

Abstract
The utility of ion mobility spectrometry (IMS) for separation of mixtures and structural characterization of ions has been demonstrated extensively, including in the biological and nanoscience contexts. A major attraction of IMS is its speed, several orders of magnitude greater than that of condensed-phase separations. Nonetheless, IMS combined with mass spectrometry (MS) has remained a niche technique, substantially because of limited sensitivity resulting from ion losses at the IMS-MS junction. We have developed a new electrospray ionization (ESI)-IMS-QToF MS instrument that incorporates electrodynamic ion funnels at both front ESI-IMS and rear IMS-QToF interfaces. The front funnel is of the novel “hourglass” design that efficiently accumulates ions and pulses them into the IMS drift tubes. Even for drift tubes of two meter length, ion transmission through IMS and on to QToF is essentially lossless across the range of ion masses relevant to most applications. The RF ion focusing at the IMS terminus does not degrade IMS resolving power, which exceeds 100 (for singly-charged ions) and is close to the theoretical limit. The overall sensitivity of present ESI-IMS-MS system is comparable to that of commercial ESI-MS, which should make IMS-MS suitable for analyses of complex mixtures with ultra-high sensitivity and exceptional throughput.
Introduction
Ion mobility spectrometry (IMS) had been known since 1970s as a tool for analysis and characterization of gas-phase ions.1–5 In particular, IMS has been used to separate mixtures6–9 (including isomeric mixtures8,9), detect and quantify the presence of specific analytes,10–12 and characterize the molecular structure of novel chemical species.2,4,13–20
IMS exploits the fact that different particles diffuse through a gas at different speeds, depending on their collision cross sections with the gas molecules. While neutrals diffuse by random Brownian motion, ions in an electric field drift in a defined direction with the velocity controlled by their mobility (K). This quantity varies with the field intensity E in general, but IMS is typically run in a low-field regime where K(E) is nearly constant. In that limit, K depends on the same ion/buffer gas cross section21,22 (rigorously the first-order orientationally averaged collision integral ). This allows a spatial separation of different ions, including structural isomers.
At the simplest, an IMS instrument consists of a tube filled with a non-reactive buffer gas (commonly He or N2), a generally uniform electric field established along the axis, and a charge/ion detector at the end. Separated species are characterized by their different mobilities, derived from the measured drift times by adjusting for E, gas number density N, and tube length L. The technology has been widely deployed to monitor the environmental air and water, track industrial processes and ensure product quality, and detect explosives or chemical warfare agents in security and defense applications.1,23–27 However, until the 1990s IMS was primarily perceived as a relatively inexpensive, low-power, compact alternative to mass-spectrometry (MS) suitable for field analyses.
The interest in coupling IMS to MS has been increasing over the last decade. This was first implemented using quadrupole MS,28,29 which necessitated either measuring separated IMS spectra for each m/z value of interest or, alternatively, scanning the m/z for each peak of interest selected from the IMS spectrum. However, real-world bioanalytical applications were limited because of low throughput and insufficient sensitivity. This situation changed with the coupling of IMS to time-of-flight (ToF) MS.30–34 The IMS-ToF combination permits the essentially simultaneous dispersion of mixture components in two dimensions: ion mobility and m/z. Since an IMS separation typically requires ~1 to 100 ms and has the resolving power of 10–200, a single species in an IMS ion packet (i.e. peak) often appears over a ~ 0.1–1 ms period. A typical ToF cycle is ~ 30–100 μs long, so one or more mass spectra can be obtained during the “elution” of an IMS peak. This enables the 2-D analysis of an ion mixture with potentially perfect ion utilization efficiency. An IMS-MS system may be enhanced to address greater levels of sample complexity by addition of an upstream condensed-phase separation such as liquid chromatography (LC), creating a 2-D separation followed by MS analysis.7,33,34 Characteristic timescales of LC and IMS also match well: a single LC peak normally elutes over 5–30 s, allowing for ~ 102–104 IMS-MS analyses.
IMS can be comparable to condensed-phase separations in resolution, but greatly superior in speed. That is, the resolving power (R) of IMS32,35,36 can exceed 100 (and 200 for multiply-charged ions32,37 that are typical of peptides from tryptic digests). However, an IMS separation requires <100 ms, while current condensed-phase separations require orders of magnitude longer. Thus, IMS may provide the basis for increasing the throughput of analyses, which is generally crucial for real proteomics, metabolomics, and other biological studies involving highly complex samples. For multidimensional separations involving IMS, for example, a LC run with peak capacity of ~ 50 in 2–3 min38 can potentially provide a combined LC-IMS separation peak capacity of >1000 on the same timescale. In comparison, a 2-D liquid-phase strong-cation exchange and reversed phase LC (MudPIT)39,40 separation with peak capacity of ~1000 to 3,000 typically requires over 24 hrs., and 2-D gel electrophoresis41 with peak capacity of 103–104 requires ~ 10 hrs.
Despite an enormous potential for high-throughput analyses of complex samples, the application of IMS-MS (and LC-IMS-MS) has been limited by low sensitivity arising primarily from ion losses at the IMS-MS interface. Ion packets traveling through the IMS experience thermal diffusion42 and (at high ion currents) Coulomb expansion43 superposed on the directed drift motion, which produces spatial broadening. The component of this broadening along the drift axis is responsible for the finite IMS resolution. The broadening perpendicular to that axis limits the fraction of ions that exit IMS, since the exit aperture is generally much smaller than the size of arriving ion packets. The vacuum needed for the subsequent MS stage and the relatively high pressure required by IMS constrain the practical diameter of the aperture connecting these stages (Θ), which results in large ion losses. To estimate them, we assume an isotropic broadening of ion packets in IMS. (In principle, an electric field renders the broadening anisotropic, preferentially augmenting the component along the drift axis.44 In the low-field regime typical for IMS, this effect is small.) Then the spherical diameter of ion packet at the IMS end W0 (the full width at half maximum - FWHM) equals its width along the drift axis. By the definition of R, W0 = L/R, and the area of ion impact on the IMS exit is ~ (π/4)(L/R)2. Since in practical IMS designs L/R >>Θ , the ion transmission efficiency (S) through IMS is given by:
| (1) |
At low E and ignoring space-charge effects, R is ideally45
| (2) |
where tP is the (temporal) width of initial packet, tD is the drift time, and Rd is the maximum IMS resolving power limited only by thermal diffusion:
| (3) |
where ze is the ionic charge, kb is the Boltzmann constant, and T is the gas temperature. (Eq. (2) is rigorous for initially spherical ion packets only.) In the limit of R ⇒ Rd, eqs. (1) and (3) yield:
| (4) |
That is, when the injected ion packet is very “thin” compared to the drift time and small compared to the broadening by IMS terminus, IMS-MS sensitivity is inversely proportional to the IMS resolving power squared. This remains approximately true for finite initial packets in realistic IMS designs, where the S(R) dependence is derived by combining eqs. (1) and (2). This circumstance had severely limited the practically achievable IMS resolution and peak capacity.
For instance, an ion packet is ~ 5 mm in diameter at the exit of 76-mm long drift tube29 in one MS-IMS-MS design operating at a pressure of P ~ 5 Torr (and providing R ~ 15). In that instrument Θ = 0.25 mm, and according to eq. (4) <1% of the ions traversing the IMS are transmitted to the following MS stage. An IMS resolving power of 15 is inadequate for many applications. Higher-resolution IMS-MS instruments require drift tubes that are longer (and thus yield broader ion packets) and/or operate at higher gas pressures (which requires even narrower apertures), thus ion transmission efficiency is even lower. For example, an instrument36 with R ~ 170 (L = 63 cm and Θ = 0.12 mm) operated at P ~ 500 Torr would have ~ 99.9% of ions projected to be lost at the exit aperture. Eq. (4) may still underestimate losses, as it ignores the finite initial size of ion packets and additional broadening by Coulomb repulsion, though this is somewhat balanced by (also neglected) gas dynamic effects near the IMS exit aperture that increase its effective diameter. Regardless, ion losses are substantial.
