

Abstract
Diverse virus families have evolved to exploit the acidification of endosomal compartments to gain entry into cells. We describe a supramolecular approach for selectively targeting and inhibiting viral infections through this central biochemical pathway. Using adenovirus as a model non-enveloped virus, we have determined that an eight-residue cyclic D,L-α-peptide, selected from a directed combinatorial library, can specifically prevent the development of low pH in endocytic vesicles, arrest the escape of virions from the endosome, and abrogate adenovirus infection without an apparent adverse effect on cell viability. The likely generality of this approach against other pH-dependent viral infections is supported by the inhibition of type-A influenza virus escape from endosomes in the presence of the same peptide. Our studies suggest that self-assembling cyclic D,L-α-peptides hold considerable potential as a new rational supramolecular approach toward the design and discovery of broad-spectrum antiviral agents.
Keywords: cyclic peptide, antiviral, adenovirus, influenza
Introduction
Most pathogenic human viruses use cellular endocytic pathways to gain entry into cells (Figure 1).1–3 However, the capacity of a virus to appropriate this pathway is intimately tied to its ability to effectively enter and vacate the endosome prior to degradation or recycling. Interestingly, to escape from endosomes many enveloped and non-enveloped viruses depend critically on the natural endosomal acidification process to trigger conformational changes in their viral proteins required for membrane fusion or lysis of endosomes.1–3 We hypothesized that membrane-associating cyclic peptides might effectively block key steps involved in virus entry into or escape from endosomes and thus provide a class of antiviral agents with broad-spectrum activity. Toward this goal, we chose to explore the utility of membrane-active cyclic D,L-α-peptides in inhibiting adenovirus (Ad) infections in mammalian cells.
Figure 1.
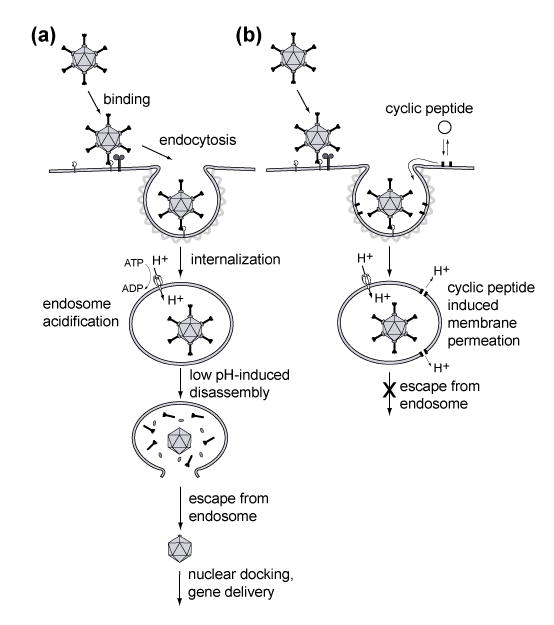
(a) Schematic depiction of the early stages of adenovirus (Ad) infection cycle. Following binding of Ad to its receptor, the virus is internalized via clathrin-mediated endocytsis. Once in the early endosome, active acidification leads to pH-dependent partial disassembly of the Ad capsid and escape from the endosome via membrane lysis. (b) Studies reported here suggest that membrane associating cyclic D,L-α-peptides incorporate into endosomal membranes during the virus internalization and exert their antiviral action by counteracting the development of low pH environment inside endosomes via the formation of membrane permeating supramolecular assemblies.
Adenoviruses are non-enveloped viruses responsible for a variety of acute respiratory, gastrointestinal, and ocular infections in humans.4 We considered adenovirus to be a particularly suitable viral infection model for testing our hypothesis because of its well-characterized life cycle that includes efficient cell entry via clathrin-mediated endocytosis and a pH-dependent endosomal escape (Figure 1).5, 6 We utilized a directed combinatorial approach to select amphiphilic cyclic D,L-α-peptides that could potentially prevent adenovirus infection. It has been shown that appropriately designed cyclic D,L-α-peptides can rapidly permeate selected cellular membranes by mechanisms that are thought to involve cyclic D,L-α-peptide uptake and self-assembly into membrane-permeating supramolecular peptide nanotubes (Figure 2c).7–9 Importantly, members of this class of synthetic membrane-active species have been shown recently to display good in vivo efficacies.9, 10
Figure 2.
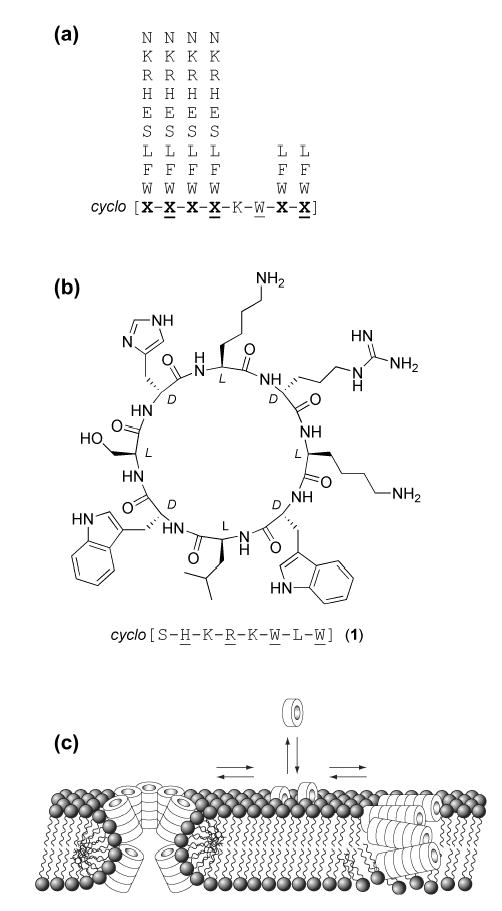
(a) Composition of the cyclic D,L-α-peptide library (brackets indicate cyclic backbone and underlined residues indicate D-amino acids). (b) Structure and sequence of peptide 1, identified in initial screens as inhibitory of adenovirus-mediated gene delivery. (c) Schematic illustration depicting plausible modes of cyclic D,L-α-peptide self-assembly into membrane permeating supramolecular peptide nanotubes.
