

Abstract
Carboranes represent a potentially rich but underutilized class of inorganic and catabolism-inert pharmacophores. The regioselectivity and ease of derivatization of carboranes allows for facile syntheses of a wide variety of novel structures. The steric bulk, rigidity, and ease of B- and C-derivatization and lack of π-interactions associated with hydrophobic carboranes may be exploited to enhance the selectivity of previously identified bioactive molecules. Transthyretin (TTR) is a thyroxine-transport protein found in the blood that has been implicated in a variety of amyloid related diseases. Previous investigations have identified a variety of nonsteroidal antiinflammatory drugs (NSAIDs) and structurally related derivatives that imbue kinetic stabilization to TTR, thus inhibiting its dissociative fragmentation and subsequent aggregation to form putative toxic amyloid fibrils. However, the cyclooxygenase (COX) activity associated with these pharmaceuticals may limit their potential as long-term therapeutic agents for TTR amyloid diseases. Here, we report the synthesis and evaluation of carborane-containing analogs of the promising NSAID pharmaceuticals previously identified. The replacement of a phenyl ring in the NSAIDs with a carborane moiety greatly decreases their COX activity with the retention of similar efficacy as an inhibitor of TTR dissociation. The most promising of these compounds, 1-carboxylic acid-7-[3-fluorophenyl]-1,7-dicarba-closo-dodecaborane, showed effectively no COX-1 or COX-2 inhibition at a concentration more than an order of magnitude larger than the concentration at which TTR dissociation is nearly completely inhibited. This specificity is indicative of the potential for the exploitation of the unique properties of carboranes as potent and selective pharmacophores.
Keywords: amyloid, cyclooxygenase, nonsteroidal antiinflammatory drug, fibril
The dicarba-closo-dodecaboranes (carboranes) are icosahedral carbon-containing boron clusters with extraordinary characteristic properties (such as resistance to catabolism, kinetic inertness to reagents, strong hydrophobicity, and well-established chemistry) that afford the opportunity for their exploitation as novel hydrophobic pharmacophores. The three isomeric dicarbon carboranes, closo-1,2-C2B10H12, closo-1,7-C2B10H12, and closo-1,12-C2B10H12, commonly known as ortho-, meta-, and para-carborane, respectively, share approximately the same volume as a rotated phenyl ring. This, as well as their structural integrity, ease of substitution, and delocalized bonding, suggests their description as three-dimensional analogs of aromatic hydrocarbons (1), allowing their facile substitution for phenyl rings as rigid scaffolding in pharmacological agents. This rigid scaffolding can be easily and selectively derivatized through deprotonation of the slightly acidic C–H vertices by a strong base (alkyllithium reagents, Grignard reagents, etc.) affording a strongly nucleophilic carboranyl anion capable of reaction with a wide range of electrophiles, including alkyl halides, carbonyl derivatives, and chlorosilanes, to mention only a few examples (1). Conversely, the B–H vertices exhibit slight nucleophilicity permitting derivatization by reactive electrophiles, leading, for example, to B-iodo derivatives. The latter, in combination with Pd-catalyzed cross-coupling reactions (2), facilitates synthesis of a diverse range of B-derivatives. This difference in reactivity makes the regioselective preparation of B- and/or C-derived carboranes possible without the need for complicated protecting group strategies or expensive reagents (for recent reviews, see refs. 3 and 4). Much of this chemistry has been used in the exploration of carboranes as agents of high-in-boron content for use in boron neutron capture therapy (BNCT), thus providing a wealth of information indicating the biocompatibility and resistance to catabolism of a variety of carborane-supported structures (5). However, this plethora of knowledge is starkly contrasted by the paucity of carborane pharmacophores used outside the field of BNCT. Recent reports of carborane pharmacophores include HIV protease inhibitors based on metallacarboranes (6) or carborane-substituted porphyrins (7). Endo and coworkers have investigated the application of carboranes as a three-dimensional hydrophobic core in the design of potent estrogen receptor agonists (8) and retinoid antagonists (9), illustrating the lipophilicity of the carborane moiety previously noted by peptide chemists when synthesizing biologically active peptides bearing (o-carboranyl) alanine (10). This hydrophobicity may be fine-tuned through choice of the carborane cage isomer and the position of substitution on the carborane cage, with the resulting structure providing unique Hansch-Fujita π parameters (11). Indeed, simple substitution of ortho-carborane for a phenyl group in an insect neuropeptide resulted in a 30-fold increase in pheromonotropic activity in vitro relative to the parent pentapeptide while showing significantly augmented resistance to saline washes (12). This same carborane derivative exhibited a 10-fold increase in potency in vivo as compared with an endogenous 33-membered pheromone biosynthesis-activating neuropeptide because of lack of vulnerability from aminopeptidase attack (12). Further success using carboranes has resulted in the discovery of powerful carboranyl analogues of the anti-estrogen tamoxifen (13) and the controversial drug thalidomide (14). In an effort to expand upon these successes, we have endeavored to identify further biological targets where the unique properties of carboranes may prove to be beneficial.
Transthyretin (TTR), also known as thyroxin-binding prealbumin, is a 55-kDa homotetrameric protein comprising 127-amino acids with an extended β-sheet conformation (15, 16). TTR is found in human plasma (0.2 mg/ml, 3.6 μM tetramer) where it binds and transports thyroxine (T4) in two funnel-shaped binding sites defined by the dimer–dimer interface and also forms a complex with retinol-binding protein, which, in turn, transports vitamin A (15–17). In 1978, Costa et al. (18) demonstrated that TTR was the major component of amyloid fibrils associated with familial amyloid polyneuropathy (FAP). Since this discovery, TTR has been implicated as the causative agent in a variety of amyloid diseases [including senile systemic amyloidosis (SSA), familial amyloid cardiomyopathy (FAC), and central nervous system selective amyloidosis (CNSA)], with SSA resulting from the deposition of wild-type TTR (WT-TTR) in the heart and the remaining diseases (FAC, FAP and CNSA) associated with the accumulation of one of >70 TTR variants in a variety of tissues (19–24). Currently, the only treatment available for FAP is gene therapy mediated by liver transplantation, in which a liver producing WT-TTR is substituted for the FAP variant-producing organ. In many cases, because of continuing deposition of WT-TTR, cardiac amyloidosis continues despite surgical intervention (25).
