indeed Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke 9. Next, we observed that glutamate-induced 12-lipoxygenase (12-Lox) activation is a critical piece of the signaling path that kills neurons 7. Glutamate-inducible c-Src and 12-Lox were identified as two key cytosolic targets of α-tocotrienol within the neuron. In the current work, we employed a microinjection approach to assess the threshold of α-tocotrienol necessary to protect a neuron against glutamate and to test whether the targets of α-tocotrienol are indeed cytosolic. Next, we sought to establish whether there is a direct connection between c-Src and 12-Lox in the neurodegenerative pathway. Finally, we examined the significance of 12-Lox and of oral α-tocotrienol in stroke-induced neurodegeneration in vivo.
MATERIAL AND METHODS
A concise description is provided here. For a more complete description see online supplement.
Cell culture
Mouse hippocampal HT4 neurons
Mouse hippocampal HT4 cells were kindly provided by D.E. Koshland Jr., University of California at Berkeley 5, 7, 10. Cells were grown in Dulbecco's modified Eagle's medium as described previously 5, 7, 11. Primary cortical neurons. Cells were isolated from the cerebral cortex of rat feti (Sprague-Dawley, day 17 of gestation) as described 7, 12.
Determination of cell viability
The viability of cells was assessed by measuring lactate dehydrogenase (LDH) leakage from cells to media 24 h following glutamate treatment 13.
Microinjection of vitamin E
Microinjection was performed using a micromanipulator Femtojet B 5247 and Injectman NI 2 (Eppendorf, Hamburg, Germany) with a 80 (HT4) or 50 (primary cells) hPa of pressure and 0.1 s of time. The compensation pressure during injection was 40 (HT4) or 30 (primary cells) hPa. The glass micropipettes (Sterile femtotip I, Eppendorf) used for injection were with 0.5 μm inner and 1 μm outer diameter.
Determination of 12-lipoxygenase phosphorylation
For immunoprecipitation, cells were treated with 0.25 mM Na3VO4 (15 min) to inhibit protein tyrosine phosphatases. Immobilized phospho-tyrosine mouse mAb was used to immunoprecipitate tyrosine-phosphorylated proteins. The immunoprecipitated proteins were separated on a 10% SDS-polyacrylamide gel electrophoresis, and probed with anti-12-Lox polyclonal antiserum (Cayman Chemical Co., Inc., Ann Arbor, MI).
Assay for c-Src dependent phosphorylation of 12-lipoxygenase in vitro
12-Lox (12.5 μl = 60μg) and 125μM ATP (0.5μl) were mixed with 5 μl of kinase assay buffer (25mM Tris-HCl, ph 7.5, 10 mM MgCl2, 0.1mM sodium orthovanadate, 2mM DTT, 5mM β-glycerophosphate) per reaction. The reaction was started with the addition of recombinant c-Src (6 units/reaction). The reaction was stopped with 5X electrophoresis buffer, boiled for 5 minutes, subjected to 10% SDS-polyacrylamide gel electrophoresis, and probed with phospho-tyrosine mouse mAb. To evaluate loading efficiency, membranes were stripped and re-probed with anti-12-Lox polyclonal antiserum.
Determination of 12-lipoxygenase activity
The in vitro activity of 12-lipoxygenase was assayed using a standard spectrophotometric method to measure the increase in the formation of conjugated dienes 14. The activity of 12-Lox was calculated from the absorbance values as n moles/min using the ε of 2.52 x 104 M−1 and normalized as % control.
Mouse stroke model
Transient focal cerebral ischemia was induced in mice by middle cerebral artery (MCA) occlusion achieved by using the intraluminal filament insertion technique previously described 15, 16. Determination of infarct volume. Brains were rapidly removed, placed in a –70° C freezer for 2 min, and then sectioned into five 2-mm-thick coronal sections. Sections were incubated for 15 min in 2,3,5-triphenyltetrazolium (TTC). TTC images were used to determine infarct size as a percentage of the contralateral hemisphere after correcting for edema, as previously described 15, 16.
Spontaneously hypertensive rat (SHR) stroke studies I and II
Study I
SHR (N=32; male; four weeks old, Harlan, Indianapolis, IN, USA) were randomly divided into control and supplemented groups. All rats were maintained in vitamin E deficient laboratory chow (TD88163, Harlan). The control group was orally gavaged with vitamin E stripped corn oil with volume matching the mean volume of the supplement in the test group. Stock supplement solution (0.3 g Tocomin per ml) was made in vitamin E stripped corn oil. The test group was orally gavaged (8 weeks at 5d/w) with the supplement oil at a dosage of 1g Tocomin per kg body weight.