Results and Discussion
We designed a single-compound-per-bead combinatorial eight-residue cyclic D,L-α-peptide library with a relatively narrow amphiphilic sequence-space to direct the potential antiviral activity toward membrane-active species (Figure 2a). Four hundred randomly selected members of the library were screened for ability to inhibit adenovirus infections of human epithelial cells (Hela). A recombinant Ad5-GFP virus expressing a green fluorescent protein (GFP) marker was used to quantitatively assess the extent of Ad-mediated transfection by the fluorescence readout of GFP transgene expression. Each peptide sample was initially screened at 10 μM for the ability to inhibit Ad5-GFP transfection and at 50 μM for potential cytotoxicity to Hela cells using a standard MTT assay of metabolic activity. Members of the peptide library that satisfied both criteria (apparent IC50<10 and LD50>50 μM) were identified using automated HPLC-MS/MS sequencing.11 Among the 20 hits identified, peptide 1 (Figure 2b) which exhibited substantial inhibition of Ad5 mediated gene delivery, was synthesized on large scale and used in the following studies.
Peptide 1 was shown to cause dose-dependent inhibition of Ad-mediated gene delivery to Hela cells (IC50 = 5 μM) (Figure 3a, S1) and was not cytotoxic in the active dose range of 5–10 μM (LD50 = 57 μM at 4 h) even after prolonged exposure (LD50 = 38 μM at 48 h). The inhibitory activity was not cell line specific with maximal inhibition (>95%) occurring at ~7 μM for the four cell lines tested (Figure 3a, Table S1). Furthermore, Hela cells remained viable when exposed to the active dose range of 1 and Ad5-GFP, ruling out the possibility of synergistic cytotoxicity as the result of the active infection process. Cells treated with peptide 1 four hours after addition of Ad5-GFP showed no change in infectivity as a function of peptide concentration (Figure 3a), suggesting that the peptide did not alter normal cellular functions including transcription or translation of Ad delivered GFP, but was required during the early stages of infection to reveal its antiviral activity. To elucidate the mechanism of antiviral activity, we systematically examined the effect of 1 on various early steps of the adenovirus infection pathway (Figure 1a).
Figure 3.
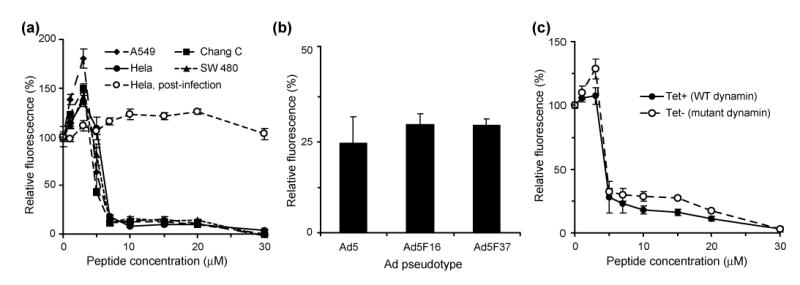
(a) Dose dependent inhibition of Adenovirus-mediated gene delivery as measured by fluorescence for four different cell lines infected with Ad5-GFP, including treatment of Hela cells after infection. (b) Percent inhibition of adenovirus mediated gene delivery for Ad5-GFP and two Ad5 fiber chimeras that use different cell receptors. (c) Dose dependent activity of peptide 1 against HeLa cells expressing either WT or Lys44Ala mutant dynamin, a protein involved in the clathrin-mediated endocytosis of the virus. All fluorescence values are normalized to untreated cells, and the mutant dynamin fluorescence is normalized to that of wild type for comparison.
Recent studies have shown a correlation between Ad capsid stability and the ability of adenovirus to escape from endosomal compartments. However, the apparent in vitro stability of Ad5 remained unchanged in the presence of 1 as judged by the temperature-dependant extent of Ad capsid permeability to the DNA intercalating dye TOTO (Figure S2).12 Moreover, when Ad particles were first incubated for 30 min with 10 μM peptide 1 and then diluted 10-fold upon addition to cells, for a final concentration well below the active dose, the extent of Ad-mediated gene delivery was similar to that of untreated virus (data not shown). Therefore, we concluded that the antiviral activity of 1 was not as a result of perturbations of virus particles.
We examined whether the peptide interferes with host cell functions including receptor engagement (Figure 1a). Although most of the 51 different Ad serotypes, including Ad5, use the Coxsackie Adenovirus Receptor (CAR) as their primary cell surface receptor, certain subgroup B (e.g. Ad16) and D adenoviruses (e.g. Ad37) use the membrane cofactor protein CD46.13–15 We reasoned that if peptide 1 selectively inhibited Ad5 interactions with CAR, it would be unlikely that it could also inhibit Ad5.F16 and Ad5.F37 which are modified Ad5-GFP viruses displaying fiber proteins that mediate receptor binding to alternate cell receptors.16 We found that in the presence of peptide 1 (10 μM), the relative inhibition of gene delivery was similar with the pseudotyped Ad-GFP vectors (Figure 3b). Therefore, peptide 1 does not inhibit receptor-mediated Ad cell attachment.
Previous studies have shown that the overexpression of a mutant form of dynamin (Lys44→Ala) significantly inhibits adenovirus infection through the clathrin-mediated endocytic pathway.17, 18 Using a cell line stably transfected with a plasmid for mutant dynamin under tetracycline-controlled promoter, we compared directly the influence of peptide 1 on Ad infectivity when this pathway was active or substantially restricted. These studies indicate that the peptide did not differentially impact virus infection in cells expressing WT or mutant dynamin (Figure 3c). Therefore, peptide 1 does not seem to interfere with the clathrin/dynamin-mediated internalization of adenovirus.