An IMS-MS interface, similar to the ESI-MS interface, should efficiently transmit incoming ions through a narrow aperture that maintains a large pressure differential. In both custom46 and commercial47 instruments, electrospray ionization (ESI) and other API sources are commonly coupled to MS analyzers using differentially-pumped orifice (or capillary)-skimmer cone (OSC) arrangements. Differential pumping between IMS and MS allows a wider IMS exit aperture and can improve ion transmission. In fact, an OSC design at the IMS terminus4,33,48–50 has been shown to improve IMS-MS sensitivity48 by a factor of 5–10. Considering the ion transmission of < 1% in earlier designs, this improvement still implies > 90–95% ion loss at the IMS-MS interface. Indeed, Clemmer and coworkers4 have pointed out that “although improvements in sensitivity have been demonstrated, the current technologies are still not as sensitive as the well-developed MS-MS strategies, however we believe that much of this difference will be diminished as additional improvements in the instruments are made.”
The device of choice for efficiently conveying ions from regions of moderately high pressure (~ 1–10 Torr) into vacuum is the electrodynamic ion funnel.51–53 The ion funnel consists of a stack of insulated electrodes carrying an axial DC gradient and a harmonic RF potential applied to adjacent electrodes with opposite phases. Each electrode features a circular aperture that narrows along the stack. An ion packet entering the funnel via its widest aperture is pushed through by the DC gradient, while confined radially by the RF field. The entrance aperture may be as large as 5 cm (or more). The small exit aperture, generally ~2 mm, provides a tight focusing of those beams through a DC-only orifice immediately following the funnel. The exceptional performance of ion funnels in ESI-MS (including quadrupole, ToF, and FTICR) interfaces has been demonstrated extensively51–56, achieving for example, zeptomole-sensitivity of ESI-FTICR.54 In a direct comparison, an ion funnel ESI-MS interface55 has provided over an order of magnitude sensitivity improvement over a conventional OSC design. The gain at the ESI-IMS interface should be comparable, and that at IMS terminus should be even greater due to a near-absence of gas dynamic disturbances. (In ESI-MS interfaces, the jet exiting the heated capillary expands inside the funnel, creating strong gas flows that can reduce the electrodynamic ion confinement and degrade efficiency.) Most importantly, an ion funnel can transmit close to 100% of incoming analyte ions over a broad m/z range,56 potentially providing the basis for nearly lossless ion transmission and much more sensitive IMS-MS instrumentation.
Here we describe a new ESI-IMS-QToF instrument incorporating ion funnels at both ESI-IMS and IMS-QToF interfaces. Despite the drift tube length of 210 cm, we show that the ion transmission through the IMS is near-perfect across a broad m/z range.
Experimental
The ESI-IMS-QToF instrument (Fig. 1) comprises three major components: ESI source, IMS drift tube, and QToF MS that analyses ion packets separated by IMS. The work reported here has used positive ions produced by ESI and the voltages on all elements are given accordingly. To analyze anions, all polarities would have to be reversed.
Fig. 1.
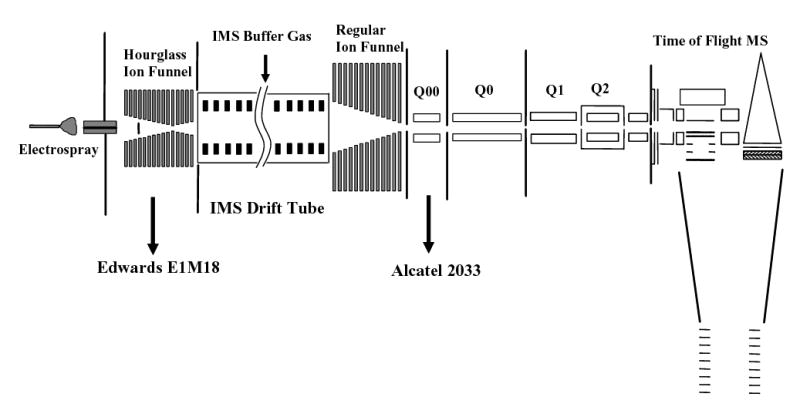
Design of the ESI-IMS-QToF interfaced using electrodynamic ion funnels.
ESI source
The source consists of an ESI emitter followed by a heated capillary inlet (both standard in modern ESI-MS) and a novel “hourglass” ion funnel for pulsed ion transmission described below. The emitter is mounted on an X-Y translation stage allowing fine position adjustment with respect to the inlet. A micro syringe driven by a pump (kd Scientific, Holliston, MA) was used to infuse samples to the ESI emitter through a metal union. The ESI source is electrically floated above the IMS voltage (see below) using adjustable RF and DC power supplies; all voltages stated in this subsection are referenced to the IMS voltage. The union and emitter are at a potential of 2–2.5 kV above the inlet. The ions generated by ESI are sampled into an inlet capillary (0.43 mm i.d. and 64 mm long) heated by two cartridge heaters.56 The temperature (~120 °C) is monitored by thermocouples and regulated using a close-loop control unit (Omega CN9000A). The DC bias on the capillary is 350 V. The ESI emitter voltage and position were adjusted to optimize instrumental sensitivity.
Ions exiting the heated inlet capillary enter the first hourglass-shaped ion funnel mounted inside a steel vacuum chamber floated at the voltage of the ESI inlet capillary. The chamber is mechanically pumped (Edwards E1M18, 5.7 L/s) through an insulating hose of reinforced plastic. A flow valve on the hose permits the control of chamber pressure down to ~ 1 Torr. While an ESI-MS interface typically transmits ion beams continuously,51–56 the ESI-IMS funnel must provide ions in discrete packets, and thus has been designed to accumulate and store ions between the pulses for IMS introduction. The capability for this step is incorporated between the funnel exit electrode and the adjacent ion gate. In a typical ion funnel design having a 2 mm exit aperture, this is a volume of only a few mm3. Such a design funnel has been previously used for interfacing ESI with IMS.55 However, in that case the IMS drift tube was of L = 4.5 cm only, allowing a high IMS injection frequency (1–10 kHz). That is, the funnel had to store ions for only 0.1–1 ms. The present high-resolution IMS is 210 cm long, and the funnel must accumulate and store ions for as long as ~50–100 ms between IMS pulses. The total charge accumulated from an ion beam arriving over that time can be substantial, and easily exceed the storage capacity of a volume of several mm3.
The present hourglass ion funnel design58 greatly increases the ion storage volume while maintaining the differential pumping required between the ESI source inlet chamber and IMS tube. Similar to our conventional ion funnel design,56 this device consists of 100 ring electrodes (machined from 0.5-mm thick brass) separated by 0.5-mm thick Teflon sheet spacers. These are divided into three sections: the front section of 24 electrodes with i.d. = 25.4 mm, the middle section of 42 electrodes with i.d. decreasing linearly from 25.4 mm to 2.0 mm, and the rear section of 34 electrodes with i.d. increasing linearly from 2.5 mm to 20.0 mm at the ion funnel exit to the IMS. The 2 mm aperture also serves as the conductance limit between the source chamber and the IMS drift tube. As in a normal funnel, RF and DC voltages are applied simultaneously to each electrode.56 The frequency and the (peak-to-peak) amplitude of RF are 500 kHz and 90 V, respectively. The DC biases of ~ 330 V on the first and ~ 90 V on the last funnel electrodes produce an axial DC field of ~ 24 V/cm in the funnel, which provides efficient ion transmission to the IMS stage. To disperse the expansion jet from the heated capillary exit and improve the uniformity of gas flow in the funnel, a “jet disrupter” element was incorporated at the funnel axis 20 mm from the entrance.59,60 This element consists of a metal disk (o.d. = 6.5 mm) that is insulated from the rest of the funnel, allowing an independent voltage control. The typical jet disrupter voltage is ~ 308 V.
