

Abstract
Reactive oxygen species are thought to be crucial for peroxisome proliferator-induced liver carcinogenesis. Free radicals have been shown to mediate the production of mitogenic cytokines by Kupffer cells and cause DNA damage in rodent liver. Previous in vivo experiments demonstrated that acute administration of the peroxisome proliferator di-(2-ethylhexyl) phthalate (DEHP) led to an increase in production of α-(4-pyridyl-1-oxide)-N-tert-butylnitrone (POBN) radical adducts in liver, an event that was dependent on Kupffer cell NADPH oxidase, but not peroxisome proliferator activated receptor (PPAR)α. Here, we hypothesized that continuous treatment with peroxisome proliferators will cause a sustained formation in POBN radical adducts in liver. Mice were fed diets containing either 4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio acetic acid (WY-14,643, 0.05% w/w), or DEHP (0.6% w/w) for up to three weeks. Liver-derived radical production was assessed in bile samples by measuring POBN-radical adducts using electron spin resonance. Our data indicate that WY-14,643 causes a sustained increase in POBN radical adducts in mouse liver and that this effect is greater than that of DEHP. To understand the molecular source of these radical species, NADPH oxidase-deficient (p47 phox-null) and PPARα-null mice were examined after treatment with WY-14,643. No increase in radicals was observed in PPARα-null mice that were treated with WY-14,643 for 3 weeks, while the response in p47 phox-nulls was similar to that of wild-type mice. These results show that PPARα, not NADPH oxidase, is critical for a sustained increase in POBN radical production caused by peroxisome proliferators in rodent liver. Therefore, peroxisome proliferator-induced POBN radical production in Kupffer cells may be limited to an acute response to these compounds in mouse liver.
INTRODUCTION
Peroxisome proliferators are a class of structurally diverse compounds that cause cancer in rodents by a non-genotoxic mechanism [1–3]. The high potential for human exposure and the known rodent carcinogenicity of these compounds has been the cause for intense debate for several decades regarding their potential adverse health effects in people [4]. The range of pleiotropic responses that these compounds induce in rodent liver includes increase in the size and number of peroxisomes in parenchymal cells, hepatomegaly, and induction of β-oxidation enzymes [5]. Considerable differences in metabolism and molecular changes induced by peroxisome proliferators in the liver, most predominantly the activation of the nuclear receptor peroxisome proliferator activated receptor (PPAR)α, have been identified between species [6]. In addition, PPARα-independent events that involve activation of Kupffer cells that involves production of reactive oxygen species have been also shown to occur after acute exposure to peroxisome proliferators in rodents [7].
Reactive oxygen species are implicated in the carcinogenesis mode of action of peroxisome proliferators. Oxidants have been shown to cause DNA damage, lipid peroxidation, and may also mediate signaling [8,9]. Within hepatocytes, these compounds activate transcription of genes encoding H2O2-generating enzymes, such as acyl-coA oxidase (ACO) and cytochrome P450 4A (CYP4A), and these events are known to be mediated by PPARα [10]. Studies using Kupffer cells demonstrated an increase in superoxide production in vitro 30 minutes after treatment with peroxisome proliferator, 4-chloro-6-(2, 3-xylidino)-2-pyrimidinylthio acetic acid (WY-14,643) [11]. In vivo studies of peroxisome proliferator-induced oxidants in rats using spin trap α-(4-pyridyl-1-oxide)-N-tert-butylnitrone (POBN) and electron spin resonance (ESR) detection reported an increase in POBN radical adducts in liver 2 hours following di-(2-ethylhexyl) phthalate (DEHP) treatment [7]. Furthermore, early reactive oxygen species generation as a result of DEHP administration was attributed to activation of NADPH oxidase in Kupffer cells, but not PPARα.
While many studies demonstrated a role for reactive oxygen species in the acute effects of peroxisome proliferators, it is not known whether Kupffer cell activation plays a role in radical generation during the long term exposure to peroxisome proliferators, and thus is a potential PPARα-independent mechanism of action of these compounds. The purpose of this study was to determine if peroxisome proliferator-derived reactive oxygen species production is in fact sustained and to identify the source of POBN radicals. These data provide direct evidence demonstrating that peroxisome proliferators cause a PPARα-dependent prolonged elevation in POBN radical adducts in rodent liver. Our findings suggest that Kupffer cell-derived POBN radical production is ephemeral and may be involved only in the acute phase of the response to these compounds in rodent liver.
MATERIALS AND METHODS
Animals
PPARα-null male mice (SV129 background; [12,13]), p47 phox -null male mice (C57BL/6J background; [14]) and corresponding wild-type counterparts (6–8 weeks of age) were used in these experiments. All animals used for this study were housed in sterilized cages in special facilities with a 12-hr night/day cycle. Temperature and relative humidity were held at 22 ± 2°C and 50 ± 5 %, respectively. The UNC Division of Laboratory Animal Medicine maintains these animal facilities, and veterinarians were always available to ensure animal health. All animals were given humane care in compliance with NIH and institutional guidelines and studies were performed according to approved protocols. Prior to experiments, animals were maintained on standard lab chow diet and purified water ad libitum.
Chemicals and Treatments
DEHP, WY-14,643, 2,2’-dipyridyl, and bathocuproinedisulfonic acid were obtained from Aldrich (Milwaukee, WI), and α-(4-pyridyl 1-oxide)-N-tert-butylnitrone (POBN) from Alexis (San Diego, CA). Control animals were given NIH-07 powdered diet. Treated animals were given the same diet blended with either DEHP or WY-14,643 at target concentrations of 0.6% w/w and 0.05% w/w, respectively. Diet was administered ad libitum for 3 days, 1 week or 3 weeks. Acutely treated mice were given one intragastric injection of either saline or DEHP at a dose of 1.2 g/kg.