It has been demonstrated that the low pH environment of the endosome induces the partial disassembly of the Ad particle, exposing a membranolytic capsid protein that lyses the endosomal membrane allowing the virus to escape to the cytoplasm.6 To investigate whether peptide 1 impacted the ability of the virus to escape from endosomes, cells were infected with varying amounts of Ad in the presence of a fixed concentration of α-sarcin, a 150-residue ribonuclease that targets 28S ribosomal RNA. During the infection process, α-sarcin is co-internalized with Ad into endosomes and subsequently escapes into the cytoplasm upon virus-mediated lysis of the endosomal membrane.19 The cytosolic α-sarcin then inactivates ribosomes, leading to decreased protein translation that can be monitored by the levels of 35S-labeled methionine incorporation into nascent protein chains. Strikingly, treatment of cells with peptide 1 (10 μM) led to a 30-fold increase in the quantity of adenovirus required to cause a 50% decrease in 35S Met incorporation as compared to cells infected in the absence of the peptide (Figure 4a). This finding strongly suggested that peptide 1 exerts its antiviral action by restricting virus escape from the endosome (Figure 1b). Given the membrane permeating activity of selected amphiphilic cyclic D,L-α-peptides (Figure 2c)7–9, it seemed plausible that peptide 1 might act by collapsing or preventing the formation of endosomal transmembrane proton gradient.20–23 However, the fact that peptide 1 in the presence or absence of Ad did not promote entry of the 17 kDa α-sarcin suggests that the putative endosomal membrane permeation is not as a direct result of membrane lysis or formation of large transmembrane pores.
Figure 4.
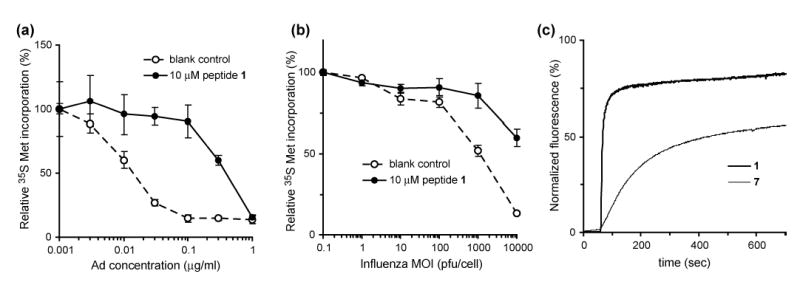
(a) Adenovirus mediated uptake of ribonuclease α-sarcin in the presence and absence of 1 as measured by radiolabeled 35S-Met incorporation. The increase in the quantity of Ad necessary for 50% incorporation of the radiolabel suggests that peptide 1 prevents virus escape from the endosome. (b) The same assay using type A influenza in place of adenovirus leads to a similar decrease in virus-induced α-sarcin uptake. (c) Peptide mediated permeation of synthetic liposomes containing sulforhodamine-B dye by peptides 1 and 7. Peptide was added at 60 sec to a final concentration of 10 μM.
The effect of peptide 1 on endosomal pH during the adenovirus infection was directly assessed by fluorescence microscopy and image analysis using carboxyfluorescein (CF)-labeled virus particles.24–26 Previous observations have revealed that adenovirus escape from endosomes is rapid and can hamper analysis of CF-labeled Ad in acidic intracellular compartments.25 To more accurately measure the pH in the virus-containing vesicles, we used Ad2-ts1, a temperature sensitive adenovirus mutant that is defective in its ability to escape from endosomes, but otherwise similar to the wild type virus with respect to CAR receptor binding and internalization.27 In the presence or absence of peptide 1 (10 μM), the intracellular localization of CF-Ad2-ts1 in cells showed similar punctuate perinuclear fluorescence (Figure 5a,b). The environmental pH values of internalized viral particles were determined using ratiometric (I488nm/I457nm) analysis of CF-Ad2-ts1 fluorescence based on a standard curve generated from images collected for CF-Ad2-ts1 infected cells incubated at various pH values (Figure S3).24, 25 In untreated cells, the environmental pH of CF-Ad2-ts1 exhibited a tendency towards acidic values with an average pH of 5.3 (Figure 5c, open symbols). This likely reflects the different stages of virus trafficking from initially coated pits at neutral pH to acidified late endosomes. In striking contrast, the environmental pH of CF-Ad2-ts1 in cells treated with 1 had a more restricted range of pH values with an average value of 6.8 (Figure 5c, filled symbols). These findings indicate that 1 inhibits endosomal acidification, a process critical for virus escape from endosomes (Figure 1).
Figure 5.
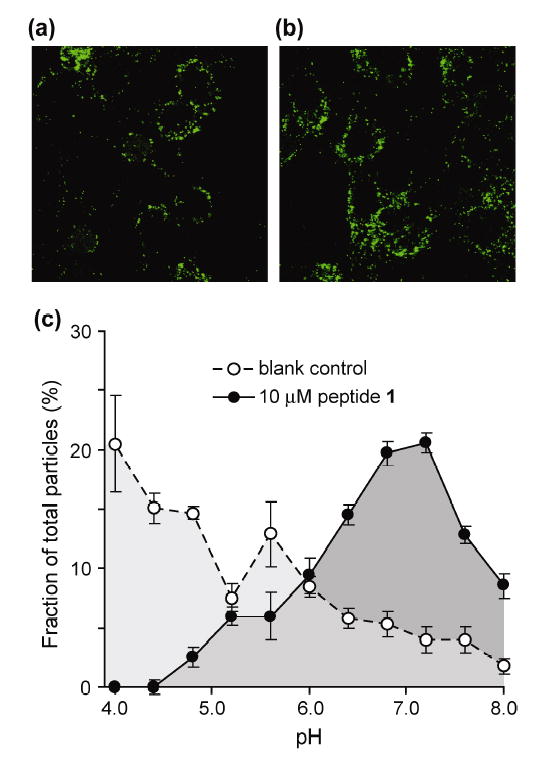
(a) Representative fluorescence microscopy image of cells infected with CF-Ad2-ts1; (b) representative image of cells infected with CF-Ad2-ts1 in the presence of 10 μM peptide 1; (c) population distribution of environmental pH for particles from images taken in the presence or absence of peptide 1.