About 2 mm after the last funnel electrode is an ion “gate” constructed of high-transmission grid (20 lines per inch, Buckbee-Mears, St. Paul, MN). Normally at 115 V, the gate confines ions to the end of the exit region of the ion funnel. To inject ions into IMS, the gate is pulsed open by a rectangular waveform that drops the grid to 42 V. The waveform is synthesized using an electrically floated generator, with the pulse frequency and duration controlled via a trigger signal sent through an optical cable. In this work, we used pulses of 50 μs duration. Between pulses, ions are accumulated in the region between the last funnel electrode and the ion gate that has a volume of ~ 600 mm3, about two orders of magnitude greater than that in the conventional funnel configuration.
The floating voltage is provided by a 60-kV DC power supply (Glassman, High Bridge, NJ) and monitored by a custom high-voltage probe with accuracy of < 0.1 %. The line AC power is fed into the floated electronic control box through a high voltage transformer (Del Electronics, Valhalla, NY) that raises the AC output to IMS voltage.
IMS drift tube
The drift tube is a modular assembly of identical units, which offers a variable IMS length and other advantages. Each unit consists of a stack of 21 ring electrodes housed in a 20-cm long cylindrical chamber (i.d = 20 cm, non-magnetic steel) with vacuum flanges on both ends. Units are electrically insulated by 1-cm thick peek disks (vacuum-sealed by O-rings) and assembled into a tube using insulating bolts and nuts (reinforced fiberglass). The disks feature center holes for ion passage and blind holes on both sides for mounting four 21-cm long solid ceramic rods (3.2 mm in diameter) that support the copper ring electrodes (i.d = 55 mm, o.d. = 80 mm) inside each unit. These electrodes are uniformly spaced and mutually insulated by tubular plastic spacers of ~ 10 mm length. All electrodes in the tube are consecutively connected by high-precision 1 MΩ resistors. The median ring of each unit is electrically shorted to its chamber, i.e. the voltages on chambers decrease along the tube in equal steps. This minimizes the voltage difference between a ring and nearest chamber wall at any point along the tube. This also minimizes the risk of electrical gas breakdown inside IMS and thus maximizes the achievable drift voltage and the obtainable resolution. The present IMS includes up to 10 units with total L = 210 cm. Further lengthening through additional units is straightforward. The DC floating voltage generated as described above is applied to the 1st electrode of front unit using a high voltage feed through on the chamber wall, while the voltage of the last electrode of back unit (normally 240 V) is set by a DC power supply referenced to ground. The resistor chain partitions the difference between those two voltages (the IMS drift voltage) across the drift tube. The front unit of IMS is interfaced to the ESI source chamber. For a smoother coupling, the first two IMS electrodes have a reduced i.d. (20 mm), the same as the aperture of last ion funnel electrode.
The second IMS-MS ion funnel operates conventionally as no ion storage step is needed. To capture all ions from the drift tube this ion funnel has an acceptance diameter of 51 mm, only slightly smaller than the IMS electrode aperture and approximately twice that of the ESI-IMS hourglass ion funnel developed here, or other ion funnels used previously for ESI-MS.51–57 Over its 84 mm funnel length, the i.d. of the 84 electrodes decreases linearly to 2.5 mm. The funnel is powered by adjustable RF and DC power supplies referenced to ground. The RF is normally 500 kHz with an (peak-to-peak) amplitude between 0 and 80 V. Typical DC biases are ~ 200 V on the first electrode and ~ 50 V on the last, creating an axial gradient of ~ 18 V/cm in the funnel. Matching this gradient to the IMS drift field may smooth the motion of ions entering the funnel. As in previous designs, following the last funnel electrode is a DC-only electrode at ~ 40 V with a 2-mm diameter orifice that serves as the IMS-MS conductance limit.
The drift tube is continuously filled with buffer gas through two gas distribution lines introduced through the back of the IMS drift tube. Any buffer gas or mixture of gases could be employed; presently we use dry N2. Needle valves installed on the distribution lines provide a fine control of gas inflow, and thus of the IMS pressure P. Two capacitance manometers (MKS, Wilmington, MA) measure the pressure in the IMS and the source chamber with 0.1% accuracy over the 0–20 Torr range. Gas must flow from the drift tube into source chamber, to prevent gases from the ESI source (here atmospheric air) from diluting the IMS buffer gas. This counter flow is also critical to prohibit volatile neutrals from entering IMS where they could become ionized via charge-transfer reactions or complex to analyte ions. Therefore the drift tube is slightly over-pressured relative to the ESI interface source chamber. This is achieved by adjusting P and/or source chamber pressure as described above.
ToF MS
The MS analysis of ion packets separated in the IMS is performed by a modified commercial QToF (Sciex Q-Star Pulsar, Concord, Canada) with MS resolving power of 6,000–10,000. At typical IMS operating pressure (P > 1 Torr), the gas outflow through the 2 mm orifice at the IMS-MS interface would significantly raise the pressure in the collisional cooling quadrupole chamber of MS (Q0 in Fig. 1). This would result in long ion transit times through that region that would unacceptably distort the IMS measurements and degrade the resolution. This issue was addressed by adding a stage of differential pumping after IMS. This has been engineered as a small chamber containing a 24-mm long quadrupole (Q00 in Fig. 1) set to pass all ions (RF only at 912 kHz and 200 V peak-to-peak). Following Q00 is a lens with 2.5-mm aperture that provides the conductance limit to MS. The DC voltages on quadrupole rods and the lens are adjustable and typically set at 35 V and 25 V, respectively. The chamber is evacuated by a mechanical pump (Alcatel 2033, 12.8 L/s). This lowers the pressure in Q00 by an order of magnitude compared to that in IMS (e.g. ~ 0.3 Torr vs. P = 4 Torr), and the gas outflow to Q0 is reduced proportionately. No significant dissociation of ions in Q00 has been observed under these conditions.
The ToF MS has a four-channel ion detector system. Ion counts from each channel stream into a related time-to-digital converter (TDC). The Q-Star TDC was upgraded to 10 GHz TDC (Ortec 9353, Oak Ridge, TN), currently the fastest commercial TDC. This however is a single-channel TDC, so the ion counts from all four detector channels were initially combined after the pre-amplifier (Fig. 2a). The timing sequences of IMS pulsing and MS data acquisition are set by an I/O control board (National Instruments PCI-6711). Both TDC and that board are installed in a PC (Dell with the Windows XP operating system). The overall instrument control software runs on the same PC.
Fig. 2.
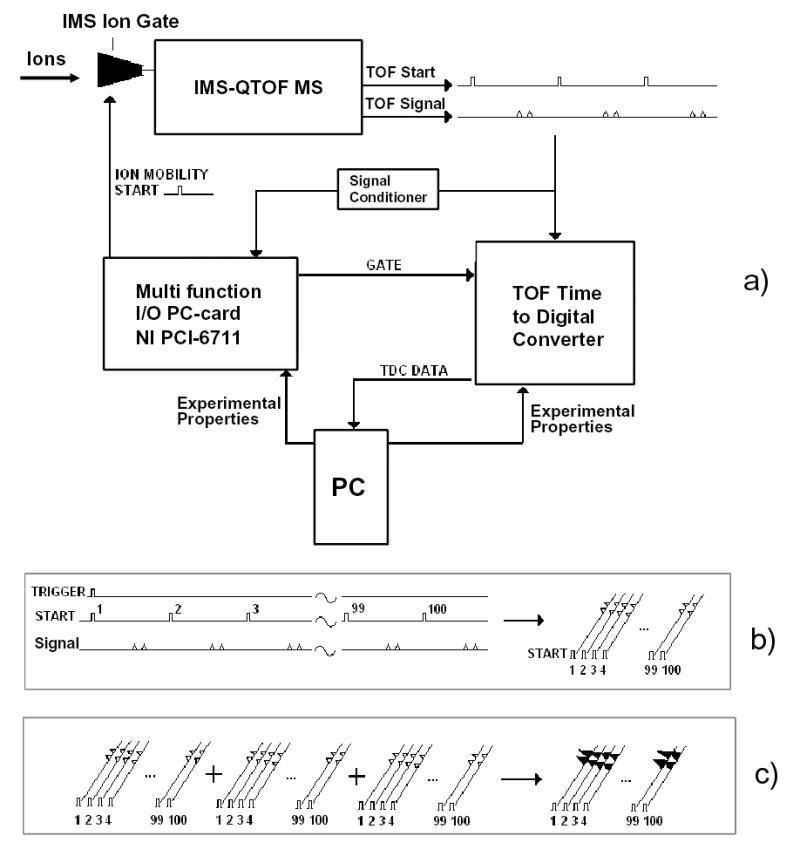
Diagram of the instrument control and data acquisition system (a), sequence of events during IMS-MS data collection (b, c).