Detection of Free Radicals in Bile
Animals were anesthetized with pentobarbital (75 mg/kg) and the spin trap POBN (1 g/kg, i.p.) was administered. The gallbladder was cannulated using a 10-cm long polyvinylchloride tube and bile samples were collected into Eppendorf tubes containing 50 μl of chelating agents, bathocupoinesulfonic acid (12 mM) and 2,2’-dipyridyl (30 mM) for 2 hrs. Bile samples were frozen immediately after collection and stored at -80°C until analyzed by electron spin resonance (ESR) spectroscopy. To consume endogenous ascorbic acid in bile which can act as reducing agent, an ascorbate oxidase spatula (Roche, Indianapolis, IN) was placed in the flat cell and O2 and N2 were bubbled through the sample in the flat cell for 10 minutes and 5 minutes, respectively. ESR spectra were recorded on a Bruker EMX ESR spectrometer with a super high Q cavity. Instrument settings were as follows: microwave power, 20 mW; modulation amplitude, 1 G; conversion time, 1.3 s; time constant, 1.3 s. Spectra were recorded on an IBM-compatible computer interfaced with the spectrophotometer and were analyzed to determine hyperfine coupling constants by computer simulation using EPR-WinSim software [15]. Graphical display of this data represents bile volume-corrected spectra amplitudes (peak-to-peak).
Acyl-coA Oxidase Activity and Expression
Acyl-coA oxidase (ACO) activity and expression are commonly used indicators of peroxisome induction [16]. The activity of ACO was determined by measuring formaldehyde, which is formed from oxidation of methanol by hydrogen peroxide. Liver tissue (100 mg) was homogenized in 10 volumes of 0.25 M sucrose buffer. A volume of 1.4 ml of reaction mixture (see [17] for details) was warmed at 37°C and mixed with 100 μl of homogenate. The reaction was terminated after 5 minutes by adding 40% trichloroacetic acid. Blanks were prepared in parallel, in which 40% trichloroacetic acid was added before homogenate. Samples and blanks were centrifuged to pellet protein and 1.0 ml of the supernatant was added to 0.4 ml of Nash reagent containing acetyl acetone, which reacts with formaldehyde to form diacetyl-dihydrolutidine [18]. The concentration of formaldehyde was measured spectrophotometrically at λ = 405 nm. Protein concentration was determined using the BCA protein assay [19].
ACO protein expression was measured by western blot analysis. Nuclear extracts were prepared from liver samples obtained from mice fed control or WY-14,643-containing diet for 3 weeks. Hepatic proteins (10 μg/lane) were separated on an SDS-PAGE and transferred to a nitrocellulose membrane. Immunodetection of ACO was performed using an anti-ACO polyclonal antibody (a generous gift from Dr. Janaradan Reddy, Northwestern University), followed by conjugation with an HRP-labeled rabbit anti-mouse secondary antibody. Chemiluminescent detection of protein was employed.
Statistics
Data are represented as mean values plus or minus the standard error for three to six animals per group. One-way ANOVA was used for statistical comparison with control (*). In cases in which more than two treatments were used, two-way ANOVA with Tukey's multiple comparison test was employed for statistical comparisons of between control-(*) or DEHP- (†) treated groups. A p value less than 0.05 was selected prior to the study to determine statistical differences between groups.
RESULTS
DEHP and WY-14,643 cause a sustained increase in free radicals
Acute treatment with DEHP has been shown to cause an increase in liver POBN radical adduct formation in vivo [7], but it has not yet been determined by direct detection methods whether such radical production remains elevated for longer than 2 hrs. To investigate whether peroxisome proliferators cause a long-term increase in radical spececies in rodent liver, mice were fed either control or DEHP-containing diet for 3 weeks. In order to establish if radical species that are produced after continuous treatment are similar to those that form after acute administration of DEHP, some mice were injected with either saline or DEHP (1.2 g/kg) intragastrically immediately before bile collection. Figure 1 shows that a DEHP-induced increase in POBN radicals is observed after both acute (2 hrs) and sub-chronic (3 weeks) treatment with the peroxisome proliferator compound. When injected in vivo, POBN forms a stable radical adduct (with a t1/2 ranging from 10–15 hrs, data not shown). Thus, the rate of radical production is proportional to the ESR spectrum amplitude and the level of induction of radical production (i.e., the amount of the radical species being produced) caused by treatment with DEHP is comparable at both time points (3.2- and 2.6-fold over control, respectively).
Figure 1. Production of POBN radical adducts caused by peroxisome proliferators is sustained.
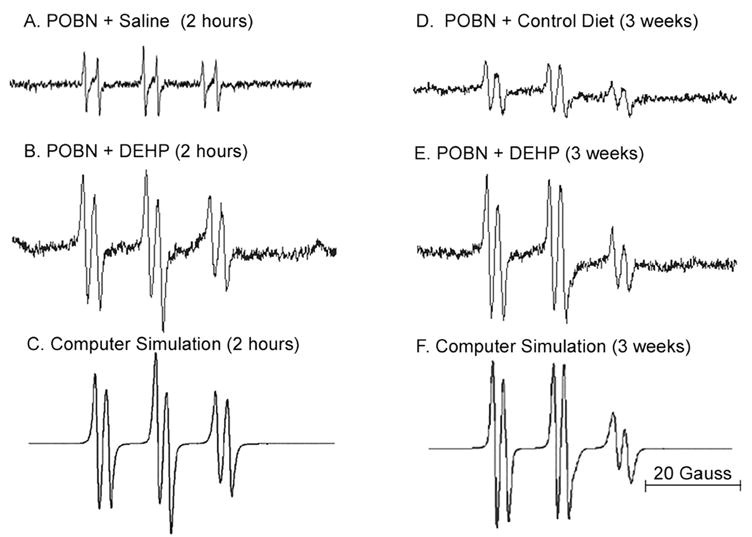
Representative spectra and computer simulations of radical adducts in C57BL/6J mouse liver following DEHP treatment are shown. Bile was collected for 2 hours after POBN administration (i.p.). Radical adducts in bile were detected using ESR. (A) ESR spectrum of radical adducts detected 2 hours following i.g. treatment with saline. (B) Same as A except treated with DEHP (1.2 g/kg). (C) Computer simulation of spectrum in panel B. (D) ESR spectrum of radical adducts detected following 3 weeks of feeding of NIH-07 diet. (E) Same as panel D, except diet contained DEHP (0.6% w/w). (F) Computer simulation of spectrum in panel E.