Studies have indicated that the mechanism of TTR amyloid fibril formation requires mildly acidic conditions, simulating the pH of lysosomes, and proceeds through tetramer dissociation to a monomeric intermediate that subsequently aggregates to form the pathogenic amyloid fibrils (26–28). However, under similar conditions, the native conformation of TTR can be stabilized in vitro by thyroid hormone and structurally similar derivatives thereof (29). As <0.5% of the two T4-binding sites within TTR are occupied in vivo, investigations have focused on small molecule inhibitors that stabilized tetrameric TTR without undesirable hormonal activity (30). This research has been successful in identifying a wide variety of structurally diverse compounds that impart kinetic stabilization to tetrameric TTR (for a recent review, see ref. 31). However, many of the most promising compounds are known nonsteriodal antiinflammatory drugs (NSAIDs), such as flufenamic acid and diflunisal (Fig. 1) or structurally related species.
Fig. 1.

Flufenamic acid and diflunisal, potent NSAID inhibitors of TTR amyloidosis.
The pharmacological effects of NSAIDs stem from their inhibition of cyclooxygenase (prostaglandin endoperoxide synthase or COX) enzymes (32). COX exists in the human body as three isozymes: COX-1, COX-2, and COX-3. Whereas the precise function of COX-3 remains elusive, inhibition of COX-1 can lead to problematic side effects such as gastrointestinal irritation, leading to ulcers and bleeding (33, 34), and, whereas COX-2-specific inhibitors have garnered much attention from the pharmaceutical industry, inhibition of COX-2 has been implicated in increased risk of cardiovascular events (35). In this way, the design of inhibitors of TTR aggregation presents the challenge of not just preventing amyloid fibril formation but also ameliorating the deleterious side effects associated with the pharmaceuticals that accomplish this challenge.
The hydrophobic binding channels in TTR seem to be ideally suited for the utilization of carboranes as a skeletal core. Crystal structures of TTR indicate that the funnel-shaped T4-binding site can be generalized into a spacious outer binding pocket large enough to bind sterically bulky substituents (such as dibenzofuran-4,6-dicarboxylic acid) and a smaller inner pocket, as shown in Fig. 2(36). We hypothesized that the three-dimensional carborane structure would fill the outer pocket while maximizing hydrophobic interactions (see Fig. 