Control software
The Q-Star data acquisition software (Sciex Analyst) does not store individual ToF spectra, but averages them over a preset period of time. This would obviously lose all information from IMS separation. The software also has no capability to control IMS operation and coordinate it with MS analyses. Hence we developed software to provide the overall instrument control, storage, and processing of the 2-D IMS-MS data. However, ToF is still operated by the Sciex Analyst resident on its original instrument PC. In particular, Analyst sets the ToF pusher period (normally in the 100–140 μs range) depending on the upper m/z limit desired. To synchronize IMS and MS operations, the IMS pulsing period is divisible by the pusher period (e.g., 500 ToF spectra lasting 100 μs each are acquired during 50 ms between IMS pulses). Beyond that constraint, the period and duration of IMS pulse are user-defined.
The software was designed in Visual Basic 6 and implemented on top of the ActiveX controls running the Ortec hardware. The system collects and stores a user-defined number of sequential 2-D maps, each derived by averaging a certain number of elementary IMS-MS frames. The acquisition of 1st frame starts from a random ToF pusher pulse (Fig. 2b). Triggered by that pulse, a call is issued to open the IMS ion gate (as described above) and simultaneously start the data acquisition process. The software then maps the ion counts recorded by the TDC onto a 2-D frame, filling a sequence of “0” and “1” in each segment of IMS drift time equal to the ToF pusher period. This continues for a pre-set integer number of periods up to the desired maximum IMS drift time (Fig. 2b). In the end, each frame pixel contains a “1” if an ion has arrived within corresponding bins of drift time and ToF flight time, and a “0” otherwise. The program then resets the experiment and starts acquiring the next frame coincidentally with a ToF pusher pulse. All following frames up to a pre-defined number (typically 100–1000) are histogrammed on top of the 1st (Fig. 2c). This process transforms a binary series in each IMS drift time segment into a ToF spectrum. This spectrum is converted into a mass spectrum using calibration and standard formulae relating m/z of ions to their flight times. A typical 2-D map generated is shown in Fig. 3a. A mass spectrum of the same sample acquired using the ToF MS data system (Fig. 3b) is clearly identical to the projection of 2-D map in Fig. 3a on the m/z axis.
Fig. 3.
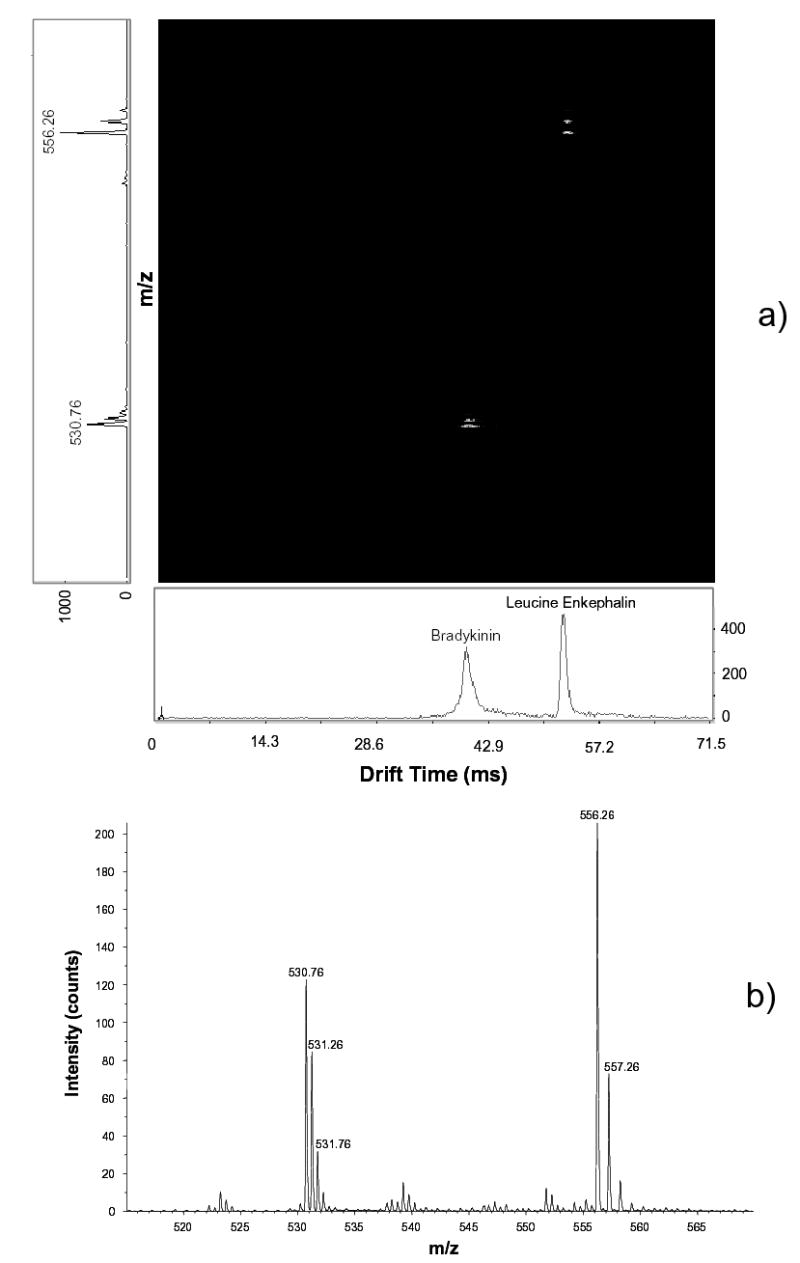
Demonstration of the data processing approach: (a) a 2-D map with projections on the drift time (horizontal) and m/z (vertical) axes; (b) the mass-spectrum obtained by Sciex Analyst for the same sample - a 1:1 mixture of Le-Enk (5 pM/μL) and Bradykinin. IMS parameters are: L = 210 cm, U = 4.76 kV, P = 4.02 Torr.
For tuning and evaluation of ion transmission efficiency, the instrument may be operated in a continuous mode. In that regime, the IMS ion gate is permanently open and ToF spectra are acquired by Analyst as usual. Peptides and other chemicals used in the instrumental evaluation are available from Sigma Aldrich.
Results and Discussion
In the low-field regime, ion mobilities are independent of the drift field. The ion mobility is derived21 from IMS measurements as
| (5) |
where U = EL is the drift voltage. Hence tD is proportional to 1/U for any IMS length and gas pressure. However, in IMS-MS one measures not tD, but the arrival time tA - the sum of times spent by an ion in all parts of the instrument between injection into IMS and detection. Previously, this was given by tA = {tD + tMS}, where tMS was the time in MS analyzer (e.g. QToF) following IMS.61 This is now modified to tA = {tD + tIF + tMS}, where tIF is the transit time through the IMS-MS ion funnel. The values of tIF and tMS are determined by conditions in respectively the funnel and QToF, and are almost independent of IMS parameters such as U or L. Thus, tA ∞ 1/U will be true only when tA ≈ tD, i.e when {tIF + tMS} << tD. The presently measured tA times are indeed linear vs. 1/U at different P, even for a shorter drift tube (L = 105 cm) and a proportionately smaller tD (Fig. 4). Hence, an ion funnel at the IMS end does not delay ions significantly and therefore does not degrade the resolution of IMS analyses.
Fig. 4.