Study II
SHR (N= 42; male; four weeks old, Harlan, Indianapolis) were randomly divided into two groups: control and supplemented. All rats were maintained in vitamin E deficient chow (TD88163, Harlan). The control group was orally gavaged with vitamin E stripped corn oil with volume matching the mean volume of the supplement in the test group. Stock solution of tocotrienol supplement solution (0.06 g tocotrienol per ml) was made in vitamin E stripped corn oil. The test group was orally gavaged (13 weeks at 5d/w) with the supplement oil at a dosage of 50 mg tocotrienol kg body weight. Compared to Study I, the longer supplementation period was aimed at improving the delivery of α-tocotrienol to the brain.
SHR stroke
Male SHR (Study I & II) weighing 250–350 grams were fasted for 24 hours prior to surgery. SHR were chosen because infarction can be reliably produced in this species with little variability in infarct size 17. Permanent focal neocortical ischemia was produced by tandem right common carotid artery (CCA) and middle cerebral artery (MCA) occlusion as described17.
12-Lipoxygenase phosphorylation in brain tissue
Rat brain tissues (100–150 mg) from study II were used to detect 12-Lox phosphorylation. The brain tissue sample was harvested from a predictable area of infarct core around the occluded vessel. This includes the primary motor cortex and the primary somatosensory cortex. Immobilized phosphotyrosine mouse mAb was used to immunoprecipitate tyrosine-phosphorylated proteins. The immunoprecipitated proteins were separated on a 10% SDS-polyacrylamide gel electrophoresis, and probed with anti-12-Lox polyclonal antiserum.
Immunohistochemistry
Tissues were collected in OCT or formalin.
Fluro-Jade B staining
To determine the neuronal degeneration, frozen brain sections (10 μm) were stained using Fluro-Jade procedure 18.
pp60srcand phspho-src staining
Formalin-fixed brain tissue were embedded in paraffin and sectioned. The sections (4 μm) were deparaffinized and stained with mouse monoclonal antibody to Src and rabbit polyclonal antiserum anti-phospho-Src.
Vitamin E extraction and analysis
Vitamin E extraction and analysis of rat brains were performed as described previously using an HPLC-coulometric electrode array detector 6, 19.
Statistics
Bar graphs represent mean ± SD. Difference between two means was tested by Students t-test. A value of p<0.05 was interpreted as a significant difference. Comparisons among multiple groups were made by analysis of variance ANOVA. P<0.05 was considered statistically significant.
RESULTS
Microinjection studies led to the observation that 10−19 moles of cytosolic, but not nuclear, α-tocotrienol into a HT4 neuron protects from subsequent glutamate-induced insult (Fig. 1A–B). This observation is consistent with our previous reports identifying that the intracellular targets of α-tocotrienol in the neuron lies in the cytosol 5, 7. Figure 1B demonstrates that after 24h of glutamate treatment all cells (Fig. 1A), except the one microinjected with α-tocotrienol, died and perished leaving the α-tocotrienol injected lone cell to survive. When repopulated with healthy cells, the surviving cell continued to live as evident by the green calcein vital stain for live cells. The reddish stain in the calcein positive cell at the center of the field marks the injected cell (Fig. 1C). Consistent with our previous report demonstrating that α-tocopherol does not share the neuroprotective effects of nanomolar α-tocotrienol, we observed that microinjection of α-tocopherol failed to protect HT4 neurons as well as primary neurons (not shown) against glutamate (Fig. 1F–I). The protective effect of sub-attomole quantity of α-tocotrienol was applicable to primary neurons as well (Fig. 1J–K).
Figure 1. Cytosolic α-tocotrienol, but not α-tocopherol, protects neurons from glutamate induced death.