2). It was further hypothesized that COX activity could be reduced by this steric bulk as well as the inability of the carborane moiety to participate in π–π stacking used by many COX inhibitors, but unnecessary for inhibition of TTR dissociation (38). This report describes the synthesis of a group of carborane-based compounds and their efficacy as potent inhibitors of TTR dissociation followed by fibril formation. We further screen those compounds recognized as promising inhibitors of TTR amyloid formation and identify a lead compound that also lacks any significant COX inhibitor activity.
Fig. 2.

Schematic representation of the tetrameric structure of TTR depicting the two symmetry related T4-binding sites. Binding mode for T4 (Left), and putative binding of a representative carborane compound (Right) showing interactions of carboxylate groups with lysines at entry of the binding site. Color coding: red, BH groups; black, carbon atom (adapted from ref. 37).
Results and Discussion
Design and Synthesis of Inhibitors.
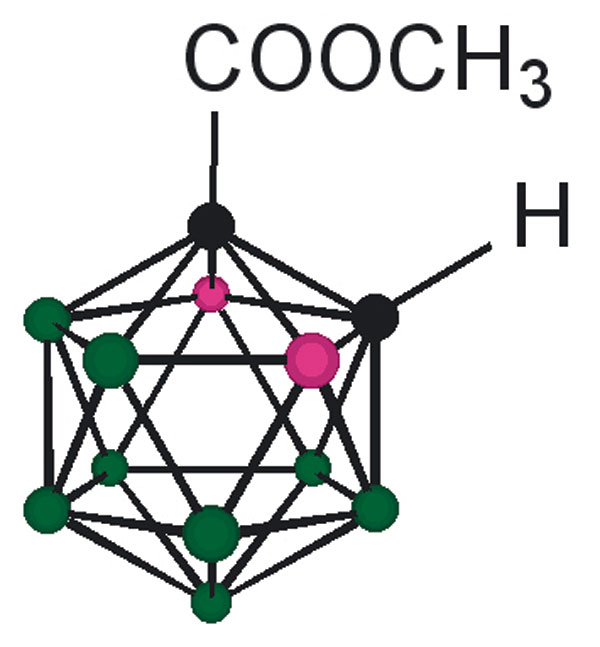
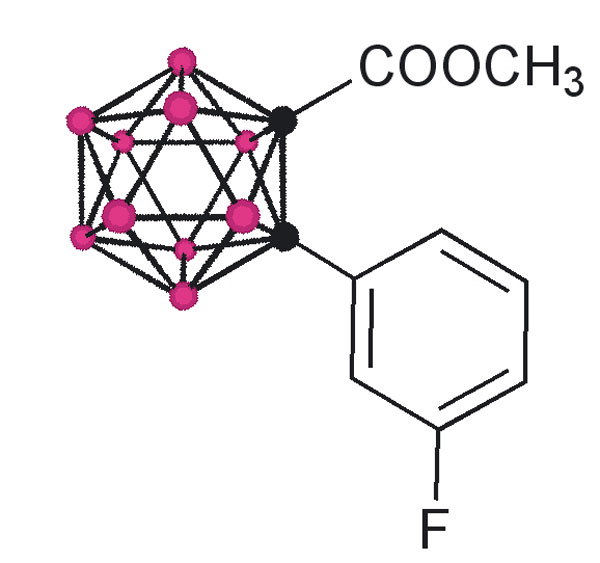
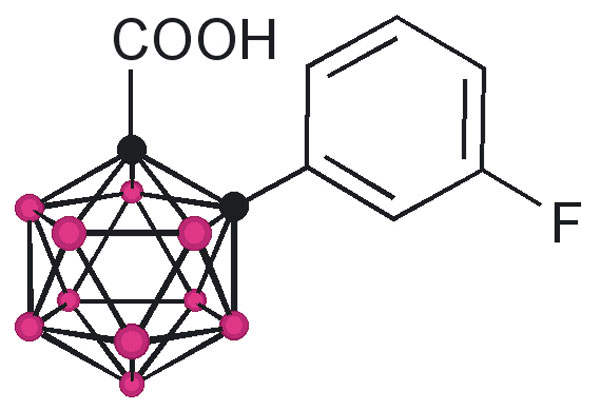
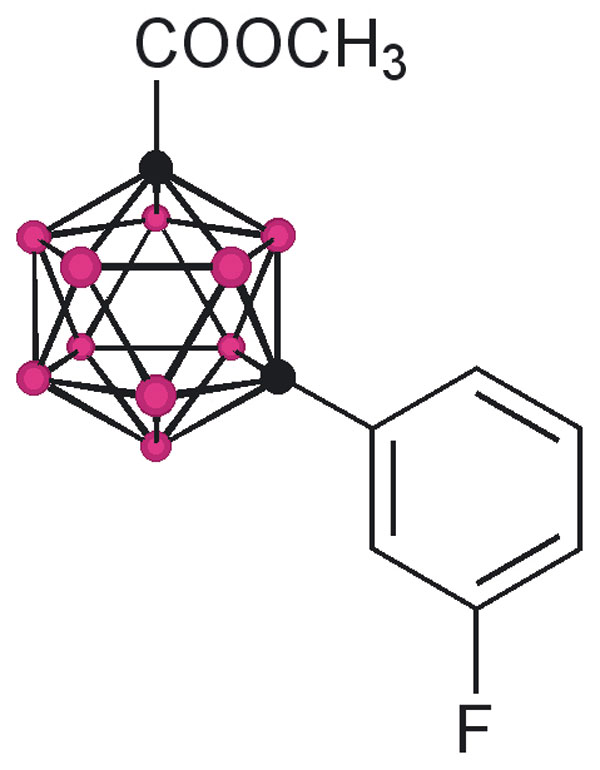
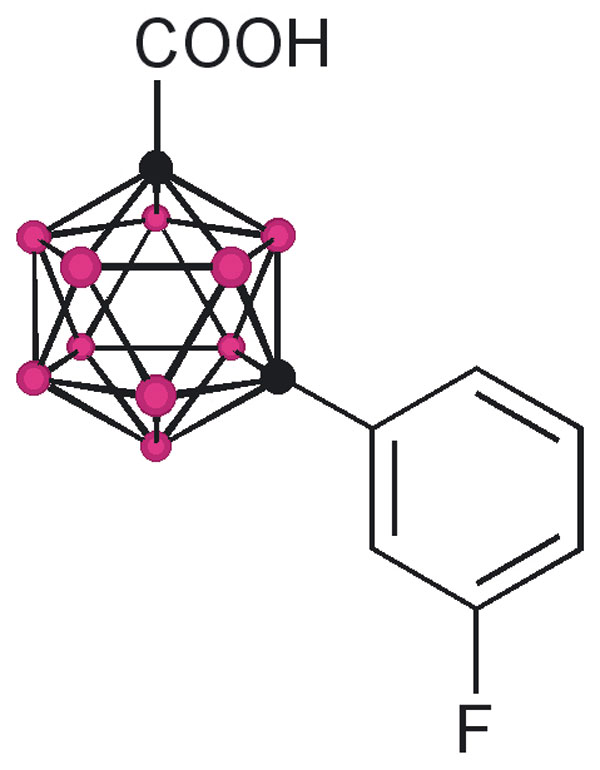
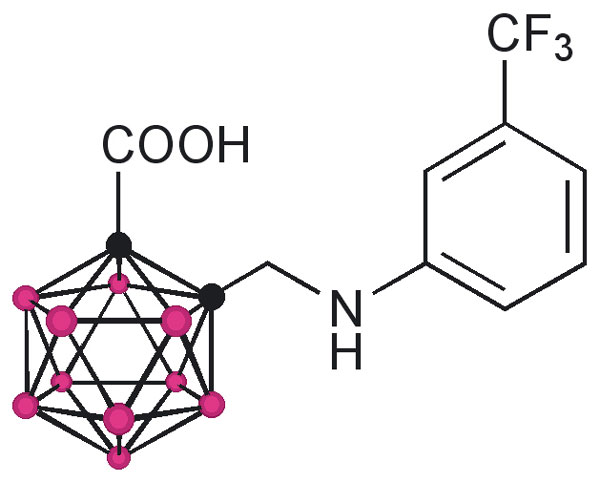
The design of the structures to be synthesized was based upon a general pharmacophore composition identified as preferable through limited screening (39, 40). Analysis of these data suggests that the carboxylic acid functionality plays an important role in the binding of amyloid fibril inhibitors to TTR. The cocrystal structures between transthyretin and T4 indicate that this carboxylic acid-functionalized moiety resides near the entry of the TTR-binding channel in the outer pocket, as represented in Fig. 2 (41). The remaining hydrophobic substituted phenyl ring of T4 is tucked into the smaller inner pocket. By using this structure as a template, it was clear that it would be efficacious to directly connect the carboxylic acid functionality to the bulky carborane moiety to fully use the increased volume of the outer pocket. Continuing to use T4 as an analog, a phenyl moiety would provide van der Waals interactions within the smaller inner pocket. With this approach in mind, potential inhibitors 1–8 were synthesized (Fig. 3).
Fig. 3.

TTR amyloid inhibitors. All assays were performed at a TTR concentration of 3.6 μM. Inhibitor concentration and percent fibril formation are given below each inhibitor.