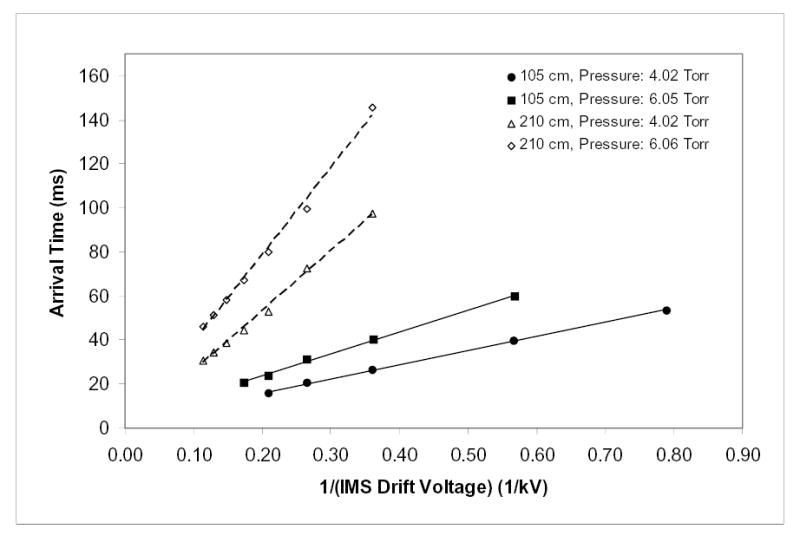
Independence of measured mobility of the IMS drift voltage under various instrumental conditions. Arrival times of Le-Enk as a function of drift voltage at two gas pressures and two drift tube lengths are as marked (10 pM/(L sample infused at 0.5 μL/min).
Lossless IMS-MS separations
As described above, ion funnels have been incorporated to our ESI-IMS-QToF design to provide more efficient ion transmission. Effectively perfect ion transmission has been achieved from the IMS to the ToF. The performance was evaluated using protonated peptide ions that cover the range of masses (m ~ 0.5–6 kDa) and charge states (z = 1–5) typical for bottom-up proteomics: Leucine Enkephalin (m = 556 amu, z = 1) - below Le-Enk, Bradykinin (m = 1061 Da, z = 2), Glufibrinopeptide B (m = 1572 Da, z = 2), and Insulin (m = 5735 Da, z = 5). In the first test, ion signals obtained for continuous operation (i.e. without IMS pulsing) were benchmarked vs. those measured on a usual ESI-QToF with no drift tube. To ensure a proper comparison, the standard OSC interface of QToF was replaced by an exact copy of the present capillary-ion funnel interface with IMS-QToF. The intensities of mass-spectra acquired on those two instruments for same samples and ESI conditions (Fig. 5a, b) differ by less than a factor of two, a customarily accepted uncertainty of such comparisons. Therefore, the cumulative ion transmission efficiency through a 147-cm long IMS stage and IMS-MS interface exceeds 50%, and likely approaches 100%. In another test, we measured ion intensities in ESI-IMS-QToF as a function of drift tube length. Combining eqs. (1) and (3), one may derive (when θ << L/R):
Fig. 5.
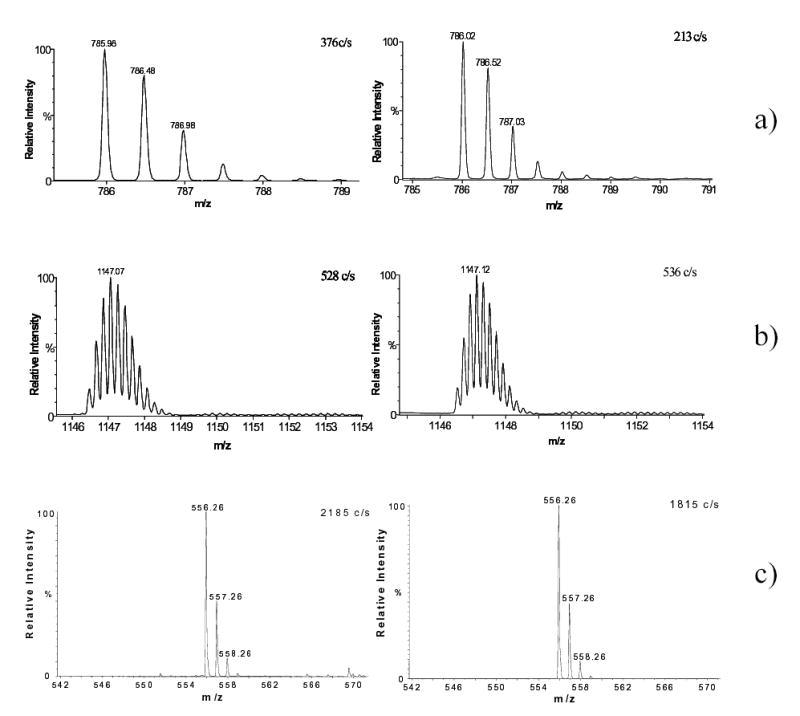
Benchmarks for different instrumental configurations prove lossless IMS transmission. In (a) and (b) are the mass-spectra for (a) (H+)2Glufibrinopeptide B (0.1 pM/μL) and (b) (H+)5Insulin (0.2 pM/μL) obtained using capillary-ion funnel interfaced with QToF (left) and IMS-QToF at L = 147 cm (right). In (c) are the mass spectra for Le-Enk (10 pM/μL) obtained using ESI-IMS-QToF at L = 105 cm (left) and L = 210 cm (right). The IMS was operated at U = 2.76 kV and P = 2 Torr (a, b), U = 1.76 kV and P = 4 Torr (c, left), and U = 5.76 kV and P = 4 Torr (c, right).
| (6) |
That is, when the ion beam inside the IMS diverges significantly beyond the reach of exit funnel, the signal scales as ~ 1/L. This is not observed experimentally: for example, sensitivities at L = 105 cm and L = 210 cm are equal within estimated uncertainty (Fig. 5c). The fact that doubling the IMS length does not measurably affect the ion intensity further supports the essentially lossless ion transmission through the IMS and to the ToF.
To realize high ion transmission efficiencies, ions exiting the IMS must obviously enter the mouth of the second larger ion funnel. When an ion packet width exceeds the funnel entrance aperture, the signal should decrease according to eqs. (1)–(3). To confirm this, we probe ion intensities as a function of E (i.e. the drift voltage) present in eq. (3). For example at U = 3.8 kV, eqs. (2) and (3) yield Rd ≈ 110 and, for the spatial width of initial ion packet equal to 20 mm (the exit aperture of hourglass funnel from which ions enter IMS), R ≈ 75. The FWHM of ion beam at IMS end (for L = 210 cm) would be ≈28 mm. Assuming a Gaussian density distribution across the ion beam (> 95% of which is found within two FWHM), the “outside” diameter of ion beam approaching the exit funnel is ~ 43 mm. That would bring the beam circumference within 4 mm of the funnel entrance ring (i.d. = 51 mm). So, when U is decreased, the sensitivity should remain constant until U ~ 3.5–4 kV and then start dropping. Experiments verify this prediction quantitatively (Fig. 6). Hence, these results indicate that ion losses through an IMS of any length can be eliminated completely as long as the IMS electrodes and ion funnel entrance are slightly larger than the ion packet width.
Fig. 6.
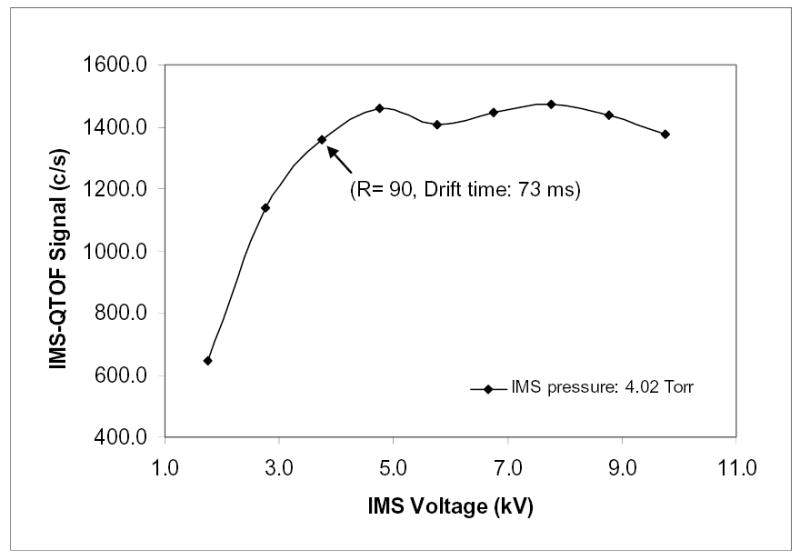
IMS-QToF sensitivity versus IMS drift voltage (for Le-Enk, 10 pM/μL solution, L = 210 cm).