Computer simulations of the ESR spectra for DEHP-induced radicals produced at 2 hours and 3 weeks (Figure 1C and 1F, respectively) suggest that the radical species responsible for POBN adduct formation were similar. For both time points, the POBN-trapped radicals produced a composite six-line spectrum. Computer simulation of radical adducts produced following acute DEHP treatment (Figure 1C) possess hyperfine coupling constants of (I) aN = 15.6 G, aβH = 3.2 G and (II) aN = 15.8 G, aβH = 2.7 G, and revealed a third species (III) aH = 2.0 G due to the ascorbate radical (19). The relative amount of each adduct species was (I) 55%, (II) 35% and (III) 11%. The predominant radical species (I) appears to be derived from POBN/•CO2− [20], while species II likely originated from a lipid-centered radical [21]. Three week treatment with DEHP produces similar radical species with coupling constants (I) aN = 15.7 G, aβH = 3.0 G, (II) aN = 15.8 G, aβH = 2.7 G, and (III), aH = 1.9 G derived from the same three species, POBN/•CO2–(49%), POBN/•L (41%), and the ascorbate radical III contributing 10%. The significant presence of formate-derived POBN/•CO2– radical adducts in both samples is likely caused by reactive species produced by oxidizing enzymes. In a Fenton-like reaction, endogenous formate is oxidized to form a carbon dioxide anion radical [21].
While our data indicates that DEHP is capable of invoking production of radical species in both acute (2 hrs) and sub-chronic (3 weeks) studies, it has been shown that DEHP is a relatively weak carcinogen that fails to produce a sustained induction of proliferative response in rodent liver [5]. To compare the level of POBN radical induction by two classical peroxisome proliferator compounds that differ in their carcinogenic potency, mice were fed a diet with either DEHP (0.6% w/w), or WY-14,643 (0.05% w/w) for 3 days, 1, or 3 weeks and radicals in bile were measured by ESR. Both DEHP and WY-14,643 caused a time-dependent increase in POBN radical adducts (Table 1 and Figure 2), with the latter treatment causing the most significant increase over control levels beginning at 1 week of treatment. These findings support the hypothesis that the carcinogenic potency of peroxisome proliferators is likely to be related to their ability to cause oxidative stress in liver [22].
Table 1.
ESR spectra amplitudes for peroxisome proliferator-treated C57BL/6J mice.
| Time course | Treatment | N | Liver Weight (% Body Wt.)a | Bile Volume (ml) | ESR Amplitude (Arbitrary Units) | Vol Corrected ESR Amplitude (Arbitrary Units)a |
|---|---|---|---|---|---|---|
| Control | 6 | 4.6 ± 0.3 | 0.15 ± 0.01 | 23.63 ± 3.47 | 3.39 ± 0.38 | |
| 3 days | DEHP | 6 | 5.4 ± 0.5b | 0.17 ± 0.02 | 9.43 ± 1.34 | 1.57 ± 0.25 |
| WY | 3 | 6.5 ± 0.9c | 0.23 ± 0.03c | 8.89 ± 3.52 | 1.94 ± 0.56 | |
| Control | 4 | 5.7 ± 0.5 | 0.18 ± 0.03 | 13.83 ± 2.16 | 2.96 ± 0.78 | |
| 1 week | DEHP | 5 | 7.1 ± 1.3 | 0.19 ± 0.04 | 14.62 ± 1.99 | 3.06 ± 0.98 |
| WY | 6 | 8.2 ± 1.0c | 0.44 ± 0.08c | 12.69 ± 2.49 | 5.48 ± 1.64b | |
| Control | 4 | 4.4 ± 0.3 | 0.17 ± 0.01 | 11.96 ± 0.72 | 1.99 ± 0.18 | |
| 3 weeks | DEHP | 6 | 6.3 ± 0.3b | 0.19 ± 0.02 | 27.55 ± 1.57 | 5.23 ± 0.65b |
| WY | 5 | 11.3 ± 1.1c | 0.76 ± 0.08c | 10.78 ± 2.00 | 7.62 ± 0.57c |
Data is represented as mean ± SEM.
Bile volume was used to determine a volume corrected ESR amplitude.
Denotes statistically significance from Control-fed animals (p<0.05).
Denotes statistical significance from DEHP-treated animals (p<0.05).
Figure 2. WY-14,643 causes a greater induction of POBN radical adducts in C57BL/6J mice.
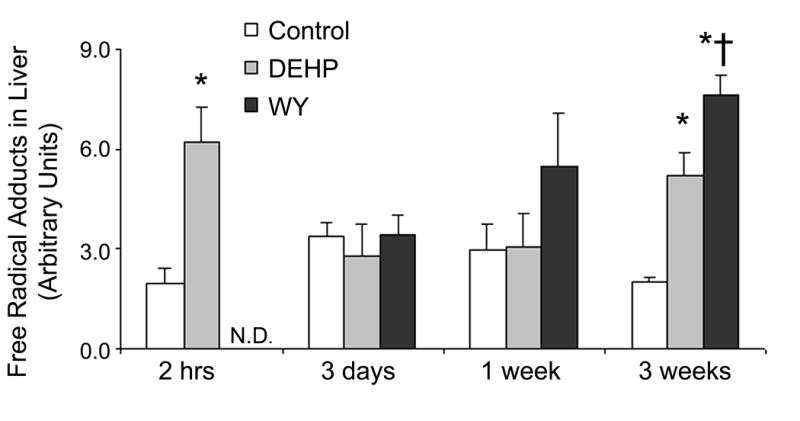
Mice were fed either a DEHP (0.6% w/w)-, or WY-14,643 (0.05% w/w)-containing diet for 3 days, 1 week, or 3 weeks. Free radical adducts in bile were measured by ESR 2 hours after POBN (i.p.) administration. Statistical significance (p<0.05) is indicated with asterisks (*, different from Control; †, different from DEHP). N.D: No Data
PPARα is essential to long-term reactive oxygen species production by peroxisome proliferators
Earlier studies demonstrated that acute production of DEHP-induced radical species in rodent liver depends on Kupffer cell NADPH oxidase, but not PPARα [7]. To determine if long-term production of POBN-detectable reactive oxygen species is mediated by either Kupffer cell- or hepatocyte-related molecular events, p47 phox- and PPARα- null mice (along with their corresponding wild-types) were treated with WY-14,643 for 3 weeks. POBN trapped radicals were collected in vivo from liver using bile and quantitated using ESR. Though the knock-out mice used in this study were on different background strains (SV129 and C57BL/6J), no strain-associated differences in radical production were observed. Both wild-type strains show a significant increase in radicals caused by dietary treatment with WY-14,643 for 3 weeks (Figure 3). Interestingly, induction of radical production by continuous treatment with WY-14,643 occurs in NADPH-oxidase deficient mice (p47 phox-null), but not in PPARα-null mice. These results clearly demonstrate that PPARα is required for prolonged formation of POBN radical adducts caused by peroxisome proliferators.
Figure 3. Prolonged radical species production is PPARα dependent.