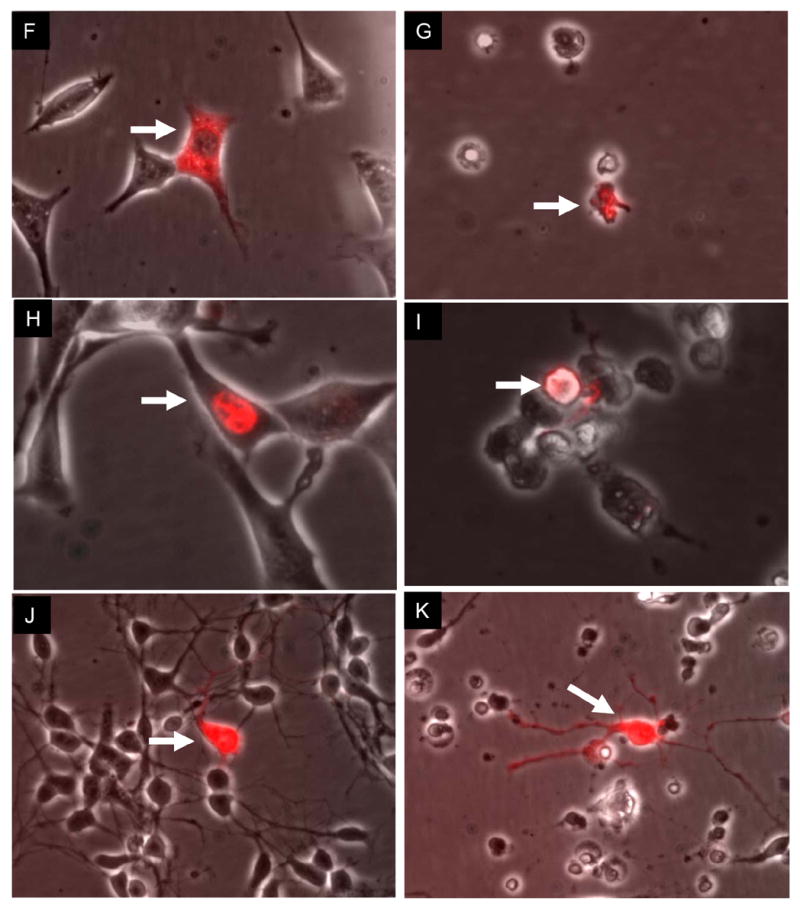
HT4 (A–I) were injected with α-tocotrienol (10−19 mole) into the cytoplasm (A; 90% survival count in six experiments) or nucleus (D). Panel B shows survival of the neuron injected with α-tocotrienol while the other neurons died and disappeared from the monolayer. Tocotrienol was co-injected with the fluorescent QDot (see in red). The culture plate containing the surviving cell was repopulated with fresh healthy HT4 cells to monitor the fate the surviving cell (arrow marked in C) over a period of 18h. Calcein AM was used to stain live cells (C). Control cells injected with QDot alone (not shown) or with tocopherol (Fig. F–G, cytosolic; H–I, nuclear) did not survive (0% survival count in five experiments) against glutamate-induced challenge. Cytosolic injection of α-tocotrienol protected primary immature cortical neurons (J–K) against glutamate challenge as well. All panels in the left column represent images at 0h of glutamate challenge. All panels in the right column represent images at 18h of 10 mM glutamate challenge. Representative illustrations of five experiments are shown. Objectively, nuclear injection of α-tocotrienol failed to protect in 100% case. α-Tocopherol (10−19 moles) failed to protect in 100% case. Cytosolic injection of α-tocotrienol protected cells in 90% of all cases. Arrows in each pair of photos (left-right)
Reduced glutathione (GSH) plays a significant role in resisting neurodegeneration 20, 21. Neurons are rich in arachidonic acid which substantially exacerbates neurotoxicity caused by lowered cellular GSH 7. In this model of neurodegeneration, pharmacological inhibitors of both c-Src as well as of 12-Lox protected HT4 neurons (Fig. 2A–B). These results suggested a direct interaction of c-Src and 12-Lox in executing neuronal death. We hypothesized that 12-Lox is subject to tyrosine phosphorylation by c-Src activated in response to glutamate challenge. Experiments directed at testing the hypothesis revealed that glutamate induces rapid tyrosine phosphorylation in 12-Lox (Fig. 2C & F). Such phosphorylation was sensitive to pharmacological inhibitors of c-Src as well as to α-tocotrienol (Fig. 2E). Tyrosine phosphorylation of neuronal 12-Lox was also observed in response to treatment of cells with GSH-depleting buthionine sulfoximine and arachidonic acid. Such phosphorylation was inhibited in the presence of nanomolar α-tocotrienol (Fig. 2D). Glutamate-induced 12-Lox phosphorylation was more pronounced in the kinase-active Src over-expressing cells than in kinase-dead K297R or control pUSE cells (Fig. 2H). This, and findings from DN c-Src transfected cells (Fig. 2G) confirmed a direct role of c-Src in phosphorylating 12-Lox in the glutamate challenged neuron. The reaction between 12-Lox and c-Src in the presence of ATP provided direct evidence that 12-Lox may be phosphorylated by c-Src. Such phosphorylation of 12-Lox was inhibited by pharmacological inhibitors of c-Src such as herbimycin and PP2 (Figs 3A&B). Previously we have published that α-tocotrienol may dock on 12-Lox to block the catalytic function of the enzyme 7. To test whether that inhibitory effect of α-tocotrienol on 12-Lox is applicable to phospho-12-Lox, an activity assay was performed. We observed that α-tocotrienol inhibits both 12-Lox as well as phospho-12-Lox activity (Fig. 3C).
Figure 2. c-Src and 12-Lox in glutamate-induced neuronal death.