We reasoned that if peptide 1 was capable of blocking Ad escape from endosomes via disruption of endosomal pH gradients, it should also restrict the cell entry of other pH-dependant viral pathogens.1–3 To probe this possibility, we analyzed the extent of α-sarcin delivery induced by type A influenza virus (WSN), an enveloped virus that enters cells via pH-dependent membrane fusion from within endocytic vesicles.28 The uptake of α-sarcin has been shown previously to correlate with entry of enveloped viruses.23 Similarly, we observed titer dependent α-sarcin uptake mediated by type A influenza virus (Figure 4b, open circles). Remarkably, peptide 1 (10 μM) significantly reduced influenza virus-mediated α-sarcin delivery (Figure 4b, closed circles) and was not toxic at this dose to the MDCK cells used in the assay (LD50 = 93 μM).
There remained a possibility that the antiviral activity of peptide 1 might not derive from the postulated pH effects caused by the peptide assembly-mediated endosomal membrane permeation but instead from the inhibition of vacuolar proton ATPases (V-ATPase). V-ATPases are large transmembrane proton pumps responsible for the production of low pH environments in a variety of intracellular vesicles including endosomes.29 The antibiotic bafilomycin is a potent inhibitor of V-ATPase and has been shown to prevent endosomal acidification and viral entry.21, 23,30 We designed and studied a number of peptide 1 analogues to probe whether the antiviral activity of 1 resulted form its specific molecular interactions with V-ATPase or other receptors (Table 1). The possibility of a receptor/ligand-mediated mechanism of action is unlikely since the enantiomeric sequence 10 and the retro-peptide 11 both display antiviral activities similar to 1 (Table 1). The antiviral activities of the single alanine-scan analogues (peptides 2–9) also support the membrane permeation mode of action. Substitution of either tryptophan residue in the putative membrane-interacting hydrophobic segment of peptide 1 with alanine (peptides 7 and 9) abolished the antiviral activity. Moreover, peptide 6 with four contiguous hydrophobic residues, instead of three in 1, showed a decrease in membrane selectivity (lower LD50 value). Alanine substitutions in the hydrophilic portion of the sequence were well tolerated in peptides 2 and 5 but not in peptides 3 and 4 providing further support to our working model that membrane partitioning is primarily mediated by hydrophobic side chains while membrane selectivity can be greatly influenced by the interactions of hydrophilic side chains with cell membrane constituents. The low cytoxicity of 1 (LD50/IC50 > 10) and its analogues and lack of apparent adverse effects on cell viability at the effective antiviral dose range could also be related to the differential uptake or self-assembly of the cyclic D,L-α-peptides in specialized membrane domains. It has been suggested that entry of some viruses are enhanced by lipid micro domains that function to increase the local concentration of molecules involved in cell entry.1, 31 Therefore, it is tantalizing to speculate that the source of observed membrane selectivity might stem from selective peptide interactions with these specialized membrane domains. Several natural antimicrobial peptides, in particular defensins, have also been shown to display in vitro antiviral activities.32–35 In some cases the binding of the peptides to viral glycoproteins (lectin-like behavior) has been implicated as the potential mechanism of antiviral action. However, in light of our findings and the well-known membrane permeating activity of many natural antimicrobial peptides, the possibility exists that some of these peptides may also operate by inhibiting virus escape from endosomes.
Table 1.
In vitro antiviral activity and cytotoxicity of cyclic D,L-α-peptides.
| # | sequence | IC50(μM)ab | LD50(μM)b |
|---|---|---|---|
| 1 | c[SHKRKWLW] | 5 | 57 |
| 2 | c[AHKRKWLW] | 8 | 54 |
| 3 | c[SAKRKWLW] | 13 | 70 |
| 4 | c[SHARKWLW] | 19 | >100 |
| 5 | c[SHKAKWLW] | 7 | 65 |
| 6 | c[SHKRAWLW] | 6 | 43 |
| 7 | c[SHKRKALW] | >30 | >100 |
| 8 | c[SHKRKWAW] | 17 | 100 |
| 9 | c[SHKRKWLA] | >30 | >100 |
| 10 | c[SHKRKWLW] | 7 | 63 |
| 11 | c[WLWKRKHS] | 5 | 35 |
Inhibition of Adenovirus (Ad-5) infections.
HeLa cells were used in both assays.
Biophysical studies in synthetic model membranes with lipid compositions selected to mimic that of early endosomes also support the membrane permeation mode of action.36 The addition of peptide 1 to sulforhodamine-B dye-entrapped liposomes led to a rapid increase in fluorescence due to release of the dye into solution as the result of liposomal membrane permeation (Figure 4c). The Trp Ala analog 7, that showed decreased ability to inhibit Ad infections, was also less active than 1 in this model (Figure 4c). Attenuated total reflectance (ATR) FT-IR spectroscopy of peptide 1 in oriented 1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine (DMPC) lipid multilayers show amide-A (NH stretch), amide-I, and amide-II bands characteristic of tightly hydrogen bonded antiparallel β-sheet structures seen in self-assembled peptide nanotubes (Figure S4 and Table S2).8, 9 Quantitative measurements indicate that the amphiphilic peptide assemblies are oriented at a 66±5° tilt angle from the membrane normal, consistent with a carpet-like mode of action (Figure 2c).