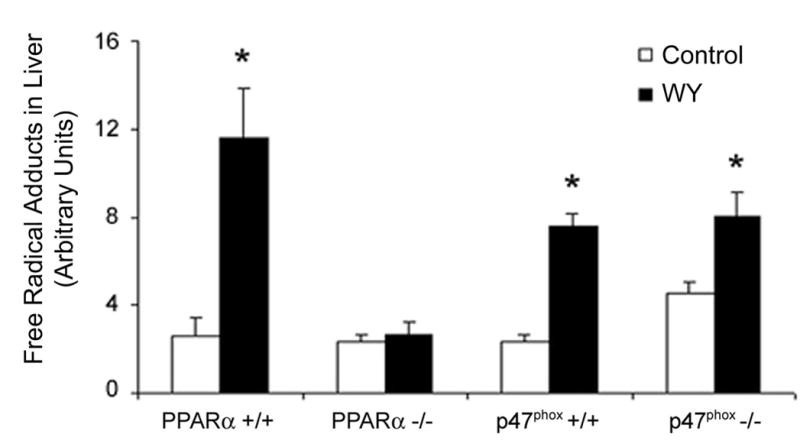
PPARα-null and p47 phox-null mice were fed a WY-containing diet (0.05% w/w) for 3 weeks. Following POBN administration, bile was collected for 2 hours. POBN radical adducts were detected in bile using ESR. Statistical significance (p<0.05) is indicated with an asterisk as compared to the data for control diet-fed animals.
It has been hypothesized that induction of peroxisomal oxidases by peroxisome proliferators is important for the mode of action of these agents since it may lead to oxidative damage of DNA, proteins and lipids in rodent liver [23]. To determine if induction of peroxisomal enzymes correlates with sustained POBN radical production observed in this study, activity of acyl-CoA oxidase (ACO) was determined in liver homogenates from wild-type mice fed either DEHP or WY-14,643-containing diets for up to 3 weeks. ACO is widely used as a marker of peroxisomal β-oxidation [10,24] and increased expression or activity is hallmark to peroxisome proliferators. Both DEHP and WY-14,643 cause a progressive increase in ACO activity, with more potent WY causing a greater increase of almost 30-fold over control as early as 3 days following diet initiation (Figure 4). Induction of ACO by DEHP and WY-14,643 persists for up to three weeks. These results corroborate that reactive oxygen species levels are sustained and progressively increase with the length of treatment. Measurements of ACO activity in liver from both knockout mouse strains revealed an induction in ACO in p47 phox-null mice (Figure 5A). No change of enzyme activity was observed in PPARα-null mice. ACO protein expression (Figure 5B) revealed a similar trend. When taken collectively, these results confirm that long term production of POBN-reactive oxygen species is PPARα–dependent, and that peroxisomal enzymes are likely a primary source.
Figure 4. DEHP and WY-14,643 cause a sustained increase acyl-CoA oxidase activity.
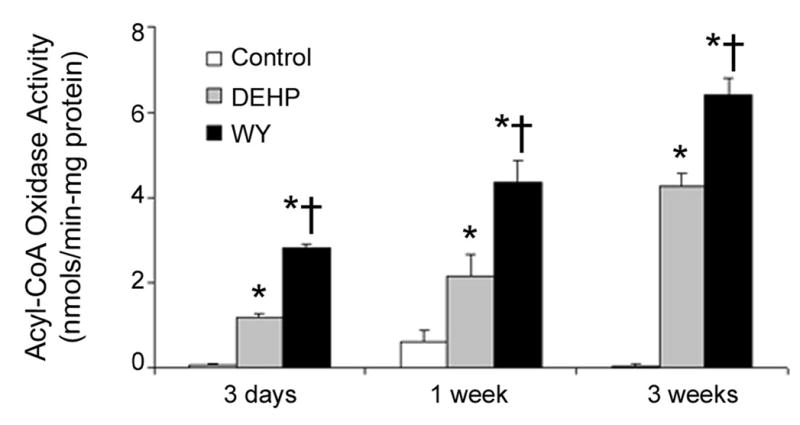
Mice were fed either a DEHP (0.6% w/w) or WY-14,643 (0.05% w/w)-containing diet for 3 days, 1 week, or 3 weeks. ACO activity was measured as described in the Experimental Procedures. Statistical significance (p<0.05) is indicated with asterisks (*, different from Control; †, different from DEHP).
Figure 5. Induction of ACO correlates with peroxisome proliferator-induced radical production.
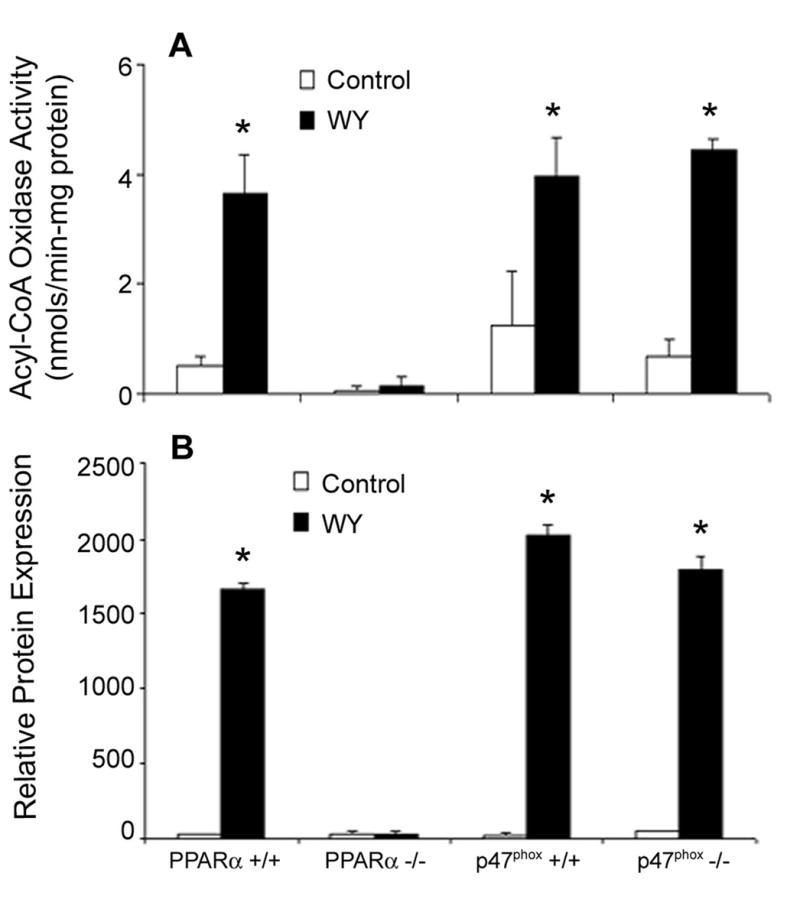
PPARα-null and p47 phox-null mice were fed WY-14,643 (0.05% w/w) containing diet for 3 weeks. (A) ACO activity was measured as described in the Experimental Procedures. (B) ACO protein expression was measured by western blot analysis. Statistical significance (p<0.05) is indicated with an asterisk as compared to the data for Control-fed animals.