HT4 cells (A–B) were either treated or not with α-tocotrienol, BL15, herbimycin or geldanamycin (as indicated) for 5 min and challenged with buthionine sulfoximine (0.15 mM; BSO, A) or BSO and arachidonic acid (0.05 mM, B) for 24 h. BL 15 is an inhibitor of 12-lipoxygenase. Both herbimycin and geldanamycin inhibit c-Src kinase activity. A, Tocotrienol, 12-Lox inhibitor as well as c-Src inhibitors protected against BSO-induced glutathione-depletion dependent loss of cell viability. †, higher than BSO non-treated; *, lower than BSO-treated. B, Tocotrienol, 12-Lox inhibitor, and c-Src kinase inhibitors protected against BSO and arachidonic acid induced loss of cell viability. †, higher than BSO (A) and non-challenged (B) groups; *, lower than BSO and arachidonic acid challenged group. p < 0.001. Glutamate (10 mM) induced 12-Lox phosphorylation was inhibited by α-tocotrienol (250 nmol/L) in HT4 cells (C) as well as in immature cortical neurons (F). Herbimycin also inhibited inducible 12-Lox phosphorylation (E). Cells were either treated or not with α-tocotrienol for 5 min and challenged with glutamate (10 mM) or BSO (0.15 mM) and arachidonic acid (0.05 mM) for 15 min (D) or 30 min as indicated. In these experiments 15 minutes before challenging, cells were treated with Na3VO4 (0.15 mM) to inhibit tyrosine phosphatases. C, control (non-treated); TCT, α-tocotrienol; H, herbimycin; G, glutamate; and AA, arachidonic acid. G–H, Glutamate-induced phosphorylation was inhibited (G) in cells expressing dominant negative c-Src (K296R/Y528F) and more prominent in kinase-active (Y529F) c-Src over-expressing cells than in wild-type (pUSE) or kinase-dead c-Src (K297R) over-expressing cells (H). Cells were activated with 10 mM glutamate for 15 mins.
Figure 3. 12-Lipoxygenase is phosphorylated by c-Src and phospho-12-lipoxygenase activity is sensitive to α-tocotrienol.

In the presence of ATP, c-Src phosphorylated 12-Lox in a herbimycin (A) and PP2 (B) sensitive manner. PP3, a negative control for PP2, did not inhibit 12-Lox phosphorylation indicating a specific role of c-Src. C, α-Tocotrienol inhibited the activity of 12-Lox as well as of phospho-12-Lox. Phospho-12-Lox was generated (as in A and B) by incubation of the enzyme with c-Src and ATP. †, p<0.05 significant increase in conjugated diene (reaction product) formation in response to 12-Lox addition to the reaction mix. *, p<0.05 lower activity of both 12-Lox as well as phospho-12-Lox in the presence of α-tocotrienol (TCT) compared to the activity observed in the absence of TCT. Values are mean ± SD.
Pursuant to our previously reported finding that 12-Lox deficient neurons are resistant to glutamate-induced death and that 12-Lox represents a key target for α-tocotrienol action 7, we observed that 12-Lox deficient mice were resistant to stroke injury (Fig. 4). The compelling neuroprotective effects of α-tocotrienol in vitro led us to question the significance of this nutrient in vivo. In SHR stroke study #1, experimental stroke outcome data revealed that the Tocomin-fed rats tended (p = 0.057, Fig. 5E) to have reduced injury following stroke than the control group. The study was repeated with a modified experimental design including a more pure form of the supplement, longer supplementation period and a larger sample size. The supplementation increased brain α-tocotrienol level without significantly changing the brain α-tocopherol (Fig. 5C–D). The brain is known to retain its vitamin E levels effectively under conditions of dietary vitamin E dedficiency 22. Consistently, we did not observe any significant drop in the α-tocopherol level of the brain of the control rats maintained on vitamin E deficiency (Fig. 5C). Thus, other antioxidant systems, such as the GSH system in the brain, are not likely to be affected under the conditions of our study.
Figure 4. 12-Lipoxygenase deficient mice are resistant to transient focal cerebral ischemia.
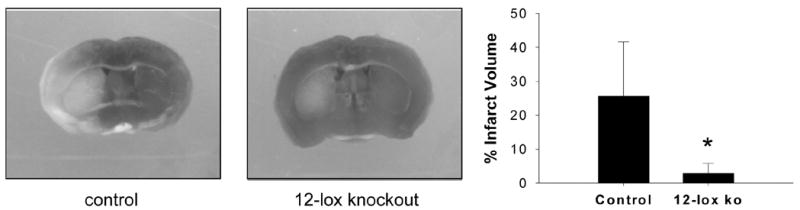
Transient focal cerebral ischemia was induced in the C57BL6/J (control) and 12-lipoxygenase knockout (12-Lox −/−) mice by middle cerebral artery occlusion. Coronal sections of brain (72h after stroke) were stained with 2,3,5-triphenyltetrazolium (TTC). Fixed sections were photographed using a Inquiry software (Loats Associates, Inc, Westminster, MD). The images were used to determine infarct size as a percentage of the contralateral hemisphere after correcting for edema. The infarct extended from caudate putamen into surrounding cortex, and was visible in 4 out of 5 slices of the brain from control wild-type mice. Data are mean ± SD. *, p<0.02.