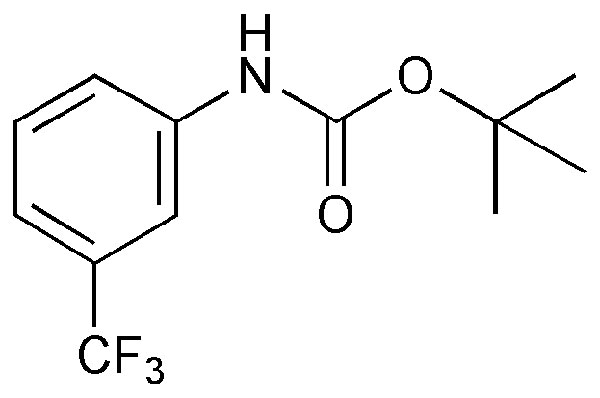
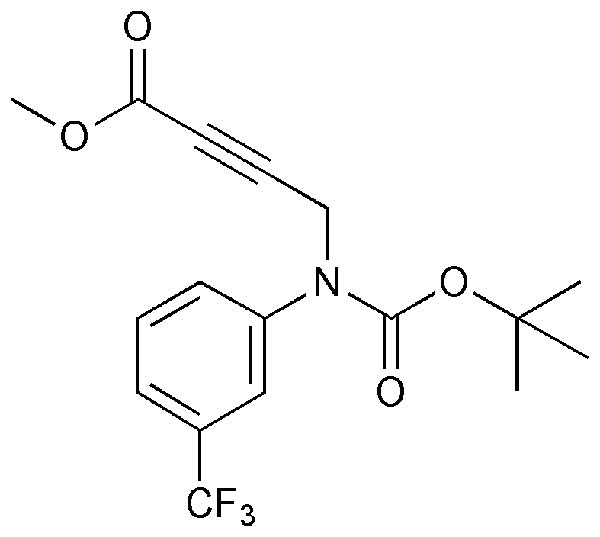
Compounds 1 and 3 were synthesized by base-mediated saponification of the corresponding methyl esters, prepared according to literature procedures (42, 43). The corresponding methyl ester of 2 produced from 4,5,7,8,9,10,11,12-octamethyl-1,2-dicarba-closo-dodecaborane, was similarly subjected to base-mediated saponification to give 2 in good yield (44). Synthesis of 5 began with the reaction of decaborane with 4-[tert-butoxycarbonyl-(3-trifluoromethyl-phenyl)-amino]-but-2-ynoic acid methyl ester [see supporting information (SI) Schemes 1–12] in refluxing toluene/acetonitrile yielding 4. Subsequent saponification of 4 produced the corresponding acid, 5, in good yield. Compounds 6 and 8 began as 1-[3-fluorophenyl]-1, 2-dicarba-closo-dodecaborane and 1-[3-fluorophenyl]-1,7-dicarba-closo-dodecaborane, respectively (45). Lithiation of the free C–H vertex followed by reaction with methyl chloroformate yielded the methyl esters of 6 and 8 (compound 7), which were saponified to provide the acids in good yield.
TTR Assay.
Inhibitors were evaluated by using a 72-h stagnant fibril formation assay previously described (29). In short, during this assay, physiological concentrations of TTR (3.6 μM) are subjected to acid-mediated partial denaturation at pH 4.4 either in the presence or absence of inhibitor. Fibril formation is measured by optical density at 400 nm, and the results are reported as percent fibril formation (% ff), with TTR in the absence of inhibitor defined as 100% ff. Therefore, the lower the % ff, the more efficacious the inhibitor.
A recent report has indicated that o-carborane, when possessing an electron-withdrawing group, such as carbonyl, α to a cage carbon, is prone to nucleophilic attack and degradation to the corresponding anionic B9 nido-carborane cage in the presence of wet polar organic compounds (46). Consequently, 1H and 13C NMR studies were performed on the methyl ester of 1 in both DMSO and acetone. Consistent with the previous report, the sample, when dissolved in DMSO, evolved a gas (presumably hydrogen, consistent with conversion to nido-carborane) in the NMR tube and showed near quantitative conversion to the nido derivative within the time required to obtain a spectrum. Fortunately, the sample prepared with acetone solvent showed no indication of degradation even after 4 h (results not reported). To demonstrate identical results with TTR assays of flufenamic acid diluted in both DMSO and acetone, analyses were performed by using both solvents to dissolve flufenamic acid, and the results were found to be strictly analogous (results not reported).
Inhibitors, positive (flufenamic acid, a known potent inhibitor) and negative controls were run in triplicate concurrently with each group of three to four compounds. The results for both of these controls were found to be quite consistent. The negative control, TTR in the absence of inhibitor, produced an OD of 0.98 ± 0.04 at 400 nm over 12 trials. Similarly, the positive control, TTR in the presence of 3.6 μM flufenamic acid, yielded 14 ± 4% ff, again over a dozen trials. Inhibitors 1–8 were synthesized to give a reasonably varied collection of structures from which promising lead compounds could be identified. The TTR assay results for inhibitors 1–8 are shown in Fig. 3. In all cases, these compounds conform to the previously expounded theory regarding the design of TTR amyloid inhibitors (39, 40).