The high sensitivity of the present instrument provided by its near-perfect ion transmission through IMS allows fast IMS-MS data acquisition. For example, a well-defined peak (S/N = 6) is obtained in just 0.6 s by averaging 10 elementary frames (Fig. 7a). The high stability of present system is evident from a linear dependence of the cumulative ion signal on acquisition time (Fig. 7b). The present sensitivity performance can be further improved (possibly several orders of magnitude) by operating electrospray in the nano-electrospray regime.62,63
Fig. 7.
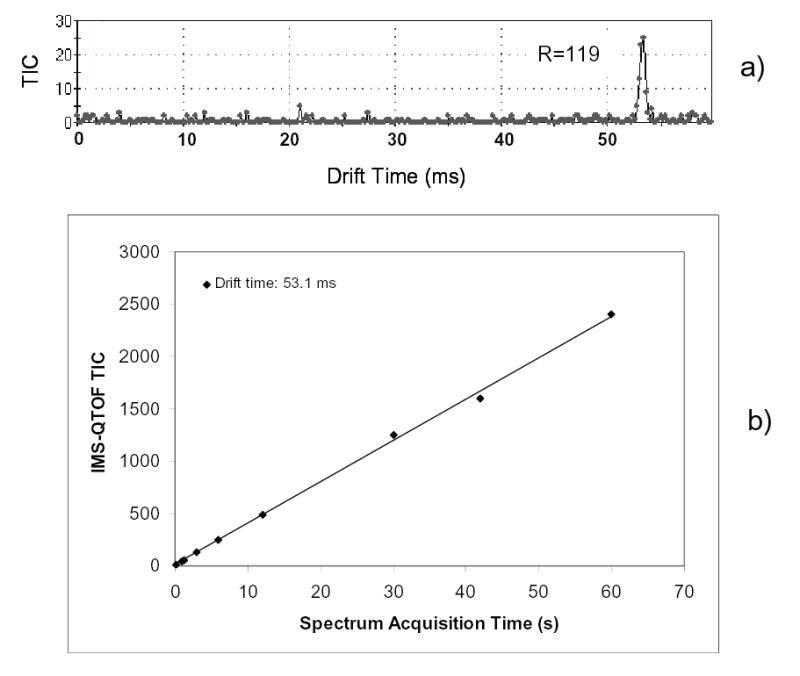
High instrumental sensitivity and stability: (a) IMS drift time distribution acquired in 0.6 s; (b) cumulative IMS-QToF signal as a function of acquisition window (for Le-Enk, 10 pM/μL solution, L = 210 cm, U = 4.76 kV, P = 4.02 Torr).
The effect of the terminal ion funnel on IMS resolution
The key question already mentioned is whether the RF fields in the IMS-MS ion funnel (or its re-focusing of ions from the IMS terminus) degrades IMS separation performance measured by the MS stage. The present measurements show convincingly that any degradation is insignificant; neither the drift time nor resolution exhibit any dependence on the RF amplitude, extending to zero V where the funnel operates in a DC-only mode that provides no ion focusing (Fig. 8). Of course, the sensitivity drops by nearly two orders of magnitude as the RF voltage decreases (Fig. 8), and the attenuation curve closely tracks that observed for an ion funnel at the API-MS interface.54 This directly demonstrates that the present virtually lossless ion transmission through IMS is achieved by RF ion focusing in electrodynamic funnels.
Fig. 8.
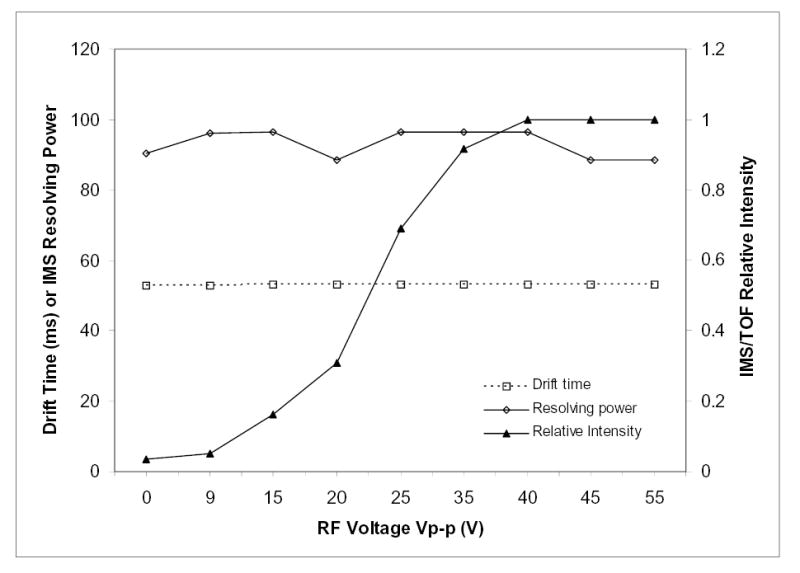
Effect of the RF amplitude on the IMS-MS funnel on IMS drift time, resolution, and sensitivity (conditions as in Fig. 7).
To gain a deeper insight into the temporal aspects of ion focusing at the IMS terminus, we have modeled the passage of realistic ion packets approaching from IMS through the IMS-MS funnel. These classical molecular dynamics simulations follow a statistically representative ion ensemble evolving under the influence of external (RF and DC) fields, thermal diffusion, and mutual Coulomb repulsion of constituent ions. This approach was successful in modeling and optimizing funnels for continuous ion transmission,52,56,61 where the kinetics of ion motion is not an issue. Here we use the same procedure to track the movements of discrete ion packets in time. The principal conclusion is that, under experimental conditions of this work, the tIF time for any ion depends almost exclusively on its mobility and the axial DC field, i.e. a funnel effectively becomes another IMS stage. In agreement with experiment, the RF fields in this regime have but a negligible effect on tIF, and thus on IMS resolution. This is not always the case: increasing the RF amplitude and/or decreasing the frequency excessively eventually creates a ladder of potential energy wells along the funnel axis that deepen toward the exit.61 This effectively turns the funnel into a sequence of ion traps. Such operation would lengthen tAR and degrade the resolution by re-mixing ions separated in IMS, as well as reduce the ion transmission efficiency of the funnel. Thus, the sensitivity and IMS resolution of IMS-MS are maximized over essentially the same operating conditions. In other words, no reason exists to operate the funnel in a mode that would degrade IMS performance.
In calculations, the IMS-MS funnel also “chokes” at very high ion currents that exceed its charge capacity in the narrowest region near the exit.64 This is, however, unlikely in reality as long as the conductance limit of ESI-IMS funnel does not significantly exceed that of IMS-MS funnel. Thus the performance of funnel as an ion collector at the IMS-MS interface should not be compromised by obtainable peak intensities.
The currently achieved IMS resolving power of 90–120 for singly-charged ions (Figs. 7a, 8) is slightly short of the best reported performance35,36 (R = 150–170). However, this is only because of a lower drift voltage: 4.76 kV at present vs. 10–14 kV in previous studies.35,36 Higher voltages could potentially cause electrical discharges in rarefied buffer gases. (Gases are most prone to electrical breakdown at pressures of a few Torr where the minima of Paschen curves are typically found.65) All high-resolution IMS spectra in the literature have been obtained at much higher P (> 200 Torr) that allow stronger electric fields. As we discussed, those instruments have lower sensitivity when coupled to MS. Also, lower-pressure drift tubes are suitable for MS-IMS-MS systems with mass-selection both before and after IMS separation,2,3 a critical capability for many applications.66 High-pressure IMS is incompatible with such designs since polyatomic ions cannot be effectively injected in the high pressure differential of the MS-IMS junction: the electric field is either insufficient to pull ions against a gas flow out of IMS or high enough to fragment them through collisional heating.