Figure 5. Oral α-tocotrienol supplementation protected against stroke induced injury and 12-lipoxygenase phosphorylation in the brain of spontaneously hypertensive rats.
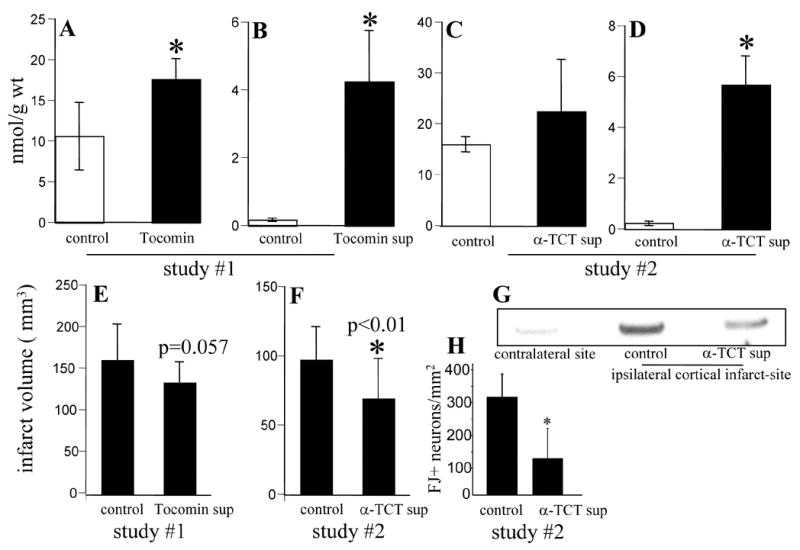
The effect of oral supplementation on brain α tocopherol (A,C) and α tocotrienol (B,D). *, p<0.01 significantly different compared to the corresponding control group. Infarct volume (E,F) in response to stroke. G, α-Tocotrienol supplementation lowered 12-lipoxygenase phosphorylation in the stroke-site brain tissue of SHR. The brain tissue sample was harvested 24h after stroke from a predictable area of infarct core around the occluded vessel. This includes the primary motor cortex and the primary somatosensory cortex. H, Histological analyses of post-stroke infarct zone cortical section (Figure 6) revealed lower count of Fluoro-Jade positive (FJ+) neurons in tocotrienol supplemented rats. *, p<0.05. These results represent the image analysis data of sections (A1, B1 & C1) shown in Figure 6. Study I, n=16; Study II, n=21.
α-Tocotrienol supplemented rats were protected against stroke-induced brain injury (Fig. 5F). Analysis of the brain tissue collected from the stroke site as well as from the contra-lateral non-stroke site of the brain demonstrated that while phospho-12-Lox was not detected in the non-stroke site, 12-Lox was clearly tyrosine phosphorylated in the tissue from the stroke site. Such stroke-associated phosphorylation of 12-Lox was lower in the brain of rats supplemented with α-tocotrienol (Fig. 5G).
The histochemical application of Fluoro-Jade results in a simple, sensitive and reliable method for staining degenerating neurons and their processes 23. More prominent Fluoro-Jade staining in brain sections from control rats compared to sections from supplemented rats (Fig. 6) was consistent with stroke-induced brain injury results shown in Fig 5F. Previously, we have reported that activation of c-Src represents a key mechanism that contributes to neurodegeneration 5. Y416 represents a major autophosphorylation site in c-Src activation loop that is also responsible for c-Src activation 24. Here we provide first in vivo evidence indicating that stroke is associated with c-Src activation at the injury site (Fig. 6). Stroke-associated c-Src activation was partly suppressed in α-tocotrienol supplemented rats (Fig. 6).
Figure 6. Histological analyses of post-stroke infarct zone cortical sections.