Compounds 1 and 2 differ only in size and were chosen to give a qualitative estimate of the steric constraints imposed by the TTR-binding channel upon the design of new inhibitors. Whereas 1 was proven to be a moderate inhibitor, 46% ff and 21% ff at 3.6 and 7.2 μM, respectively, 2 exhibited poor inhibition, 72% ff at the higher 7.2 μM concentration. Furthermore, the superior potency of 1 compared with that of 3 substantiated the preference of a carboxylic acid functionality α- (as opposed to β-) with respect to the aromatic platform in agreement with previous studies (39). These results were encouraging; they definitively indicated that the carborane skeletal structure was able to enter the TTR-binding channel and inhibit dissociation as well as validate the suggestion that the design of carborane-containing TTR inhibitors would follow known theory. This enabled the focus of inhibitor designs upon carborane analogs of previously identified inhibitors, with the reasonable expectation that similar efficacy could be achieved: thus, the synthesis of compounds 4 and 5, which can be viewed as carborane analogs of flufenamic acid, as well as 6–8, which share structural similarities with diflunisal. Assays of 4 and 7 indicated that these methyl esters, unable to interact electrostatically with the protonated ammonium groups on the two symmetry related Lys-15 located at the mouth of the binding channel, were poor inhibitors of amyloid formation at all concentrations. However, anions of the corresponding carboxylic acids, 5 and 8, capable of interacting with the ammonium group, were both found to be potent inhibitors of aggregation. Whereas 6 exhibited poor inhibition, it affords the opportunity for comparison with both 5 and 8. Inhibitor 8 is the meta isomer of 6, which provides a more linear configuration, maximizing interactions between its carboxylate functionality and the ammonium groups of Lys-15. Similarly, whereas 5 shares being an ortho isomer with 6, the increased degrees of freedom imparted through the aminomethyl spacer of 5 allows a conformation that again would maximize the electrostatic interactions. In summary, both 5 and 8 have been identified as potent inhibitors of TTR amyloid formation, on the order of the known inhibitor, flufenamic acid.
Further testing with compounds 1, 5, and 8 was initiated to verify that these compounds inhibited aggregation in a manner similar to that of the known inhibitor, flufenamic acid. All compounds exhibited dose-dependent effectiveness, as shown in Fig. 4, with IC50 values of 4.4, 2.6, 2.1, and 2.9 μM for 1, 5, 8, and flufenamic acid, respectively. As these compounds are presumed to inhibit TTR fibril formation through raising the tetramer dissociation barrier through ground-state stabilization, good inhibitors of fibril formation should slow tetramer dissociation and thereby prevent aggregation, leading to fibril formation (47). Therefore, the rate of fibril formation was monitored by turbidity at a final pH of 4.4 over 120 h (Fig. 5), demonstrating a slowing of aggregation proportional to the efficacy shown by the inhibitors. Compounds 5 and 8 consistently showing decreased turbidity at all time points as compared with 1, as would be expected. In summary, both 5 and 8 have been identified as potent inhibitors of TTR amyloid formation, on the order of the known inhibitor, flufenamic acid.
Fig. 4.

Stagnant TTR (3.6 μM) amyloid formation assay (pH 4.4) with compounds 1, 5, and 8 and flufenamic acid (FLU) at various concentrations over 72 h.
Fig. 5.

TTR (3.6 μM) fibril formation (pH 4.4) in the presence of 3.6 μM inhibitors 1, 5, 8, and flufenamic acid (FLU) plotted with WT-TTR control.
COX-1/COX-2 Assays.
Three TTR inhibitors were chosen to be screened for cyclooxygenase activity, compounds 1, 5, and 8. Compounds 5 and 8 were chosen because of their effectiveness in inhibiting TTR amyloid aggregation whereas 1 was chosen to establish a baseline of inhibition, which could be attributed to the carborane carboxylic acid moiety itself. Inhibition of COX-1 (ovine) and COX-2 (human recombinant) was performed by using a commercially available enzyme immunoassay according to the manufacturer's instructions. Stock solutions of compounds 1, 5, and 8 were dissolved in absolute ethanol. As in the case of the TTR assay stock solutions, there was concern that dissolution of the compounds in polar organic solvents might lead to degradation of their carborane cages. However, 11B NMR spectra of ethanol solutions of 1 showed no perceptible degradation after 24 h (results not reported). The compounds were initially screened at 100 μM to give the results reported in Table 1. The cyclooxygenase active site of both COX-1 and COX-2 is located at the end of a long hydrophobic channel that is broad near the membrane-binding domain (the lobby) and narrows as it extends toward the active site (48). A network of hydrogen bonds involving Arg-120, Tyr-355, His-513 (Arg in COX-2), and Glu-524 form a constriction, which functions as a gate for ligand entrance to the COX active site (49). [For the sake of simplicity, the numbering of amino acid residues for both COX-1 and COX-2 isoforms is based on the sheep seminal vesicle COX-1 sequences as described in ref. 50.] Additionally, the active site, itself, consists of several aromatic residues (including Phe-381, Tyr-385, and Trp-387) that can contribute to both hydrophobic and π–π interactions with the substrate (49). It was hypothesized that both the steric bulk and lack of π–π stacking of the carborane skeleton would produce a poor COX inhibitor and enhance TTR selectivity. Compound 1 exhibits minimal COX inhibition, with COX-1 retaining 83 ± 10% of initial activity and COX-2 retaining 93 ± 13%, implying that the carborane moiety is a poor substrate for COX, through either of the above mechanisms. Similarly, 8 is an exceptionally poor inhibitor of both COX isoforms, with COX-1 and COX-2 retaining 93 ± 13% and 94 ± 16% initial activity, respectively. Curiously, compound 5 is an inhibitor of COX-1 (6.0 ± 5.9% initial activity) and less so of COX-2 (57 ± 10% initial activity) at this elevated dose. Again, this difference between 5 and 8 could presumably be due to the enhanced distance between aromatic platforms leading to increased flexibility of 5 as compared with 8. Presuming that 8 is being excluded from the active site because of steric constraints at the constriction, one could conclude that a longer, more flexible linker would enable the phenyl ring of 5 to slip past this roadblock and participate in hydrophobic interactions slightly inhibiting COX.
Table 1.
TTR fibril formation and COX assay data for selected compounds
| Compound | TTR,* % ff | % Initial activity† |
|
|---|---|---|---|
| COX-1 | COX-2 | ||
| 1 | 46 ± 6 | 83 ± 10 | 93 ± 13 |
| 5 | 22 ± 2 | 6 ± 5.9 | 57 ± 10 |
| 8 | 15 ± 5 | 93 ± 13 | 94 ± 16 |
*WT-TTR (3.6 μM) fibril formation assay (pH 4.4) in the presence of 3.6 μM inhibitors.