The key metric of IMS design quality is the measured resolving power as a fraction of theoretical limit.45 At the conditions of Figs. 7 and 8, Rd = 129 by eq. (3) and more precisely Rd = 127 correcting for the high-field anisotropic diffusion.44 Taking into account finite widths45 of the ion injection pulse (50 μs) and ToF acquisition window (100 μs) reduces R to 120, and the measured R of ~90–119 is ~75–99% of that value. As a benchmark, the highest performance drift tubes with flat endplates exhibit 80–88% of the theoretical limit in stand-alone IMS,45 ~90% in IMS/quadrupole MS,36 and ~70–100% in IMS/ToF.32 This again demonstrates that an ion funnel at IMS-MS interface does not degrade IMS resolution.
Conclusions
We have developed new ESI-IMS-ToF instrumentation for rapid, sensitive analyses of complex mixtures based on ion mobility separations followed by mass spectrometry. The instrumentation has two major attractions - an extremely high throughput of gas-phase separation and the simultaneous dispersion of all ions in IMS and MS dimensions. The key distinction of present instrument is the usage of electrodynamic ion funnels at both the ESI-IMS and IMS-ToF MS interfaces. Other innovations include (a) an hourglass ion funnel for effective ion transmission in the pulsed mode (employed here to accumulate ions arriving from the source and inject them into IMS), (b) a modular IMS design that provides experimental flexibility, permits a record drift tube length (here 210 cm), and minimizes the electrical breakdown hazard, and (c) the new wide ion funnel with acceptance aperture over 5 cm. (double that of the largest earlier designs).
A near-perfect ion collection by the IMS terminal ion funnel results in virtually lossless (> 50%) transmission of ions across a broad m/z range relevant to most biological analyses through an IMS of any length and to the ToF MS stage. The ion drift time (mobility) and IMS resolution are not degraded by ion funnel operation. In particular, the measured IMS resolving power exceeds 100, which is > 80% of the theoretical limit, and comparable to the best reported IMS or IMS/MS performance. The sensitivity of the ESI-IMS-QToF was close to that of commercial ESI-MS, i.e. the IMS separation capability is added “for free” in terms of either sensitivity or throughput. This separation power allows one to handle a greater sample complexity within the same time and/or increase the throughput without sacrificing quality, which should make IMS-MS an attractive practical platform for biological analyses and other demanding high-throughput applications. Further increases in overall separation power can be obtained by the combination with prior fast condensed phase separations (e.g. using capillary electrophoresis or LC) or use of another gas phase separation stage based upon differential IMS or FAIMS, as will be described elsewhere. Finally, further increases in sensitivity are anticipated from optimization of the ion accumulation process in the first ion funnel and operation using low nL/min flow rates for ESI where ionization efficiencies can approach 100%.62,67
Acknowledgments
The authors thank the Laboratory Directed Research and Development Program and the NIH National Center for Research Resources (RR18522) for supporting portions of this research. We also thank Professor David Clemmer (Indiana University) for helpful discussions. Pacific Northwest National Laboratory is operated by the Battelle Memorial Institute for the U.S. Department of Energy through Contract DE-ACO6-76RLO1830.
References
- 1.Hill HH, Siems WF, St Louis RH, McMinn DG. Anal Chem. 1990;62:A1201. doi: 10.1021/ac00222a001. [DOI] [PubMed] [Google Scholar]
- 2.Bowers MT, Kemper PR, von Helden G, van Koppen PAM. Science. 1993;260:1446. doi: 10.1126/science.260.5113.1446. [DOI] [PubMed] [Google Scholar]
- 3.Shvartsburg AA, Hudgins RR, Dugourd P, Jarrold MF. Chem Soc Rev. 2001;30:26. [Google Scholar]
- 4.Hoaglund-Hyzer CS, Lee YJ, Counterman AE, Clemmer DE. Anal Chem. 2002;74:992. doi: 10.1021/ac010837s. [DOI] [PubMed] [Google Scholar]
- 5.Collins DC, Lee ML. Anal Bioanal Chem. 2002;372:66. doi: 10.1007/s00216-001-1195-5. [DOI] [PubMed] [Google Scholar]
- 6.Barnes CAS, Hilderbrand AE, Valentine SJ, Clemmer DE. Anal Chem. 2002;74:26. doi: 10.1021/ac0108562. [DOI] [PubMed] [Google Scholar]
- 7.Moon MH, Myung S, Plasencia M, Hilderbrand AE, Clemmer DE. J Proteome Res. 2003;2:589. doi: 10.1021/pr034018v. [DOI] [PubMed] [Google Scholar]
- 8.Wu C, Siems WF, Klasmeier J, Hill HH. Anal Chem. 2000;72:391. doi: 10.1021/ac990601c. [DOI] [PubMed] [Google Scholar]
- 9.Fromherz R, Gantefor G, Shvartsburg AA. Phys Rev Lett. 2002;89 doi: 10.1103/PhysRevLett.89.083001. # 083001. [DOI] [PubMed] [Google Scholar]
- 10.Beegle LW, Kanik I, Matz L, Hill HH. Anal Chem. 2001;73:3028. doi: 10.1021/ac001519g. [DOI] [PubMed] [Google Scholar]
- 11.Srebalus CA, Li JW, Marshall WS, Clemmer DE. J Am Soc Mass Spectrom. 2000;11:352. doi: 10.1016/s1044-0305(00)00099-4. [DOI] [PubMed] [Google Scholar]
- 12.Counterman AE, Clemmer DE. Anal Chem. 2002;74:1946. doi: 10.1021/ac011083k. [DOI] [PubMed] [Google Scholar]
- 13.Lee SH, Gotts NG, von Helden G, Bowers MT. Science. 1995;267:999. doi: 10.1126/science.267.5200.999. [DOI] [PubMed] [Google Scholar]
- 14.Wyttenbach T, Bushnell JE, Bowers MT. J Am Chem Soc. 1998;120:5098. [Google Scholar]
- 15.Wyttenbach T, Bowers MT. Top Curr Chem. 2003;225:207. [Google Scholar]
- 16.Shvartsburg AA, Hudgins RR, Gutierrez R, Jungnickel G, Frauenheim T, Jackson KA, Jarrold MF. J Phys Chem A. 1999;103:5275. [Google Scholar]
- 17.Shvartsburg AA, Hudgins RR, Dugourd Ph, Gutierrez R, Frauenheim T, Jarrold MF. Phys Rev Lett. 2000;84:2421. doi: 10.1103/PhysRevLett.84.2421. [DOI] [PubMed] [Google Scholar]
- 18.Jackson KA, Horoi M, Chaudhuri I, Frauenheim T, Shvartsburg AA. Phys Rev Lett. 2004;93 doi: 10.1103/PhysRevLett.85.546. # 013401. [DOI] [PubMed] [Google Scholar]