Conclusion
Despite remarkable achievements in recent years, the most commonly employed strategy of inhibiting species-specific viral proteins typically affords narrow-spectrum antiviral therapeutics that are subject to deactivation by the rampant evolution of drug resistant viral species. We speculate that the supramolecular approach described here, by virtue of acting on a common cellular pathway involved in many virus infections, might offer a new opportunity in the rational design of broad-spectrum antiviral agents that are less prone to the development of resistance. However, despite the promising initial findings reported here, for the practical utility of this new class of antiviral agents to be seriously contemplated, cyclic D,L-α-peptides with significantly wider therapeutic indices need to be fashioned.
Materials and Methods
General
2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), 2-Cl-trityl chloride resin, and all protected amino acids were purchased from Novabiochem. (7-azabenzotriazole-1-tloxy)tripyrrolidinophosphonium hexafluorophosphate (PyAOP) was purchased from Aldrich. Lipids were purchased from Avanti Polar Lipids. Sulforhodamine B was obtained from Molecular Probes. Dulbecco's Modified Eagle Media (DMEM) was obtained from Invitrogen and supplemented with 10% FBS. Solvents and all other reagents were purchased from Aldrich or Fisher unless otherwise noted. All peptide stock solutions used for biological assays were prepared in 5% DMSO/saline unless otherwise noted. Ad5 E1,E3 deteleted containing GFP under control of CMV promoter was prepared according to a literature procedure.6
Synthesis of Cyclic D,L-α-Peptides.11
Fmoc-Lys(Boc)-OAllyl37 (1.0 mmol/g of resin to be loaded) was treated with 1:1 TFA/CH2Cl2 for 1 hr. The solution was concentrated to dryness by rotary evaporation, and subjected to high vacuum to remove the residual TFA. The crude material was dissolved in CH2Cl2, and DIEA was added (4.0 eq relative to amino acid). This solution was transferred to a sintered peptide synthesis vessel containing 2-Cl-trityl chloride polystyrene resin. The mixture was agitated on a shaker for 4 hr. The vessel was then drained, and the resin washed with CH2Cl2 (3 × 1 min), 8:2:1 CH2Cl2/MeOH/DIEA (2 × 10 min), CH2Cl2 (3 × 1 min), and MeOH. After drying under vacuum, loading was measured by UV quantitation of Fmoc release after treatment with 20% piperidine/DMF.
The orthogonally protected Fmoc-Lys-OAllyl loaded trityl resin prepared above (0.10 mmol) was placed in a sintered glass peptide synthesis vessel and swollen in CH2Cl2 for 45 min. The resin was washed twice with DMF, treated with 20% piperidine/DMF (2 × 8 min) to remove the Fmoc group, and washed again with DMF (3×). For each coupling, a solution of the Fmoc-protected amino acid (0.40 mmol), HBTU (148 mg, 0.39 mmol), and diisopropylethylamine (175 μl, 1.0 mmol) in DMF (2 ml) was allowed to prereact for 2 min then added to the resin. The resin suspension was agitated for 30 min, drained, and washed with DMF (3×). This deprotection-coupling cycle was repeated until the desired linear peptide sequence was obtained.
The resin bearing full length linear peptide was washed with CH2Cl2, then suspended in 8:2:1 CH2Cl2/N-methylmorpholine/acetic acid. The mixture was degassed by bubbling Ar for 10 min. Pd(PPh3)4 (58 mg, 0.05 mmol) was added and the mixture was degassed for an additional 5 min. The vessel was then sealed and agitated for 4 hr. The vessel was then drained, and the resin washed with CH2Cl2, DMF, 5% DIEA/DMF, 0.5% w/v sodium diethyldithiocarbamate in DMF, and finally once more with DMF. The N-terminal Fmoc group was then removed by treatment with 20% piperidine/DMF (2 × 8 min), and the resin washed with DMF (3×) and 5% DIEA in DMF (3×).
PyAOP (307 mg, 0.6 mmol) was dissolved in DMF (2 ml). DIEA (0.209 ml, 1.2 mmol) was added and the solution was transferred to the vessel containing the linear peptide on resin. The mixture was agitated for 4 hr, drained, and washed with DMF (3×). In some cases, the cyclization was repeated with fresh reagents to drive the reaction to completion. The resin was washed with DMF, CH2Cl2, and MeOH and dried under vacuum. The peptide was cleaved from the resin by treatment with a mixture of 81.5% TFA : 5% thioanisole : 5% phenol : 5% water : 2.5% 1,2-ethanedithiol : 1% triisopropylsilane for 4 hr. The cleavage solution was collected by filtration and the crude peptide precipitated by addition of cold ether. The suspension was centrifuged, decanted, and the solid washed twice more with ether. The solid was dried under vacuum to yield crude peptide. All peptides were purified by preparative RP-HPLC on a C18 column to better than 95% purity as determined by analytical HPLC. Identity of the purified products was confirmed by MALDI- or ESI-MS.
Preparation of Peptide Library
A library of peptides was prepared using a split-and-pool approach based as previously reported.11 Briefly, the method is based on the general protocol for peptide synthesis described above with the following modifications. Trityl-chloride loaded polystyrene macrobead resin (Peptides International) was used as the solid support. Reaction times were extended to 3 min for each wash, 15 min for each deprotection, and 90 min for each coupling. Coupling reactions were carried out with 5 eq of Fmoc-amino acid, 4.95 eq HBTU, and 12.5 eq diisopropylethylamine (quantities based on approximate resin quantity after splitting). Allyl removal and cyclization reactions were each carried out twice for 5–6 hours. After washing as described above, the beads were dried under high vacuum and stored at 4°C.