†COX enzyme immunoassay in the presence of 100 μM inhibitors.
To directly compare the COX activity of 8 relative to flufenamic acid, a full COX inhibition curve was run with 8 and flufenamic acid (Fig. 6) by using a commercially available colorimetric COX screening assay. Inhibition assays for flufenamic acid and 8 were run for ovine COX-1 (Fig. 6A) and COX-2 (Fig. 6B) at various concentrations. Compound 8 proved to be a virtually imperceptible inhibitor of both COX-1 (IC50 > 200 μM) and COX-2 (IC50 > 100 μM) isoforms at a concentration more than an order of magnitude larger than the concentration necessary for 8 to perform exceptionally well with regard to the inhibition of TTR amyloid formation, making it an ideal lead compound for further study.
Fig. 6.

Colorimetric COX-1 (A) and COX-2 (B) inhibition assays using 8 and flufenamic acid (FLU).
Conclusion
Our results have shown that carborane analogs of NSAIDs bestow kinetic stabilization of transthyretin in vitro. The hydrophobicity, steric bulk, and lack of π-interactions make the carborane functionality an ideal scaffolding for use in the binding channel of TTR. Similarly, these same properties are detrimental when applied to cyclooxygenase inhibitors. Consequently, we have shown that substitution of a carborane moiety for a phenyl ring in NSAIDs with known TTR activity retains the TTR potency of the compound while dramatically reducing the detrimental COX activity. The unique properties noted above as well as their biological and chemical stability make carboranes very attractive pharmacophores in the design and synthesis of potent and selective pharmaceuticals.
Materials and Methods
Synthesis of Inhibitors.
The synthesis and characterization of compounds 1–8 are described in SI Materials and Methods. Flufenamic acid was used as purchased (Lancaster Synthesis, Pelham, NH).
TTR Cloning.
The transthyretin cDNA was amplified by PCR by using a human cDNA clone obtained from ATCC (American Type Culture Collection, Manassas, VA) as the template. The primers GGGGCCATGGCTGGCCCTACGGGCACCGGTG and GG- GGCTCGAGATTATTCCTTGGGATTGGTGACGACAG-CCGTGGTGG were used to amplify the entire gene as well as to introduce NcoI and XhoI sites at the 5′ and 3′ ends of the product, respectively. The PCR products were cloned into pCRBluntII-TOPO (Invitrogen, Carlsbad, CA) cloning vectors. After sequence confirmation, the gene was subcloned into pETM-11 (EMBL). Again, the construct was confirmed by nucleotide sequencing. This construct added a methionine and an alanine to the N terminus of the protein as well as a hexa-histidine tag.
Expression, Purification, and Characterization.
Human transthyretin was expressed in BL21–Gold (DE3) cells grown in a shake flask in LB media with 0.1 mM isopropyl β-d-thiogalactoside (IPTG) induction for 16 h. Cells were harvested by centrifugation. Each gram of cell pellet was resuspended and lysed in 5 ml of lysis buffer (20 mM Tris, pH 8.0/0.3 M NaCl/10 mM imidazole/2 mM 2-mercaptoethanol (β-ME)/2 μg/ml DNase I/0.2 mg/ml lysozyme/1:100 protease inhibitor mixture; Sigma, St. Louis, MO). The lysate was clarified by centrifugation at 27,000 × g for 30 min. The soluble, recombinant protein was purified by binding to Ni-NTA Superflow resin (Qiagen, Valencia, CA) and elution with 300 mM imidazole. The purified protein was ≥99% pure, estimated by SDS/PAGE. Mass spectrometry confirmed the molecular weight of the recombinant protein and the purity of the sample.
TTR Assay.
Stagnant assay.
Concentrated stock solutions of the inhibitors tested were freshly prepared by dissolving the compounds in acetone. The vibration-free amyloid fibril inhibition assay was performed as described (29) with the following changes. A series of microfuge tubes were prepared, each containing 490 μl of a 200 mM sodium acetate-buffered (pH 4.2, 100 mM KCl, 1 mM EDTA) solution. To these tubes was added 500 μl of a 10 mM phosphate-buffered (pH 7.4, 100 mM KCl, 1 mM EDTA) solution containing 7.2 μM tetrameric TTR to yield a final pH of 4.4 and TTR concentration of 3.6 μM. A 10-μl aliquot of inhibitor stock solution was then added (360 or 720 μM), resulting in a final inhibitor concentration of 3.6 or 7.2 μM in a total volume of 1 ml. The Eppendorf tubes were then incubated while motionless at 37°C for 72 h, after which the extent of fibril formation was measured by OD at 400 nm. Results are reported as percent fibril formation, essentially the ratio of the ODs for TTR in the presence of inhibitor to TTR in the absence of inhibitor. Inhibitors were generally assayed in groups of three to four at both concentrations and in triplicate. In addition, three controls were also included with each group. A positive control of protein with flufenamic acid, known to be a potent inhibitor of TTR amyloid formation, as well as a negative control of protein in the absence of inhibitor were both assayed in triplicate in addition to a sample of inhibitor alone to give the results shown in Fig. 3.
Concentration vs. inhibition assay.
Samples were prepared identical to the stagnant assay above with the following change. Inhibitors 1, 5, 8, and flufenamic acid were serially diluted to yield final concentrations of 0.36, 1.8, 3.6, 7.2, and 10.8 μM. Protein concentration remained at 3.6 μM, and turbidity measurements were again taken after 72 h of incubation at 37°C to provide the results shown in Fig. 4.
Time course assay.
Samples were prepared according to the protocol described for the stagnant assay above with the following changes. Compounds 1, 5, and 8 and flufenamic acid were diluted to provide final concentrations of 3.6 μM, equimolar to the protein concentration. Turbidity measurements were then performed at 12, 25, 48, 72, 96, and 120 h in triplicate. Samples at each time point were vortexed and measured as described above and then discarded, providing a separate set of samples for each time point shown in Fig. 5.