- 19.Leavell MD, Gaucher SP, Leary JA, Taraszka JA, Clemmer DE. J Am Soc Mass Spectrom. 2002;13:284. doi: 10.1016/S1044-0305(01)00362-2. [DOI] [PubMed] [Google Scholar]
- 20.Myung S, Badman ER, Lee YJ, Clemmer DE. J Phys Chem A. 2002;106:9976. [Google Scholar]
- 21.Mason EA, McDaniel EW. Transport Properties of Ions in Gases. Wiley; N.Y.: 1988. [Google Scholar]
- 22.Mesleh MF, Hunter JM, Shvartsburg AA, Schatz GC, Jarrold MF. J Phys Chem. 1996;100:16082. [Google Scholar]
- 23.Eiceman GA, Leasure CS, Vandiver VJ. Anal Chem. 1986;58:76. [Google Scholar]
- 24.Lawrence AH, Neudorfl P. Anal Chem. 1988;60:104. [Google Scholar]
- 25.Lopez-Avila V, Hill HH. Anal Chem. 1997;69:289R. [Google Scholar]
- 26.Przybylko ARM, Thomas CLP, Anstice PJ, Fielden PR, Brokenshire J, Irons F. Anal Chim Acta. 1995;311:77. [Google Scholar]
- 27.Wu C, Siems WF, Hill HH, Hannan RM. J Chromatogr A. 1998;811:157. [Google Scholar]
- 28.von Helden G, Hsu MT, Kemper PR, Bowers MT. J Chem Phys. 1991;95:3835. [Google Scholar]
- 29.Jarrold MF, Constant VA. Phys Rev Lett. 1991;67:2994. doi: 10.1103/PhysRevLett.67.2994. [DOI] [PubMed] [Google Scholar]
- 30.Guevremont R, Siu KWM, Wang JY, Ding LY. Anal Chem. 1997;69:3959. doi: 10.1021/ac970359e. [DOI] [PubMed] [Google Scholar]
- 31.Hoaglund CS, Valentine SJ, Sporleder CR, Reilly JP, Clemmer DE. Anal Chem. 1998;70:2236. doi: 10.1021/ac980059c. [DOI] [PubMed] [Google Scholar]
- 32.Srebalus CA, Li J, Marshall WS, Clemmer DE. Anal Chem. 1999;71:3918. doi: 10.1021/ac9903757. [DOI] [PubMed] [Google Scholar]
- 33.Lee YJ, Hoaglund-Hyzer CS, Srebalus Barnes CA, Hilderbrand AE, Valentine SJ, Clemmer DE. J Chromatogr B. 2002;782:343. doi: 10.1016/s1570-0232(02)00569-x. [DOI] [PubMed] [Google Scholar]
- 34.Myung S, Lee YJ, Moon MH, Taraszka J, Sowell R, Koeniger S, Hilderbrand AE, Valentine SJ, Cherbas L, Cherbas P, Kaufmann TC, Miller DF, Mechref Y, Novotny MV, Ewing MA, Sporleder CR, Clemmer DE. Anal Chem. 2003;75:5137. doi: 10.1021/ac030107f. [DOI] [PubMed] [Google Scholar]
- 35.Asbury GR, Hill HH. J Microcolumn Sep. 2000;12:172. [Google Scholar]
- 36.Dugourd Ph, Hudgins RR, Clemmer DE, Jarrold MF. Rev Sci Instrum. 1997;68:1122. [Google Scholar]
- 37.Wu C, Siems WF, Asbury GR, Hill HH. Anal Chem. 1998;70:4929. doi: 10.1021/ac980414z. [DOI] [PubMed] [Google Scholar]
- 38.Issaeva T, Kourganov A, Unger K. J Chromatogr A. 1999;846:13. [Google Scholar]
- 39.Washburn MP, Wolters D, Yates JR. Nature Biotechnol. 2001;19:242. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 40.Wolters DA, Washburn MP, Yates JR. Anal Chem. 2001;73:5683. doi: 10.1021/ac010617e. [DOI] [PubMed] [Google Scholar]
- 41.Gygi SP, Corthals GL, Zhang Y, Rochon Y, Aebersold R. Proc Natl Acad Sci USA. 2000;97:9390. doi: 10.1073/pnas.160270797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rokushika S, Hatano H, Baim MA, Hill HH. Anal Chem. 1985;57:1902. [Google Scholar]
- 43.Spangler GE. Anal Chem. 1992;64:1312. [Google Scholar]
- 44.Verbeck GF, Ruotolo BT, Gillig KJ, Russell DH. J Am Soc Mass Spectrom. 2004;15:1320. doi: 10.1016/j.jasms.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 45.Siems WF, Wu C, Tarver EA, Hill HH, Larsen PR, McMinn DG. Anal Chem. 1994;66:4195. [Google Scholar]
- 46.Smith RD, Loo JA, Edmonds CG, Barinaga CJ, Udseth HR. Anal Chem. 1990;62:882. doi: 10.1021/ac00208a002. [DOI] [PubMed] [Google Scholar]
- 47.Sannes-Lowery K, Griffey RH, Kruppa GH, Speir JP, Hofstadler SA. Rapid Commun Mass Spectrom. 1998;12:1957. doi: 10.1002/(SICI)1097-0231(19981215)12:23<1957::AID-RCM418>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 48.Lee YJ, Hoaglund-Hyzer CS, Taraszka JA, Zientara GA, Counterman AE, Clemmer DE. Anal Chem. 2001;73:3549. doi: 10.1021/ac010295e. [DOI] [PubMed] [Google Scholar]
- 49.Valentine SJ, Koeniger SL, Clemmer DE. Anal Chem. 2003;75:6202. doi: 10.1021/ac030111r. [DOI] [PubMed] [Google Scholar]
- 50.Weis P, Gilb S, Gerhardt P, Kappes MM. Int J Mass Spectrom. 2002;216:59. [Google Scholar]
- 51.Shaffer SA, Tang K, Anderson GA, Prior DC, Udseth HR, Smith RD. Anal Chem. 1998;70:4111. doi: 10.1021/ac9802170. [DOI] [PubMed] [Google Scholar]
- 52.Shaffer SA, Tolmachev A, Prior DC, Anderson GA, Udseth HR, Smith RD. Anal Chem. 1999;71:2957. doi: 10.1021/ac990346w. [DOI] [PubMed] [Google Scholar]
- 53.Smith RD, Shaffer SA. # 6,107,628. US Patent. 2000
- 54.Belov ME, Gorshkov MV, Udseth HR, Anderson GA, Smith RD. Anal Chem. 2000;72:2271. doi: 10.1021/ac991360b. [DOI] [PubMed] [Google Scholar]
- 55.Kim T, Udseth HR, Smith RD. Anal Chem. 2000;72:5014. doi: 10.1021/ac0003549. [DOI] [PubMed] [Google Scholar]
- 56.Kim T, Tolmachev AV, Harkewicz R, Prior DC, Anderson GA, Udseth HR, Smith RD, Bailey TH, Rakov S, Futrell JH. Anal Chem. 2000;72:2247. doi: 10.1021/ac991412x. [DOI] [PubMed] [Google Scholar]
- 57.Wyttenbach T, Kemper PR, Bowers MT. Int J Mass Spectrom. 2001;212:13. [Google Scholar]
- 58.Smith RD, Tang K, Shvartsburg AA. US Patent. granted 05/2004.
- 59.Kim T, Tang K, Udseth HR, Smith RD. Anal Chem. 2001;73:4162. doi: 10.1021/ac010174e. [DOI] [PubMed] [Google Scholar]
- 60.Smith RD, Kim T, Tang K, Udseth HR. 6,583,408. US Patent. 2003
- 61.Tolmachev AV, Kim T, Udseth HR, Smith RD, Bailey TH, Futrell JH. Int J Mass Spectrom. 2000;203:31. [Google Scholar]
- 62.Schmit A, Karas M, Dulcks T. J Am Soc Mass Spectrom. 2003;14:492. doi: 10.1016/S1044-0305(03)00128-4. [DOI] [PubMed] [Google Scholar]
- 63.Tang K, Page JS, Smith RD. J Am Soc Mass Spectrom. 2004;15:1416. doi: 10.1016/j.jasms.2004.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shvartsburg AA, Tang K, Tolmachev AV, Anderson GA, Smith RD. Proceedings of the 52nd ASMS Meeting; Nashville. 2004. [Google Scholar]
- 65.Meek JM, Craggs JD, editors. Electrical Breakdown of Gases. Wiley; New York: 1978. [Google Scholar]
- 66.Shvartsburg AA, Jarrold MF. Phys Rev Lett. 2000;85:2530. doi: 10.1103/PhysRevLett.85.2530. [DOI] [PubMed] [Google Scholar]
- 67.Smith RD, Shen Y, Tang K. Acc Chem Res. 2004;37:269. doi: 10.1021/ar0301330. [DOI] [PubMed] [Google Scholar]