DISCUSSION
Involvement of reactive oxygen species in the mode of action of peroxisome proliferators
Reactive oxygen species are thought to be intimately associated with the mechanism of tumorigenesis by peroxisome proliferators. This assumption is based to a large degree on the fact that various proteins that are induced by these chemicals in liver parenchymal cells (peroxisomes, mitochondria and microsomes) are prone to formation of hydrogen peroxide and free radical species. Thus, it was hypothesized that such overproduction of oxidants might cause DNA damage and lead to mutations and cancer [10,23]. In addition, recent discoveries show that reactive oxygen species play an important signaling role in a rapid increase of parenchymal cell proliferation caused by peroxisome proliferators {Rusyn, 2000 4357 /id}. Collectively, it appears that oxidant-related molecular events are intertwined with other pathways activated by peroxisome proliferators in vivo in rodent liver.
It was initially hypothesized that fatty acyl-CoA oxidase in the peroxisome is the enzyme responsible for oxidative stress by peroxisome proliferators [26,27]; however, mice lacking this protein, instead of being protected from chemically induced liver cancer, develop liver tumors spontaneously, possibly as a result of a hyper-activation of PPARα by unmetabolized lipids [28]. A number of indirect confirmations for peroxisome proliferator-initiated increases in reactive oxygen species have been collected over past two decades; however, the causative relevance of some of this evidence to the carcinogenic effect of peroxisome proliferators has been questioned and contrasting views have been presented (reviewed in [29]). Therefore, we provide direct in vivo evidence for sustained production of oxidants after treatment with peroxisome proliferators, as well as information on whether POBN radical adducts are produced by PPARα-dependent mechanisms, both of which are critical for understanding the mode of action of these agents.
Direct evidence for peroxisome proliferator-induced sustained production of POBN radical adducts in rodent liver
Despite the fact that few question a role of oxidative stress in the mechanism of action of these compounds, direct evidence that radicals are produced under conditions of continuous exposure to peroxisome proliferators, as well as knowledge on precise molecular source(s) of reactive oxygen species that are involved were still lacking. It has been previously reported that PPARα is not required for generation of reactive oxygen species in mouse liver after acute exposure to peroxisome proliferators [7]. The same study demonstrated that Kupffer cell NADPH oxidase was the source of radical species production in rodent liver within hours after administration of DEHP. Thus, Kupffer cells have been suggested to be a potential source of radicals in rat and mouse liver after treatment with peroxisome proliferators [7,30].
In this study, we provide the first direct evidence of peroxisome proliferator-induced sustained radical production in vivo in mouse liver. It appears that ability of peroxisome proliferators to increase radical production in mouse liver correlates with the carcinogenic potency. Specifically, WY-14,643, which is known to be highly tumorigenic in rodents, causes greater radical production than DEHP, a weaker rodent liver carcinogen.
We also show that irrespective of the duration of treatment with peroxisome proliferators when the cellular source of radicals (i.e., Kupffer cell or hepatocyte) may differ (see below), the ultimate macromolecule-reactive species produced in mouse liver are similar and consist of roughly equal amounts of •CO2– and •L. While Kupffer cell-derived radicals (e.g., superoxide anion) can either react directly with the surrounding lipid membrane (at low pHs [31]), or be converted to a hydroxyl radical that will react with lipids; in hepatocytes, excess peroxisomal H2O2 conversion to a reactive hydroxyl radical in the presence of iron is the presumed mechanism of formation of lipid peroxides [32]. It should be noted that the utility of spin trapping with POBN for direct detection of hydroxyl radical, superoxide or nitric oxide is limited [33]; thus, the results of this study should be interpreted with caution. However, the high reactivity of the hydroxyl radical, abundance of lipids in liver and the chain reaction nature of lipid peroxidation are likely to facilitate the formation of •L as a key terminal radical detected here by POBN.
Our results indicate that continuous treatment with peroxisome proliferators causes a time-dependent significant increase in POBN radical adducts in mouse liver. It is interesting, however, that the initial increase in POBN radical adducts following acute exposure to DEHP was short-lived, as an appreciable increase in radical production is not observed until after 3 weeks of treatment with DEHP and after 1 week of treatment with WY-14,643. This result implies that the early, Kupffer cell-mediated, effect on increased reactive oxygen species is not sustained and we suggest that Kupffer cell activation by peroxisome proliferators is short lived (see below).
The peroxisome proliferator-induced PPARα-dependent (see below) prolonged radical production in liver, detected here by POBN spin trapping and ESR, may result from several sources in the parenchymal cell. A number of peroxisomal (e.g., fatty acyl-CoA oxidase) and microsomal (e.g., 4A superfamily of cytochrome P450 enzymes) oxidases are regulated by PPARα and are involved in the catabolism of long chain and very long chain fatty acids by β- and ω-oxidation, respectively. These enzyme systems are "leaking" electrons and are known to generate considerable amounts of secondary reactive oxygen species even under normal physiologic conditions [10].
Disproportionately large increases in expression of hydrogen peroxide-generating fatty acyl-CoA oxidase, as compared to hydrogen peroxide-degrading catalase, have been reported following treatment with peroxisome proliferators in rodents [24,27]. However, given the extremely high rate at which peroxisomal catalase converts H2O2 into H2O and O2, it should not escape peroxisomes [34]. Furthermore, peroxisomal β-oxidation is limited by substrate availability. In fact, it has been shown that treatment with peroxisome proliferators increased H2O2 in vitro, but not in the perfused liver because fatty acid supply is rate-limiting in intact cells [35,36]. Indeed, the timing of the increases in radical production observed in this study did not correlate with that for the induction of ACO protein level and activity. However, peroxisomes devoid of catalase but capable of production of H2O2 via beta-oxidation of fatty acyl CoA compounds or via the activity of urate oxidase have been observed following clofibrate treatment and massive liver regeneration [37,38]. Thus, with continuous peroxisome proliferator treatment, there may be increased formation of microperoxisomes that leak catalase; hydrogen peroxide produced in the microperoxisome would not be scavenged immediately, but rather may be available for reaction with cellular components ultimately leading to the formation of POBN radical adducts. Marked induction of CYP4A [39] may be another likely source of oxidants under condition of continuous treatment with peroxisome proliferators.