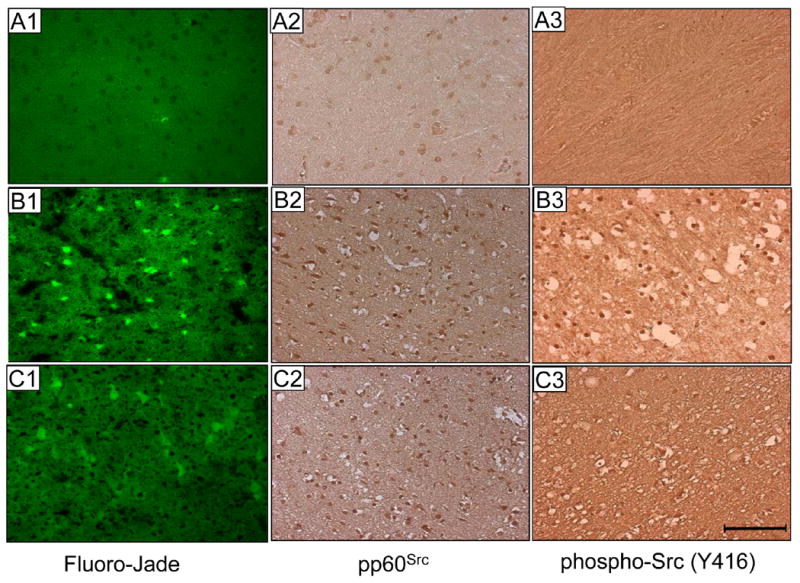
Fluoro-Jade (FJ) stain. In the contralateral hemisphere of the brain from control rats (A1) there was hardly any FJ-positive neuron. The ipsilateral hemisphere of the brain from α-tocotrienol supplemented rats (C1) contained fewer (see Figure 5H) FJ-positive cells compared to such cells in the ipsilateral hemisphere of the brain from control rats (B1). Src and phospho-Src staining. In non-stroke site of the brain section, Src was present (A2) but not in a phosphorylated form (A3). Stroke did not influence Src expression but clearly induced Src phosphorylation and activation. Such stroke-induced Src activation was less in the brain sections of tocotrienol-supplemented rats compared to control rats that were subjected to stroke (C3 versus B3). For all sections shown, brain was harvested 24h after stroke. Bar = 100 micron.
DISCUSSION
Tocotrienols differ from tocopherols by possessing a farnesyl (isoprenoid) rather than a saturated phytyl side chain. The unsaturated side chain of tocotrienol allows for more efficient penetration into tissues that have saturated fatty layers such as the brain and liver 25. Micromolar amounts of tocotrienol, not tocopherol, have been shown to suppress the activity of hydroxy-3-methylglutaryl coenzyme A reductase 26, 27. Our finding that cytosolic, but not nuclear, α-tocotrienol is neuroprotective is in line with our previous observations characterizing that the molecular targets of α-tocotrienol in the neuron are cytosolic 5, 7. Furthermore, the results showing that at sub-attomole levels α-tocotrienol, but not α-tocopherol, is neuroprotective is consistent with our previous reports claiming that at low doses the neuroprotective property of α-tocotrienol is not shared by α-tocopherol 5. Efforts to elucidate the mechanisms underlying the neuroprotective properties of α-tocotrienol led to the finding that glutamate-induced rapid c-Src activation is prevented in the presence of nanomolar concentrations of α-tocotrienol. This function of α-tocotrienol was not shared by α-tocopherol 5. The significance of α-tocotrienol as inducible c-Src inhibitor in neuronal cells was further enhanced by a subsequent study reporting that Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke 9. Next, we identified 12-lipoxygenase (12-Lox) as another α-tocotrienol sensitive molecular checkpoint that proved to be critical in executing death of neurons in response to glutamate and other GSH lowering agents 7. Following glutamate challenge, 12-Lox was rapidly activated and migrated from the cytosol to the membrane 7. Here, we tested the efficacy of inhibitors of both c-Src as well as 12-Lox in models of cell death other than glutamate-induced but related to GSH lowering. Based on past experience with such experimental systems 5, 7, we challenged cells with either BSO alone or with a combination of BSO and arachidonic acid. α-Tocotrienol protected the neurons under both challenging conditions. Of interest, inhibitors of c-Src as well as of 12-Lox protected the cells against both challenging conditions. These observations led us to question whether 12-Lox may act as a substrate for c-Src. Results presented herein suggest that 12-Lox is subject to rapid tyrosine phosphorylation in neuronal cells challenged with glutamate or GSH-lowering agents. Such phosphorylation is rapid and coincides with the timeline of c-Src activation 5, 11. Inhibitors of c-Src abrogated such inducible 12-Lox tyrosine phosphorylation supporting the notion that c-Src may directly phosphorylate 12-Lox in challenged neurons. To test this hypothesis we utilized genetic approaches of over-expressing kinase-active, kinase-dead or dominant negative c-Src in neuronal cells. Current findings from cell biology studies as well as from the study of c-Src and 12-Lox in cell-free systems indicate that in response to challenge by glutamate or GSH-lowering agents, c-Src is rapidly activated and phosphorylates 12-Lox.
