

Abstract
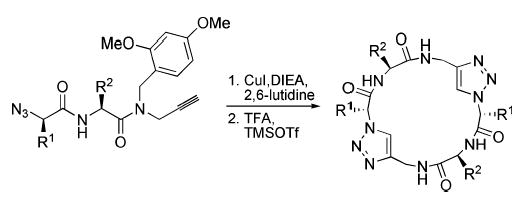
A tandem dimerization–macrocyclization approach using 1,3-dipolar azide–alkyne cycloaddition reactions has been employed in the facile and convergent solution phase syntheses of C2 symmetric cyclic peptide scaffolds bearing triazole ε2-amino acids as dipeptide surrogates.
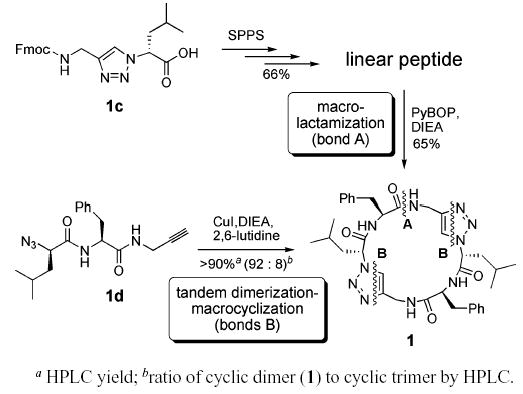
Hydrogen bond-directed self-assembly of appropriately designed cyclic peptides is an effective method for the preparation of organic nanotubes1,2 for applications in the solid state, solution phase, membrane environments, and biological settings.3 Although the structural and functional properties of self-assembling peptide nanotubes can often be modulated by varying the cyclic peptide sequence and ring size, their functional repertoire can be significantly expanded using cyclic architectures bearing unnatural amino acids or alternative backbone structures.2 We have recently explored the utility of 1,4-disubstituted 1,2,3-triazole ε-amino acids as trans-amide dipeptide surrogates and reported the high-resolution structural features of these substitutions in the context of α-helical coiled-coils,4 rigid β-turn scaffolds,5 and extended β-sheet-like hollow tubular assemblies.2h Considering that triazole ε-amino acids can be conveniently synthesized via Cu(I)-catalyzed 1,3-dipolar cycloaddition6 between amino acid-derived azides and alkynes, we wished to explore whether C2 symmetric triazole-substituted cyclic peptides, useful in the fabrication of self-assembling peptide nanotubes, could be efficiently synthesized in solution via tandem dimerization–macrocyclization cycloaddition reactions (Scheme 1).7 We reported recently that peptide 1 has high affinity for self-association in solution and peptide nanotube formation in the solid state.2h The original synthesis of 1 was accomplished in 43% overall yield by employing Fmoc-protected ε2-amino acid 1c in the solid phase peptide synthesis of the linear peptide precursor, which was subsequently cyclized via macrolactamization to afford the desired ring structure (Scheme 1, top route). We envisioned an alternative strategy for the synthesis of macrocycle 1 (Scheme 1, bottom route) using azido–alkyne functionalized peptide 1d via 1,3-dipolar cycloaddition reactions involving intermolecular dimerization followed by an intramolecular ring forming reaction.7 Although this approach could, in principle, produce a number of linear and cyclic oligomers, we hypothesized that the backbone conformational preference imposed by the alternating R,S-amino acid chirality would kinetically favor formation of the desired cyclic structure, which is also the smallest unconstrained cyclic product possible. This route was expected to provide a facile entry into a series of self-assembling pseudo-hexapeptide scaffolds via an expedient solution phase method.
Scheme 1.