COX-1/COX-2 Assay.
COX enzyme immunoassay.
Compounds 1, 5, and 8 were screened for their ability to inhibit COX-1 and COX-2 by using a commercially available COX inhibitor screening assay kit (Catalog No. 560131; Cayman Chemical, Ann Arbor, MI) with provided reagents and according to the manufacturer's instructions. Stock solutions of the test compounds were made by dissolving a weighed amount of each inhibitor in absolute ethanol to yield final inhibitor concentrations of 100 μM and then assayed to provide the results reported in Table 1.
COX colorimetric assay.
Compound 8 and flufenamic acid were subjected to a full inhibition curve versus COX-1 and COX-2 by using a commercially available colorimetric COX inhibitor screening assay kit (Catalog No. 760111; Cayman Chemical, Ann Arbor, MI) with provided reagents and according to the manufacturer's instructions. Flufenamic acid and 8 were serially dissolved in ethanol to yield a spectrum of final concentrations and assayed, providing the results shown in Fig. 6.
Supplementary Material
Acknowledgments
We thank Dr. Gunter Stier (European Molecular Biology Laboratory, Heidelberg, Germany) for pET-M11; and Dr. Beth Lazazzera and Rebecca Terra (Department of Microbiology, Immunology, and Molecular Genetics, University of California, Los Angeles) and Jim Bixby (University of Missouri) for assistance with and use of their microplate readers. We thank the National Science Foundation (NSF) (Grant CHE-0515928; Equipment Grants CHE-9871332, CHE-98713320, CHE-9610080, CHE-9808175, CHE-9974928, and CHE-0078299) for their support of this research as well as for a NSF Graduate Research Fellowship (to O.K.F.). This work has been further supported by the Department of Energy (L.J.P.) and the National Institutes of Health (L.J.P.).
Abbreviations
- TTR
transthyretin
- FAP
familial amyloid polyneuropathy
- NSAID
nonsteroidal antiinflammatory drug
- COX
cyclooxygenase % ff percent fibril formation.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0700316104/DC1.
References
- 1.Grimes RN. Carboranes. New York: Academic; 1970. [Google Scholar]
- 2.Zheng Z, Jiang W, Zinn AA, Knobler CB, Hawthorne MF. Inorg Chem. 1995;34:2095–2100. [Google Scholar]
- 3.Valliant JF, Guenther KJ, King AS, Morel P, Schaffer P, Sogbein OO, Stephenson KA. Coor Chem Rev. 2002;232:173–230. [Google Scholar]
- 4.Bregradze VI. Chem Rev. 1992;92:209–233. [Google Scholar]
- 5.Hawthorne M. F. Angew Chem Int Ed Engl. 1993;32:950–984. [Google Scholar]
- 6.Cígler P, Kožíšek M, Řezáčová P, Brynda J, Otwinowski Z, Pokorná J, Plešek J, Grüner B, Dolečková-Marešová L, Máša M, Sedláček J, Boden J, Kräusslich H-G, Král V, Konvalinka J. Proc Natl Acad Sci USA. 2005;102:15394–15399. doi: 10.1073/pnas.0507577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Debnath AK, Jiang SB, Strick N, Lin K, Kahl SB, Neurath AR. Med Chem Res. 1999;9:267–275. [Google Scholar]
- 8.Endo Y, Iijima T, Yamakoshi Y, Yamaguchi M, Fukasawa H, Shudo K. J Med Chem. 1999;42:1501–1504. doi: 10.1021/jm9900725. [DOI] [PubMed] [Google Scholar]
- 9.Ohta K, Iijima T, Kawachi E, Kagechika H, Endo Y. BioMed Chem Lett. 2004;14:5913–5918. doi: 10.1016/j.bmcl.2004.09.035. [DOI] [PubMed] [Google Scholar]
- 10.Fischli W, Leukart O, Schwyzer R. Helv Chim Acta. 1977;60:959–963. doi: 10.1002/hlca.19770600326. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto K, Endo Y. BioMed Chem Lett. 2001;11:2389–2392. doi: 10.1016/s0960-894x(01)00438-3. [DOI] [PubMed] [Google Scholar]
- 12.Nachman RJ, Teal PET, Radel PA, Holman GM, Abernathy RL. Peptides. 1996;17:747–752. doi: 10.1016/0196-9781(96)00111-8. [DOI] [PubMed] [Google Scholar]
- 13.Valliant JF, Schaffer P, Stephenson KA, Britten JF. J Org Chem. 2002;67:383–387. doi: 10.1021/jo0158229. [DOI] [PubMed] [Google Scholar]
- 14.Tsuji M, Koiso Y, Takahashi H, Hashimoto Y, Endo Y. Biol Pharm Bull. 2000;23:513–516. doi: 10.1248/bpb.23.513. [DOI] [PubMed] [Google Scholar]
- 15.Blake CCF, Geisow MJ, Swan IDA, Rerat C, Rerat B. J Mol Biol. 1974;88:1–12. doi: 10.1016/0022-2836(74)90291-5. [DOI] [PubMed] [Google Scholar]
- 16.Blake CCF, Geisow MJ, Oatley SJ, Rerat B, Rerat C. J Mol Biol. 1978;121:339–356. doi: 10.1016/0022-2836(78)90368-6. [DOI] [PubMed] [Google Scholar]
- 17.Peterson PA. J Biol Chem. 1971;246:34–43. [PubMed] [Google Scholar]
- 18.Costa PP, Figueira AS, Bravo FR. Proc Natl Acad Sci USA. 1978;75:4499–4503. doi: 10.1073/pnas.75.9.4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saraiva MJ. Human Mutat. 1995;5:191–196. doi: 10.1002/humu.1380050302. [DOI] [PubMed] [Google Scholar]