In support of a central role of 12-Lox in glutamate-induced neurodegeneration, we have previously reported that inhibitors of 12-Lox prevent death of neuronal cells caused in response to glutamate or GSH-lowering agents 7. Our case for 12-Lox as a critical mediator of glutamate-induced neurodegeneration was strengthened by the finding that compared to neurons from corresponding wild-type mice, cortical neurons from 12-Lox deficient mice are resistant to glutamate-induced death 7. Our current findings demonstrate that 12-Lox deficiency protects against stroke injury. This builds a compelling case to look at 12-Lox as a therapeutic target for the management of stroke-related injury in the brain. Functional outcomes in in vivo stroke models have been proposed to be valuable while building the case for clinical trials to test the effect of any neuroprotective agent 28. Although the incorporation of cognitive and sensorimotor functional outcome assessment represents an important step forward in stroke research, reports of MCAO induced behavioral deficits often conflict 29. The effect of tocotrienol on stroke-induced changes in functional outcome remains to be investigated. In glutamate-challenged neurons, α-tocotrienol effectively modulates both 12-Lox as well as c-Src activity to favor survival 5, 7. This study demonstrated that oral α-tocotrienol supplementation may protect against stroke in vivo.
Supplementary Material
Acknowledgments
Supported by NIH NS42617 to CKS and in part by NS40267 to ACD.
References
- 1.Brigelius-Flohe R, Traber MG. Vitamin e: Function and metabolism. FASEB Journal. 1999;13:1145–1155. [PubMed] [Google Scholar]
- 2.Traber MG, Packer L. Vitamin e: Beyond antioxidant function. Am J Clin Nutr. 1995;62:1501S–1509S. doi: 10.1093/ajcn/62.6.1501S. [DOI] [PubMed] [Google Scholar]
- 3.Traber MG, Sies H. Vitamin e in humans: Demand and delivery. Annu Rev Nutr. 1996;16:321–347. doi: 10.1146/annurev.nu.16.070196.001541. [DOI] [PubMed] [Google Scholar]
- 4.Sen CK, Khanna S, Roy S. Tocotrienol: The natural vitamin e to defend the nervous system? Ann New York Acad Sci. 2004;1031:127–142. doi: 10.1196/annals.1331.013. [DOI] [PubMed] [Google Scholar]
- 5.Sen CK, Khanna S, Roy S, Packer L. Molecular basis of vitamin e action. Tocotrienol potently inhibits glutamate-induced pp60(c-src) kinase activation and death of ht4 neuronal cells. Journal of Biological Chemistry. 2000;275:13049–13055. doi: 10.1074/jbc.275.17.13049. [DOI] [PubMed] [Google Scholar]
- 6.Roy S, Lado BH, Khanna S, Sen CK. Vitamin e sensitive genes in the developing rat fetal brain: A high-density oligonucleotide microarray analysis. FEBS Letters. 2002;530:17–23. doi: 10.1016/s0014-5793(02)03309-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khanna S, Roy S, Ryu H, Bahadduri P, Swaan PW, Ratan RR, Sen CK. Molecular basis of vitamin e action: Tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. Journal of Biological Chemistry. 2003;278:43508–43515. doi: 10.1074/jbc.M307075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Byrne D, Grundy S, Packer L, Devaraj S, Baldenius K, Hoppe PP, Kraemer K, Jialal I, Traber MG. Studies of ldl oxidation following alpha-, gamma-, or delta-tocotrienyl acetate supplementation of hypercholesterolemic humans. Free Radical Biology & Medicine. 2000;29:834–845. doi: 10.1016/s0891-5849(00)00371-3. [DOI] [PubMed] [Google Scholar]
- 9.Paul R, Zhang ZG, Eliceiri BP, Jiang Q, Boccia AD, Zhang RL, Chopp M, Cheresh DA. Src deficiency or blockade of src activity in mice provides cerebral protection following stroke. Nature Medicine. 2001;7:222–227. doi: 10.1038/84675. [DOI] [PubMed] [Google Scholar]
- 10.Tirosh O, Sen CK, Roy S, Packer L. Cellular and mitochondrial changes in glutamate-induced ht4 neuronal cell death. Neuroscience. 2000;97:531–541. doi: 10.1016/s0306-4522(00)00028-2. [DOI] [PubMed] [Google Scholar]
- 11.Khanna S, Venojarvi M, Roy S, Sen CK. Glutamate-induced c-src activation in neuronal cells. Methods in Enzymology. 2002;352:191–198. doi: 10.1016/s0076-6879(02)52019-x. [DOI] [PubMed] [Google Scholar]
- 12.Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB Journal. 1990;4:1624–1633. [PubMed] [Google Scholar]
- 13.Han D, Sen CK, Roy S, Kobayashi MS, Tritschler HJ, Packer L. Protection against glutamate-induced cytotoxicity in c6 glial cells by thiol antioxidants. American Journal of Physiology. 1997;273:R1771–1778. doi: 10.1152/ajpregu.1997.273.5.R1771. [DOI] [PubMed] [Google Scholar]