An alternative explanation for lack of increase in radicals until after 1 week of treatment could also be related to peroxisome proliferator-induced changes in iron homeostasis in rodent liver. Reactive species produced at early time points (2 hours or less) are presumably Kupffer-cell derived radicals (e.g., superoxide anion) that would not depend on availability of excess transition metals for conversion to POBN/ESR-detectable species. However, a sufficient level of iron in the liver is critical for conversion of non-radical oxidants, like H2O2, to POBN-reactive radical species. Several studies have shown that dietary treatment with peroxisome proliferators in rodents creates a condition of iron overload in liver [40–42]. An increase in hepatic iron stores is thought to be one of the potential reasons for chronic oxidative stress in liver by peroxisome proliferators. In fact, an enhancement in lipid peroxidation as a result of hepatic iron overload after treatment with WY-14,643 was observed recently [42]. In addition to increased intracellular pools of iron, altered expression of proteins responsible for iron transport from the liver to plasma (i.e., transferrin receptors, ferritin, and iron regulatory protein 1) has been reported [40,43].
NADPH oxidase is not a source of POBN radical adducts under condition of chronic administration of peroxisome proliferators
Several studies used p47 phox-null mouse model to demonstrate that Kupffer cell NADPH oxidase-derived reactive species, as detected using POBN and ESR, are critical for the pathogenesis of liver and lung injury [44,45], as well as the early effects of peroxisome proliferators in mouse liver [7,46]. Since it was not known whether Kupffer cell-specific events play a role in long-term effects of peroxisome proliferators, this study determined if this cell type may be involved in peroxisome proliferator-induced reactive oxygen species production when animals are administered these compounds for up to 3 weeks. We show here that radical species formation still occurs in the absence of active NADPH oxidase (as observed in p47 phox-null mice), but is PPARα-mediated. The importance of PPARα for hepatocarcinogenicity of peroxisome proliferators is well known [47]. Importantly, here we demonstrate, by direct measurements of POBN radical adducts in liver, that PPARα-dependent pathways are responsible for the long-term increase in oxidants after continuous treatment with peroxisome proliferators. This work, together with measurements of the functional outcome of increased production of reactive species such as oxidative DNA damage under similar conditions [48], establishes a link between activation of PPARα, radical production and DNA damage, a key step in the mechanism of carcinogenesis.
In conclusion, this study is the first to provide direct in vivo evidence that increased production of POBN radical adducts in mouse liver is sustained following dietary treatment with peroxisome proliferators. Despite the apparent temporal shift in the cellular source of radicals (from Kupffer cells to hepatocytes) as peroxisome proliferator treatment is continued, there appears to be no difference in the radical species that are produced. Finally, we have demonstrated that long term reactive oxygen species production is mediated by PPARα and that NADPH oxidase-derived reactive species may only be important as early responses to peroxisome proliferators.
Acknowledgments
We thank Dr. Wonyoung Tak with the University of North Carolina-Chapel Hill for providing assistance with mouse bile collection.
ABBREVIATIONS
- WY-14,643
4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio acetic acid
- POBN
α-(4-pyridyl-1-oxide)-N-tert-butylnitrone
- ACO
fatty acyl-coA oxidase
- CYP4A
cytochrome P450 4A
- DEHP
di-(2-ethylhexyl) phthalate
- ESR
electron spin resonance spectroscopy
- PPAR
peroxisome proliferator activated receptor
- PP
peroxisome proliferators
Footnotes
Financial support for these studies was provided, in part, by grants from the National Institutes of Health (NIH): R01-ES12686, P30-ES10126, U19-ES11391, K22-ES11660 and F32-ES13342.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lake BG. Peroxisome proliferation: current mechanisms relating to nongenotoxic carcinogenesis. Toxicol Lett. 1995;82–83:673–681. doi: 10.1016/0378-4274(95)03513-3. [DOI] [PubMed] [Google Scholar]
- 2.Reddy JK, Azarnoff DL, Hignite CE. Hypolipidaemic hepatic peroxisome proliferators form a novel class of chemical carcinogens. Nature. 1980;283:397–398. doi: 10.1038/283397a0. [DOI] [PubMed] [Google Scholar]
- 3.Reddy JK, Rao MS. Peroxisome proliferation and hepatocarcinogenesis. IARC Sci Publ. 1992:225–235. [PubMed] [Google Scholar]
- 4.Cattley RC, DeLuca J, Elcombe C, Fenner-Crisp P, Lake BG, Marsman DS, Pastoor TA, Popp JA, Robinson DE, Schwetz B, Tugwood J, Wahli W. Do peroxisome proliferating compounds pose a hepatocarcinogenic hazard to humans? Regul Toxicol Pharmacol. 1998;27:47–60. doi: 10.1006/rtph.1997.1163. [DOI] [PubMed] [Google Scholar]
- 5.Marsman DS, Cattley RC, Conway JG, Popp JA. Relationship of hepatic peroxisome proliferation and replicative DNA synthesis to the hepatocarcinogenicity of the peroxisome proliferators di(2-ethylhexyl)phthalate and [4-chloro-6-(2,3-xylidino)-2- pyrimidinylthio]acetic acid (Wy-14,643) in rats. Cancer Res. 1988;48:6739–6744. [PubMed] [Google Scholar]
- 6.Peters JM, Cheung C, Gonzalez FJ. Peroxisome proliferator-activated receptor-alpha and liver cancer: where do we stand? J Mol Med. 2005;83:774–785. doi: 10.1007/s00109-005-0678-9. [DOI] [PubMed] [Google Scholar]