Library Stock Plate Preparation
Dried macrobeads from the above library were individually arrayed into wells of a 96-well polypropylene plate. A mixture of 95:2.5:2.5 TFA/water/triethylsilane (~100 μl/well) was added, and the plate was covered. After 4 hr, the cleavage mixture was removed under a stream of nitrogen followed by additional drying in a vacuum desiccator. To this crude residue and the remaining bead was added DMSO (10 μl/well) followed by PBS, pH 7.0 (90 μl/well). Based on resin loading, the concentration of these stock solutions was approximately 1 mM.
Peptide Sequencing
Unknown peptides were sequenced using HPLC-MS/MS with software for automated analysis of fragmentation spectra as previously described.11
Ad infectivity by bulk fluorescence
Cells were plated into tissue culture treated, clear bottom, black 96-well plates (200 ul/well, 5000 cells/well). The cells were incubated in DMEM +10% FBS for 24 hr at 37°C under an atmosphere of 5% CO2. Peptide stock solution (20 ul/well, triplicate for each concentration) was added followed by virus (20 ul/well, 1.25x109 particles/ml in TBS). The cells were then incubated for 4 hr at 37°C. After 4 hr, the media was removed, fresh media was added, and the cells were allowed to incubate for an additional 24 hr at 37°C. After 24 hr, the media was removed and the cells washed twice with HBS and left under HBS (100 ul/well). The plates were then scanned on a Molecular Dynamics Fluorimager 595. The integrated intensity of GFP fluorescence for each well was determined using the accompanying software. These values were background corrected against wells containing uninfected cells, and triplicate conditions averaged.
MTT toxicity assay
Cells were plated into tissue culture treated, clear bottom, black 96-well plates (200 ul/well, 5000 cells/well). The cells were incubated for 48 hr at 37°C. Peptide stock solution (20 ul/well, triplicate for each concentration) was added and the cells were incubated in the presence of peptide for 4 hr at 37°C. After 4 hr, the media was removed and fresh media added along with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (100 μl/well of a 0.2 mg/ml solution in DMEM). The cells were incubated for 2 hr at 37°C, washed, and dissolved in DMSO (100 ul/well). The plate was allowed to stand at 37°C for 20 min to completely dissolve the formazan precipitate. The absorbance at 560 nm was then measured on a Molecular Devices SpectraMAX 250 microplate reader. Replicate wells were background corrected, averaged, and divided by the value obtained for untreated cells on the same plate to obtain relative metabolic activity for peptide treated cells.
Measurements of Ad Capsid Stability
The thermal stability of the Ad capsid following exposure to 1 was measured by the changes in accessibility of the viral DNA to a fluorescent interacalating dye, TOTO-1 (Molecular Probes), as previously described with minor modifications.6, 12 Briefly, 100 μg/ml Ad5 was incubated with 60 nM TOTO-1 in 50 mM HEPES, 100 mM NaCl pH 7.5. The fluorescence emission of TOTO-1 was monitored as a function of temperature using an ABI Prism 7900HT real-time PCR machine (Applied Biosystems) programmed to measure fluorescence (λex 488 nm, λem 540 nm) every 2.5 ºC between 20 and 70 ºC. Samples were equilibrated at each temperature for 2 minutes prior to measurement.
Sarcin Assay
Cells (Hela for Ad5; MDCK for influenza) were plated in 96 well plates at a density of 10,000 cells/well 24 hr. prior to infection. One hour prior to infection, cells were washed once with DMEM without cysteine or methionine and supplemented with 10 mM HEPES, 2 mM glutamine and 0.5% BSA with penicillin and streptomycin (abbreviated DMEM- hereafter) and incubated in DMEM- for one hour at 37 ºC. The media was then removed and cells were incubated with DMEM- containing 0.1 mg/ml α-sarcin (Sigma) and either Ad5 ranging from 1 to 1×10−3 μg/ml or Influenza A ranging from 0.1 to 1×104 PFU/ml, in the presence or absence of 10 μM of 1, for 2 hrs at 37 ºC. The cells were then washed once with DMEM- and incubated with DMEM- containing 0.1 μCi of 35S-L-Methionine (Amersham Biosciences) per well for an additional 2 hrs at 37 ºC. The cells were then washed twice with 100 μL of 20 mM TBS pH 7.4, incubated with 50 μL of ice cold 5% TCA at 4 ºC for one hour and then centrifuged at 1000 g for 15 min. at 4 ºC and washed twice with cold ethanol. The pellet was then dissolved overnight in 5 μL of 1% SDS, 0.1 N NaOH at 4 ºC on a rotary shaker. The resulting solutions were neutralized with 1 μl 0.6 N HCl and 34 μl of MicroScint 20 liquid scintillation cocktail (Packard) was added to each well followed by vortexing. Radioactivity was measured using a Topcount (Packard) 96 well liquid scintillation counter.
Microscopy with Fluorescently Labeled Adenovirus
The Ad temperature sensitive mutant, ts1, was labeled with approximately three molecules of carboxyfluorescein (CF) succimidyl ester (Molecular Probes) per hexon protein as previously described.25 Similar labeling of wild type Ad2 maintained ~85% of the original infectivity. Hela cells (7×104 cells/well) were grown overnight on glass coverslips in 24 well plates in DMEM. Cells were washed with ice cold DMEM and incubated on ice for 10 minutes before adding 5×104 particles per cell of CF-ts1 and incubated further on ice for 1 hr. Unbound virus was removed with three washes of ice cold DMEM and then warmed media was added with or without 10 μM peptide 1 and incubated at 37 ºC for 20 minutes. Cells were then placed on ice and washed 3 times with ice cold PBS. Coverslips were mounted with PBS containing 2.5% (w/v) DABCO pH 7.4 and examined by laser scanning confocal microscopy. In additional samples, cells were infected with CF-ts1 in conditions identical to those above, fixed for 10 min with 4% paraformaldehyde in PBS, and mounted in PBS containing 2.5% w/v DABCO at specific pH values in the presence of the ionophores nigericin (20 μM) and monensin (10 μM). These samples were used as standards to generate a curve for pH calibration.