- 20.Gustavsson A, Jhar H, Tobiassen R, Jacobson DR, Sletten K, Westermark P. Lab Invest. 1995;73:703–708. [PubMed] [Google Scholar]
- 21.Westermark P, Sletten K, Johansson B, Cornwell GG., III Proc Natl Acad Sci USA. 1990;87:2843–2845. doi: 10.1073/pnas.87.7.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCutchen SL, Lai Z, Miroy GJ, Kelly JW, Colon W. Biochemistry. 1995;34:13527–13536. doi: 10.1021/bi00041a032. [DOI] [PubMed] [Google Scholar]
- 23.Hammarstrom P, Sekijima Y, White JT, Wiseman RL, Lim A, Costello CE, Atland K, Garzuly F, Budka H, Kelly JW. Biochemistry. 2003;42:6656–6663. doi: 10.1021/bi027319b. [DOI] [PubMed] [Google Scholar]
- 24.Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, Balch WE, Kelly JW. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 25.Olofsson BO, Backman C, Karp K, Suhr OB. Transplantation. 2002;73:745–751. doi: 10.1097/00007890-200203150-00015. [DOI] [PubMed] [Google Scholar]
- 26.Kelly JW. Curr Opin Struct Biol. 1996;6:11–17. doi: 10.1016/s0959-440x(96)80089-3. [DOI] [PubMed] [Google Scholar]
- 27.Kelly JW, Lansbury PTJ. Amyloid. 1994;1:186–205. [Google Scholar]
- 28.Colon W, Kelly JW. Biochemistry. 1992;31:8654–8660. doi: 10.1021/bi00151a036. [DOI] [PubMed] [Google Scholar]
- 29.Miroy GJ, Lai Z, Lashuel H, Peterson SA, Strang C, Kelly JW. Proc Natl Acad Sci USA. 1996;93:15051–15056. doi: 10.1073/pnas.93.26.15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamilton JA, Benson MD. Cell Mol Life Sci. 2001;58:1491–1521. doi: 10.1007/PL00000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson SM, Wiseman RL, Sekijima Y, Green NS, Adamski-Werner SL, Kelly JW. Acc Chem Res. 2005;38:911–921. doi: 10.1021/ar020073i. [DOI] [PubMed] [Google Scholar]
- 32.Marnett LJ, Kalgutkar AS. Trends Pharmacol Sci. 1999;20:465–469. doi: 10.1016/s0165-6147(99)01385-1. [DOI] [PubMed] [Google Scholar]
- 33.Wallace J. Best Pract Res Clin Gastroenterol. 2001;15:691–703. doi: 10.1053/bega.2001.0229. [DOI] [PubMed] [Google Scholar]
- 34.Cryer B. Gastroenterol Clin North Am. 2001;30:877–894. doi: 10.1016/s0889-8553(05)70218-1. [DOI] [PubMed] [Google Scholar]
- 35.Mukherjee D, Nissen SE, Topol EJ. J Am Med Assoc. 2001;286:954–959. doi: 10.1001/jama.286.8.954. [DOI] [PubMed] [Google Scholar]
- 36.Petrassi HM, Johnson SM, Purkey HE, Chiang KP, Walkup T, Jiang X, Powers ET, Kelly JW. J Am Chem Soc. 2005;127:6662–6671. doi: 10.1021/ja044351f. [DOI] [PubMed] [Google Scholar]
- 37.Green NS, Foss TR, Kelly JW. Proc Natl Acad Sci USA. 2005;102:14545–14550. doi: 10.1073/pnas.0501609102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hood WF, Gierse JK, Isakson PC, Keifer JR, Kurumbail RG, Seibert K, Monahan JB. Mol Pharmacol. 2003;63:870–877. doi: 10.1124/mol.63.4.870. [DOI] [PubMed] [Google Scholar]
- 39.Barnes PW, Oza VB, Peterson SA, Kelly JW. Bioorg Med Chem. 1999;7:1339–1347. doi: 10.1016/s0968-0896(99)00066-8. [DOI] [PubMed] [Google Scholar]
- 40.Barnes PW, Peterson SA, Kelly JW. Bioorg Med Chem. 1998;6:1389–1401. doi: 10.1016/s0968-0896(98)00130-8. [DOI] [PubMed] [Google Scholar]
- 41.Wojtczak A, Cody V, Luft JR, Pangborn W. Acta Crystallogr D. 1996;52:758–765. doi: 10.1107/S0907444996003046. [DOI] [PubMed] [Google Scholar]
- 42.Heying TL, Ager JW, Jr, Clark SL, Mangold DJ, Goldstein HL, Hillman R, Polak RJ, Szymanski JW. Inorg Chem. 1963;2:1089–1092. [Google Scholar]
- 43.Heying TL, Ager JW, Jr, Clark SL, Alexander RP, Papetti S, Reid JA, Trotz SI. Inorg Chem. 1963;2:1097–1102. [Google Scholar]
- 44.Herzog A, Maderna A, Harakas GN, Knobler CB, Hawthorne MF. Chem Eur J. 1999;5:1212–1217. [Google Scholar]
- 45.Hawthorne MF, Berry TE, Wegner PA. J Am Chem Soc. 1965;87:4746–4750. doi: 10.1021/ja00949a014. [DOI] [PubMed] [Google Scholar]
- 46.Schaeck JJ, Kahl SB. Inorg Chem. 1999;38:204–206. [Google Scholar]
- 47.Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Science. 2003;299:713–716. doi: 10.1126/science.1079589. [DOI] [PubMed] [Google Scholar]
- 48.Kurumbail RG, Keifer JR, Marnett LJ. Curr Opin Struct Biol. 2001;11:752–760. doi: 10.1016/s0959-440x(01)00277-9. [DOI] [PubMed] [Google Scholar]
- 49.Llorens O, Perez JJ, Palomer A, Mauleon D. Bioorg Med Chem Lett. 1999;9:2779–2784. doi: 10.1016/s0960-894x(99)00481-3. [DOI] [PubMed] [Google Scholar]
- 50.Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning JD, Seibert K, et al. Nature. 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}