- 7.Rusyn I, Kadiiska MB, Dikalova A, Kono H, Yin M, Tsuchiya K, Mason RP, Peters JM, Gonzalez FJ, Segal BH, Holland SM, Thurman RG. Phthalates rapidly increase production of reactive oxygen species in vivo: role of Kupffer cells. Mol Pharmacol. 2001;59:744–750. doi: 10.1124/mol.59.4.744. [DOI] [PubMed] [Google Scholar]
- 8.Conway JG, Tomaszewski KE, Olson MJ, Cattley RC, Marsman DS, Popp JA. Relationship of oxidative damage to the hepatocarcinogenicity of the peroxisome proliferators di(2-ethylhexyl)phthalate and Wy-14,643. Carcinogenesis. 1989;10:513–519. doi: 10.1093/carcin/10.3.513. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki YJ, Forman HJ, Sevanian A. Oxidants as stimulators of signal transduction. Free Radic Biol Med. 1997;22:269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- 10.Yeldandi AV, Rao MS, Reddy JK. Hydrogen peroxide generation in peroxisome proliferator-induced oncogenesis. Mutat Res. 2000;448:159–177. doi: 10.1016/s0027-5107(99)00234-1. [DOI] [PubMed] [Google Scholar]
- 11.Rose ML, Rivera CA, Bradford BU, Graves LM, Cattley RC, Schoonhoven R, Swenberg JA, Thurman RG. Kupffer cell oxidant production is central to the mechanism of peroxisome proliferators. Carcinogenesis. 1999;20:27–33. doi: 10.1093/carcin/20.1.27. [DOI] [PubMed] [Google Scholar]
- 12.Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. Targeted disruption of the alpha isoform of the peroxisome proliferator- activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995;15:3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akiyama TE, Nicol CJ, Fievet C, Staels B, Ward JM, Auwerx J, Lee SS, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-alpha regulates lipid homeostasis, but is not associated with obesity: studies with congenic mouse lines. J Biol Chem. 2001;276:39088–39093. doi: 10.1074/jbc.M107073200. [DOI] [PubMed] [Google Scholar]
- 14.Jackson SH, Gallin JI, Holland SM. The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med. 1995;182:751–758. doi: 10.1084/jem.182.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong Z, Froh M, Connor HD, Li X, Conzelmann LO, Mason RP, Lemasters JJ, Thurman RG. Prevention of hepatic ischemia-reperfusion injury by green tea extract. Am J Physiol Gastrointest Liver Physiol. 2002;283:G957–G964. doi: 10.1152/ajpgi.00216.2001. [DOI] [PubMed] [Google Scholar]
- 16.Tomaszewski KE, Derks MC, Melnick RL. Acyl CoA oxidase is the most suitable marker for hepatic peroxisomal changes caused by treatment of F344 rats with di(2-ethylhexyl)phthalate. Toxicol Lett. 1987;37:203–212. doi: 10.1016/0378-4274(87)90133-0. [DOI] [PubMed] [Google Scholar]
- 17.Rose ML, Germolec D, Arteel GE, Schoonhoven R, Thurman RG. Dietary glycine prevents increases in hepatocyte proliferation caused by the peroxisome proliferator WY-14,643. Chem Res Toxicol. 1997;10:1198–1204. doi: 10.1021/tx970079u. [DOI] [PubMed] [Google Scholar]
- 18.Nash T. The colorimetric estimation of formaldehyde by means of the Hantzsch reaction. Biochem J. 1953;55:416–421. doi: 10.1042/bj0550416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 20.Dikalova AE, Kadiiska MB, Mason RP. An in vivo ESR spin-trapping study: free radical generation in rats from formate intoxication--role of the Fenton reaction. Proc Natl Acad Sci U S A. 2001;98:13549–13553. doi: 10.1073/pnas.251091098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayashi Y, Ueda Y, Nakajima A, Mitsuyama Y. EPR evidence of hydroxyl radical generation as an initiator of lipid peroxidation in amyloid beta-protein-stimulated PC12 cells. Brain Res. 2004;1025:29–34. doi: 10.1016/j.brainres.2004.07.067. [DOI] [PubMed] [Google Scholar]
- 22.Rusyn I, Denissenko MF, Wong VA, Butterworth BE, Cunningham ML, Upton PB, Thurman RG, Swenberg JA. Expression of base excision repair enzymes in rat and mouse liver is induced by peroxisome proliferators and is dependent upon carcinogenic potency. Carcinogenesis. 2000;21:2141–2145. doi: 10.1093/carcin/21.12.2141. [DOI] [PubMed] [Google Scholar]
- 23.Reddy JK, Rao MS. Oxidative DNA damage caused by persistent peroxisome proliferation: its role in hepatocarcinogenesis. Mutat Res. 1989;214:63–68. doi: 10.1016/0027-5107(89)90198-x. [DOI] [PubMed] [Google Scholar]
- 24.Nemali MR, Reddy MK, Usuda N, Reddy PG, Comeau LD, Rao MS, Reddy JK. Differential induction and regulation of peroxisomal enzymes: predictive value of peroxisome proliferation in identifying certain nonmutagenic carcinogens. Toxicol Appl Pharmacol. 1989;97:72–87. doi: 10.1016/0041-008x(89)90056-2. [DOI] [PubMed] [Google Scholar]
- 25.Rusyn I, Rose ML, Bojes HK, Thurman RG. Novel role of oxidants in the molecular mechanism of action of peroxisome proliferators. Antiox Redox Signal. 2000;2:607–621. doi: 10.1089/15230860050192350. [DOI] [PubMed] [Google Scholar]
- 26.Nemali MR, Usuda N, Reddy MK, Oyasu K, Hashimoto T, Osumi T, Rao MS, Reddy JK. Comparison of constitutive and inducible levels of expression of peroxisomal beta-oxidation and catalase genes in liver and extrahepatic tissues of rat. Cancer Res. 1988;48:5316–5324. [PubMed] [Google Scholar]
- 27.Reddy JK, Goel SK, Nemali MR, Carrino JJ, Laffler TG, Reddy MK, Sperbeck SJ, Osumi T, Hashimoto T, Lalwani ND. Transcription regulation of peroxisomal fatty acyl-CoA oxidase and enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase in rat liver by peroxisome proliferators. Proc Natl Acad Sci U S A. 1986;83:1747–1751. doi: 10.1073/pnas.83.6.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan CY, Pan J, Usuda N, Yeldandi AV, Rao MS, Reddy JK. Steatohepatitis, spontaneous peroxisome proliferation and liver tumors in mice lacking peroxisomal fatty acyl-CoA oxidase. Implications for peroxisome proliferator-activated receptor alpha natural ligand metabolism. J Biol Chem. 1998;273:15639–15645. doi: 10.1074/jbc.273.25.15639. [DOI] [PubMed] [Google Scholar]