Imaging of CF-labeled virions was performed using a Biorad 2100 confocal laser scanning microscope. Images were sequentially scanned at 457 and 488 nm using a 60x 1.4N.A. objective. The environmental pH of internalized CF-ts1 was determined by employing the pH-dependence of the I488nm/I457nm ratio. Images were analyzed using ImageJ (NIH). Regions of interest (ROIs) were selected using 488nm excitation images by thresholding and selecting regions composed of a minimum of 10 pixels to minimize contributions from noise. Typically, 150–200 ROIs were identified per field. Based on the standard curve generated from cells mounted in different pH media, ratios in peptide untreated and treated samples were converted to a pH value. Histograms were generated with ROI’s binned in 0.4 pH unit increments. Two fields were analyzed for each sample, and three replicate samples were averaged for each experimental condition.
Liposome Dye Release
Liposomes were prepared using the technique of reverse phase evaporation. A 16 mM solution in chloroform containing phosphatidylcholine (PC, egg), phosphatidylethanolamine (PE, egg), phosphatidylinositol (PI, soy), phosphatidylserine (PS, egg), sphingomyelin (SM, egg), gangliosides (porcine), and cholesterol was prepared in molar ratio of 5 PC : 1 PE : 1 PI : 1 PS : 1 SM : 1 gangliosides : 3 cholesterol. 2.5 ml of this solution was transferred to a degassed flask under argon. Ether (1 ml) was added followed by 2 ml of a 100 mM solution of sulforhodamine B in Dulbecco's PBS. The flask was sealed under argon, and sonicated for 7 min in an ice cooled bath. Most of the organics were removed by rotary evaporation, and an additional 1.5 ml of PBS was added. After an additional 5 minutes of rotary evaporation, the solution was transferred to a syringe and passed through stacked 200 μm and 400 μm filters. The solution was then transferred to a Sephadex G-25 column and eluted with PBS. The liposomes which eluted as a pink band were collected and used within 24 hr.
The lipid preparation from above (12.5 μl) was added to PBS (1 ml) in a fluorescence cuvette with equipped stir bar. Time dependent fluorescence measurements were carried out on an Aminco Bowman Series 2 Luminescence Spectrometer at λex = 535 nm and λem = 585 nm. Measurements were taken in 1 second intervals with stirring. After 1 min of background measurement, peptide (10 μl of a 10 mM solution in DMSO) was added to the sample. After the fluorescence stabilized (~10 min), Triton X-100 (25 μl of a 10% solution in H2O) was added. Normalized fluorescence values (In) were calulated as follows:
where It, I0, and I∞ are the fluorescence values at time t, time 0, and after addition of the triton X-100, respectively.
ATR-FTIR sample preparation8
1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine (DMPC, Avanti Polar Lipids) (20 mg) was transferred to a degassed flask. Chloroform (1 ml) was added followed by ether (1 ml) and water (2 ml). The flask was sealed under argon and placed in an ice cooled sonicator for 7 minutes. The flask was then transferred to a rotary evaporator and evaporated until foaming was no longer observed. Water (2 ml) was added, and the suspension was placed on the rotary evaporator for an additional 10 minutes. Peptide (16 μl of a 10 mM solution in DMSO) was added to a portion of this lipid preparation (184 μl) for a final concentration of 6.8 mM DMPC, 0.8 mM peptide in 8% DMSO/water. This mixture was vortexed periodically over a 10 minute period and passed through a pre-wetted column of Sephadex-G25. 100 μl of the resulting mixture was deposited on a 45° Ge-ATR mirror (Spectra Tech) and allowed to dry several hours in air then for 3 hours in a vacuum dessicator.
ATR-FTIR spectroscopy
Spectra were collected on a Nicolet 550 Magna Series II FT-IR instrument with a liquid nitrogen cooled mercury-cadmium-telluride detector under a nitrogen atmosphere. The multibilayer coated mirror prepared above was fitted on a baseline horizontal ATR optical bench with a ZnSe polarizer (Spectra Tech). Each spectrum represents an average of 800 scans obtained at a resolution of 2 cm−1. The spectra were processed with Omnic (Nicolet) with ATR correction against a background consisting of the empty mirror at the same polarization angle, and further baseline corrected manually. The final spectra were subjected to exponential smoothing using Mathematica (Wolfram Research). Calculation of lipid and cyclic peptide orientation angles were carried out as previously described for this class of compound.8
Supplementary Material
Acknowledgments
We thank our colleagues Prof. S. Schmid for providing samples of the K44A dynamin transfected Hela cells and Prof. L. Sherman for samples of influenza A virus. We are grateful to NSF for a Predoctoral Fellowship to WSH and NIH for a NRSA Postdoctoral Fellowship to CMW (AI007354). This work was supported by grants from the National Institutes of Health (GM52190 to MRG) and (HL54352 and EY11431 to GRN).