- 14.Beierschmitt WP, McNeish JD, Griffiths RJ, Nagahisa A, Nakane M, Amacher DE. Induction of hepatic microsomal drug-metabolizing enzymes by inhibitors of 5-lipoxygenase (5-lo): Studies in rats and 5-lo knockout mice. Toxicological Sciences. 2001;63:15–21. doi: 10.1093/toxsci/63.1.15. [DOI] [PubMed] [Google Scholar]
- 15.DeVries AC, Joh HD, Bernard O, Hattori K, Hurn PD, Traystman RJ, Alkayed NJ. Social stress exacerbates stroke outcome by suppressing bcl-2 expression. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:11824–11828. doi: 10.1073/pnas.201215298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Blizzard KK, Zeng Z, DeVries AC, Hurn PD, McCullough LD. Chronic behavioral testing after focal ischemia in the mouse: Functional recovery and the effects of gender. Experimental Neurology. 2004;187:94–104. doi: 10.1016/j.expneurol.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 17.Brint S, Jacewicz M, Kiessling M, Tanabe J, Pulsinelli W. Focal brain ischemia in the rat: Methods for reproducible neocortical infarction using tandem occlusion of the distal middle cerebral and ipsilateral common carotid arteries. Journal of Cerebral Blood Flow & Metabolism. 1988;8:474–485. doi: 10.1038/jcbfm.1988.88. [DOI] [PubMed] [Google Scholar]
- 18.Schmued LC, Albertson C, Slikker W., Jr Fluorojade: A novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Research. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- 19.Roy S, Venojarvi M, Khanna S, Sen CK. Simultaneous detection of tocopherols and tocotrienols in biological samples using hplc-coulometric electrode array. Methods in Enzymology. 2002;352:326–332. doi: 10.1016/s0076-6879(02)52029-2. [DOI] [PubMed] [Google Scholar]
- 20.Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biological Chemistry. 2003;384:505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- 21.Fonnum F, Lock EA. The contributions of excitotoxicity, glutathione depletion and DNA repair in chemically induced injury to neurones: Exemplified with toxic effects on cerebellar granule cells. Journal of Neurochemistry. 2004;88:513–531. doi: 10.1046/j.1471-4159.2003.02211.x. [DOI] [PubMed] [Google Scholar]
- 22.Goss-Sampson MA, MacEvilly CJ, Muller DP. Longitudinal studies of the neurobiology of vitamin e and other antioxidant systems, and neurological function in the vitamin e deficient rat. J Neurol Sci. 1988;87:25–35. doi: 10.1016/0022-510x(88)90051-2. [DOI] [PubMed] [Google Scholar]
- 23.Schmued LC, Hopkins KJ. Fluoro-jade b: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Research. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- 24.Shah OJ, Kimball SR, Jefferson LS. The src-family tyrosine kinase inhibitor pp1 interferes with the activation of ribosomal protein s6 kinases. Biochemical Journal. 2002;366:57–62. doi: 10.1042/BJ20020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki YJ, Tsuchiya M, Wassall SR, Choo YM, Govil G, Kagan VE, Packer L. Structural and dynamic membrane properties of alpha-tocopherol and alpha-tocotrienol: Implication to the molecular mechanism of their antioxidant potency. Biochemistry. 1993;32:10692–10699. doi: 10.1021/bi00091a020. [DOI] [PubMed] [Google Scholar]
- 26.Pearce BC, Parker RA, Deason ME, Dischino DD, Gillespie E, Qureshi AA, Volk K, Wright JJ. Inhibitors of cholesterol biosynthesis. 2. Hypocholesterolemic and antioxidant activities of benzopyran and tetrahydronaphthalene analogues of the tocotrienols. Journal of Medicinal Chemistry. 1994;37:526–541. doi: 10.1021/jm00030a012. [DOI] [PubMed] [Google Scholar]
- 27.Pearce BC, Parker RA, Deason ME, Qureshi AA, Wright JJ. Hypocholesterolemic activity of synthetic and natural tocotrienols. Journal of Medicinal Chemistry. 1992;35:3595–3606. doi: 10.1021/jm00098a002. [DOI] [PubMed] [Google Scholar]
- 28.Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke. 1999;30:2752–2758. doi: 10.1161/01.str.30.12.2752. [DOI] [PubMed] [Google Scholar]
- 29.DeVries AC, Nelson RJ, Traystman RJ, Hurn PD. Cognitive and behavioral assessment in experimental stroke research: Will it prove useful? Neurosci Biobehav Rev. 2001;25:325–342. doi: 10.1016/s0149-7634(01)00017-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.