- 29.Rusyn I, Peters JM, Cunningham ML. Modes of action and species-specific effects of di-(2-ethylhexyl)phthalate in the liver. Crit Rev Toxicol. 2006;36:459–479. doi: 10.1080/10408440600779065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rose ML, Rusyn I, Bojes HK, Belyea J, Cattley RC, Thurman RG. Role of Kupffer cells and oxidants in signaling peroxisome proliferator- induced hepatocyte proliferation. Mutat Res. 2000;448:179–192. doi: 10.1016/s0027-5107(99)00235-3. [DOI] [PubMed] [Google Scholar]
- 31.Fridovich I. Biological effects of the superoxide radical. Arch Biochem Biophys. 1986;247:1–11. doi: 10.1016/0003-9861(86)90526-6. [DOI] [PubMed] [Google Scholar]
- 32.Dunford HB. Free radicals in iron-containing systems. Free Radic Biol Med. 1987;3:405–421. doi: 10.1016/0891-5849(87)90019-0. [DOI] [PubMed] [Google Scholar]
- 33.Towner RA, Qian SY, Kadiiska MB, Mason RP. In vivo identification of aflatoxin-induced free radicals in rat bile. Free Radic Biol Med. 2003;35:1330–1340. doi: 10.1016/j.freeradbiomed.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 34.Nichols P, Schonbaum GR. Catalases. In: Boyer PD, editor. The Enzymes. New York: Academic Press; 1963. pp. 147–225. [Google Scholar]
- 35.Handler JA, Thurman RG. Catalase-dependent ethanol oxidation in perfused rat liver. Requirement for fatty-acid-stimulated H2O2 production by peroxisomes. Eur J Biochem. 1988;176:477–484. doi: 10.1111/j.1432-1033.1988.tb14305.x. [DOI] [PubMed] [Google Scholar]
- 36.Handler JA, Seed CB, Bradford BU, Thurman RG. Induction of peroxisomes by treatment with perfluorooctanoate does not increase rates of H2O2 production in intact liver. Toxicol Lett. 1992;60:61–68. doi: 10.1016/0378-4274(92)90047-n. [DOI] [PubMed] [Google Scholar]
- 37.Klucis E, Crane DI, Hughes JL, Poulos A, Masters CJ. Identification of a catalase-negative sub-population of peroxisomes induced in mouse liver by clofibrate. Biochim Biophys Acta. 1991;1074:294–301. doi: 10.1016/0304-4165(91)90167-f. [DOI] [PubMed] [Google Scholar]
- 38.Oikawa I, Novikoff PM. Catalase-negative peroxisomes: transient appearance in rat hepatocytes during liver regeneration after partial hepatectomy. Am J Pathol. 1995;146:673–687. [PMC free article] [PubMed] [Google Scholar]
- 39.Milton MN, Elcombe CR, Gibson GG. On the mechanism of induction of microsomal cytochrome P450IVA1 and peroxisome proliferation in rat liver by clofibrate. Biochem Pharmacol. 1990;40:2727–2732. doi: 10.1016/0006-2952(90)90594-b. [DOI] [PubMed] [Google Scholar]
- 40.Powanda MC, Blackburn BS, Bostian KA, Fowler JP, Hauer EC, Pekarek RS. Clofibrate-induced alterations in zinc, iron and copper metabolism. Biochem Pharmacol. 1978;27:125–127. doi: 10.1016/0006-2952(78)90270-8. [DOI] [PubMed] [Google Scholar]
- 41.Huang HL, Shaw NS. Role of hypolipidemic drug clofibrate in altering iron regulatory proteins IRP1 and IRP2 activities and hepatic iron metabolism in rats fed a low-iron diet. Toxicol Appl Pharmacol. 2002;180:118–128. doi: 10.1006/taap.2002.9378. [DOI] [PubMed] [Google Scholar]
- 42.Fischer JG, Glauert HP, Yin T, Sweeney-Reeves ML, Larmonier N, Black MC. Moderate iron overload enhances lipid peroxidation in livers of rats, but does not affect NF-kappaB activation induced by the peroxisome proliferator, Wy-14,643. J Nutr. 2002;132:2525–2531. doi: 10.1093/jn/132.9.2525. [DOI] [PubMed] [Google Scholar]
- 43.Hertz R, Seckbach M, Zakin MM, Bar-Tana J. Transcriptional suppression of the transferrin gene by hypolipidemic peroxisome proliferators. J Biol Chem. 1996;271:218–224. doi: 10.1074/jbc.271.1.218. [DOI] [PubMed] [Google Scholar]
- 44.Kono H, Rusyn I, Yin M, Gabele E, Yamashina S, Dikalova A, Kadiiska MB, Connor HD, Mason RP, Segal BH, Bradford BU, Holland SM, Thurman RG. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest. 2000;106:867–872. doi: 10.1172/JCI9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sato K, Kadiiska MB, Ghio AJ, Corbett J, Fann YC, Holland SM, Thurman RG, Mason RP. In vivo lipid-derived free radical formation by NADPH oxidase in acute lung injury induced by lipopolysaccharide: a model for ARDS. FASEB J. 2002;16:1713–1720. doi: 10.1096/fj.02-0331com. [DOI] [PubMed] [Google Scholar]
- 46.Rusyn I, Yamashina S, Segal BH, Schoonhoven R, Holland SM, Cattley RC, Swenberg JA, Thurman RG. Oxidants from nicotinamide adenine dinucleotide phosphate oxidase are involved in triggering cell proliferation in the liver due to peroxisome proliferators. Cancer Res. 2000;60:4798–4803. [PubMed] [Google Scholar]
- 47.Peters JM, Cattley RC, Gonzalez FJ. Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis. 1997;18:2029–2033. doi: 10.1093/carcin/18.11.2029. [DOI] [PubMed] [Google Scholar]
- 48.Rusyn I, Asakura S, Pachkowski B, Bradford BU, Denissenko MF, Peters JM, Holland SM, Reddy JK, Cunningham ML, Swenberg JA. Expression of base excision DNA repair genes is a sensitive biomarker for in vivo detection of chemical-induced chronic oxidative stress: Identification of the molecular source of radicals responsible for DNA damage by peroxisome proliferators. Cancer Res. 2004;64:1050–1057. doi: 10.1158/0008-5472.can-03-3027. [DOI] [PubMed] [Google Scholar]