References
- 1.Dimitirov DS. Nat Rev Microbiol. 2004;2:109–122. doi: 10.1038/nrmicro817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sieczkarski SB, Whittaker GR. Curr Top Microbiol. 2005;285:1–23. doi: 10.1007/3-540-26764-6_1. [DOI] [PubMed] [Google Scholar]
- 3.Carrasco L. FEBS Lett. 1994;350:151–154. doi: 10.1016/0014-5793(94)00780-2. [DOI] [PubMed] [Google Scholar]
- 4.Lukashok SA, Horwitz MS. Curr Clin Top Infect Dis. 1998;18:286–305. [PubMed] [Google Scholar]
- 5.Stewart PL, Dermody TS, Nemerow GR. Adv Protein Chem. 2003;64:455–491. doi: 10.1016/s0065-3233(03)01013-1. [DOI] [PubMed] [Google Scholar]
- 6.Wiethoff CM, Wodrich H, Gerace L, Nemerow GR. J Virol. 2005;79:1992–2000. doi: 10.1128/JVI.79.4.1992-2000.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghadiri MR, Granja JR, Buehler LK. Nature. 1994;369:301–304. doi: 10.1038/369301a0. [DOI] [PubMed] [Google Scholar]
- 8.Kim HS, Hartgerink JD, Ghadiri MR. J Am Chem Soc. 1998;120:4417–4424. [Google Scholar]
- 9.Fernandez-Lopez S, Kim HS, Choi EC, Delgado M, Granja JR, Khasanov A, Kraehenbuehl K, Long G, Weinberger DA, Wilcoxen KM, Ghadiri MR. Nature. 2001;412:452–456. doi: 10.1038/35086601. [DOI] [PubMed] [Google Scholar]
- 10.Dartois V, Sanchez-Quesada J, Cabezas E, Chi E, Dubbelde C, Gritzen C, Weinberger D, Ghadiri MRTRP., Jr submitted. [Google Scholar]
- 11.Redman JE, Wilcoxen KM, Ghadiri MR. J Comb Chem. 2003;5:33–40. doi: 10.1021/cc0200639. [DOI] [PubMed] [Google Scholar]
- 12.Rexroad J, Wiethoff CM, Green AP, Kierstead TD, Scott MO, Middaugh CR. J Pharm Sci. 2003;92:665–678. doi: 10.1002/jps.10340. [DOI] [PubMed] [Google Scholar]
- 13.Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. Science. 1997;275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 14.Sirena D, Lilienfeld B, Eisenhut M, Kaelin S, Boucke K, Beerli RR, Vogt L, Ruedl C, Bachmann MF, Greber UF, Hemmi S. J Virol. 2004;78:4454–4462. doi: 10.1128/JVI.78.9.4454-4462.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu E, Trauger SA, Pache L, Mullen TM, Von Seggern DJ, Siuzdak G, Nemerow GR. J Virol. 2004;78:3897–3905. doi: 10.1128/JVI.78.8.3897-3905.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Von Seggern DJ, Huang S, Fleck SK, Stevenson SC, Nemerow GR. J Virol. 2000;74:354–362. doi: 10.1128/jvi.74.1.354-362.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Damke H, Baba T, Warnock DE, Schmid SL. J Cell Biol. 1994;127:915–934. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang K, Huang S, Kapoor-Munshi A, Nemerow G. J Virol. 1998;72:3455–3458. doi: 10.1128/jvi.72.4.3455-3458.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez-Puentes C, Carrasco L. Cell. 1980;20:769–775. doi: 10.1016/0092-8674(80)90323-2. [DOI] [PubMed] [Google Scholar]
- 20.Irurzun A, Nieva JL, Carrasco L. Virology. 1997;227:488–492. doi: 10.1006/viro.1996.8340. [DOI] [PubMed] [Google Scholar]
- 21.Merion M, Schlesinger P, Brooks RM, Moehring JM, Moehring TJ, Sly WS. Proc Natl Acad Sci U S A. 1983;80:5315–5319. doi: 10.1073/pnas.80.17.5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tartakoff AM. Cell. 1983;32:1026–1028. doi: 10.1016/0092-8674(83)90286-6. [DOI] [PubMed] [Google Scholar]
- 23.Perez L, Carrasco L. J Gen Virol. 1994;75:2595–2606. doi: 10.1099/0022-1317-75-10-2595. [DOI] [PubMed] [Google Scholar]
- 24.Ohkuma S, Poole B. Proc Natl Acad Sci U S A. 1978;75:3327–3331. doi: 10.1073/pnas.75.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyazawa N, Crystal RG, Leopold PL. J Virol. 2001;75:1387–1400. doi: 10.1128/JVI.75.3.1387-1400.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakano MY, Greber UF. J Struct Biol. 2000;129:57–68. doi: 10.1006/jsbi.1999.4201. [DOI] [PubMed] [Google Scholar]
- 27.Greber UF, Webster P, Weber J, Helenius A. EMBO J. 1996;15:1766–1777. [PMC free article] [PubMed] [Google Scholar]
- 28.Skehel JJ, Wiley DC. Annu Rev Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 29.Kawasaki-Nishi S, Nishi T, Forgac M. FEBS Lett. 2003;545:76–85. doi: 10.1016/s0014-5793(03)00396-x. [DOI] [PubMed] [Google Scholar]
- 30.Bayer N, Schober D, Prchla E, Murphy RF, Blaas D, Fuchs R. J Virol. 1998;72:9645–9655. doi: 10.1128/jvi.72.12.9645-9655.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson RGW, Jacobson K. Science. 2002;296:1821–1825. doi: 10.1126/science.1068886. [DOI] [PubMed] [Google Scholar]
- 32.Lehrer RI. Nat Rev Microbiol. 2004;2:727–738. doi: 10.1038/nrmicro976. [DOI] [PubMed] [Google Scholar]
- 33.Matanic VCA, Castilla V. Int J Antimicrob Ag. 2004;23:382–389. doi: 10.1016/j.ijantimicag.2003.07.022. [DOI] [PubMed] [Google Scholar]
- 34.Bastian A, Schafer H. Regul Pept. 2001;101:157–161. doi: 10.1016/s0167-0115(01)00282-8. [DOI] [PubMed] [Google Scholar]
- 35.Daher KA, Selsted ME, Lehrer RI. J Virol. 1986;60:1068–1074. doi: 10.1128/jvi.60.3.1068-1074.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Simone C, Zandonatti MA, Buchmeier MJ. Virology. 1994;198:455–465. doi: 10.1006/viro.1994.1057. [DOI] [PubMed] [Google Scholar]
- 37.Alsina J, Rabanal F, Giralt E, Albericio F. Tetrahedron Lett. 1994;35:9633–9635. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.