Alternative Routes to Cyclic Peptide 1
We set out initially to resynthesize previously characterized cyclic peptide 1 in order to evaluate the viability of the proposed route and its efficiency relative to the macrolac-tamization method (Scheme 1). Addition of copper(I) iodide, diisopropylethylamine, and 2,6-lutidine to a millimolar solution of 1d in acetonitrile led to the gradual formation of a white precipitate from an initially homogeneous reaction mixture. Analysis of the reaction mixture by RP-HPLC indicated formation of cyclic pseudo-hexapeptide 1 as the major product (>90%) together with small amounts of the cyclic trimer and larger oligomers. Despite the excellent yield and product selectivity, the insolubility of the reaction products severely complicated isolation of 1. Self-assembling cyclic peptides lacking ionizable side chains generally exhibit low solubility due to their inherent propensity to form large tubular assemblies. Although this property is desirable from the nanotube supramolecular design perspective, it nevertheless adds practical difficulty to product isolation. To alleviate this shortcoming, we took advantage of an amide backbone N-alkylation strategy which has been shown to prevent formation of nanotubular aggregates by breaking the network of extended intermolecular backbone–backbone hydrogen bonding.8 Our synthetic strategy thus was modified to employ peptide precursors bearing N-(2,4-dimethoxy)benzyl (Dmb) amide substituents.9
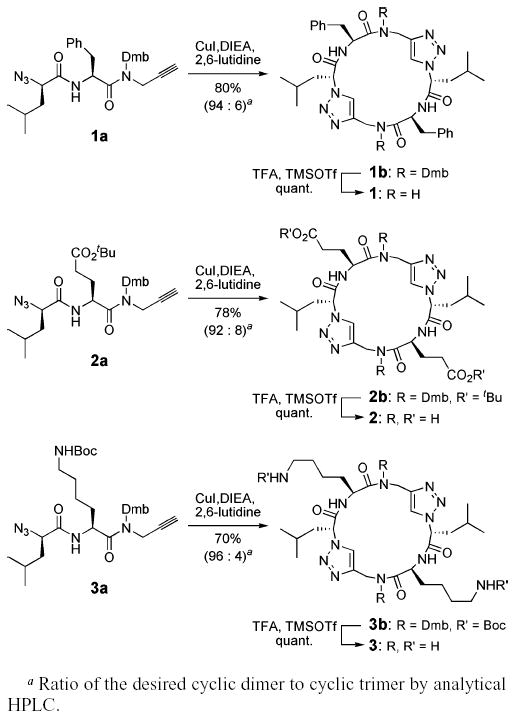
The Dmb backbone modifications were expected to render the peptides highly soluble in most typical organic solvents and be readily removed under strongly acidic conditions. Subjecting N-Dmb-substituted azido–alkyne 1a (Scheme 2) to a similar reaction condition used in the tandem dimerization–macrocyclization of 1d gave a homogeneous reaction mixture that was >90% cyclic product 1b by HPLC analysis. The improved solubility of the heterocyclic peptide allowed facile purification by preparative HPLC providing 80% isolated yield of 1b. The Dmb groups were removed by treatment with TFA/TMSOTf/anisole (8:1:1) to afford free peptide 1, which could be isolated quantitatively by trituration upon addition of diethyl ether to the reaction mixture. Similarly, azido–alkynes 2a and 3a were used to prepare heterocyclic products 2 and 3 in good isolated yields and product selectivity (Scheme 2), suggesting the likely generality of this approach.
Scheme 2.
Synthesis of C2 Symmetric Heterocyclic Peptides
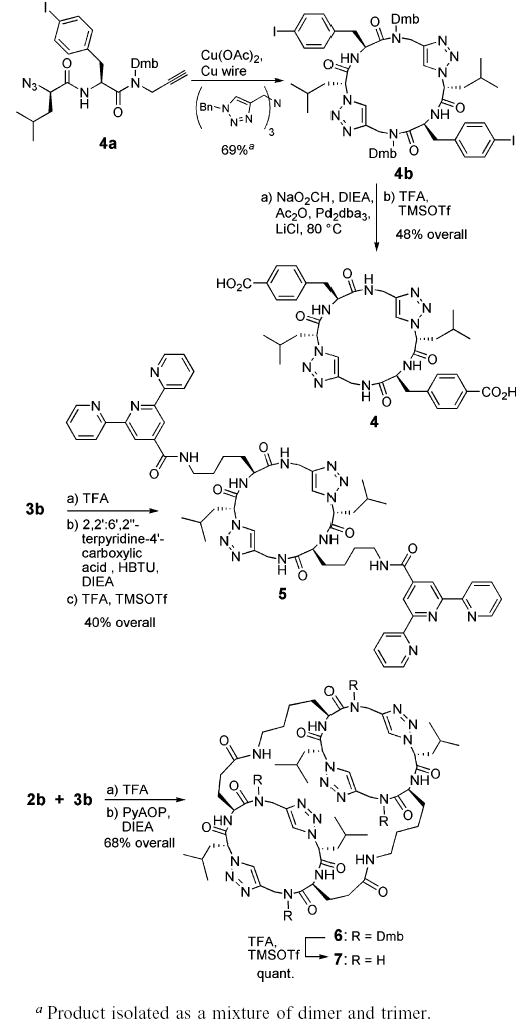
The use of Dmb substituents and the resulting enhanced solubility of heterocyclic peptides also enabled facile postsynthetic modifications (Scheme 3). For example, cyclic peptide 4 having p-carboxyphenylalanine side chains could be readily prepared starting from azido–alkyne 4a bearing a p-iodo-phenylalanine residue by its conversion to 4b using copper with a tris(benzyltriazolylmethyl)amine ligand10 and subsequent Pd0-catalyzed carboxylation of the aryl iodides with acetic anhydride and formate.11 Selective removal of the side chain protective groups in 2b and 3b by treatment with TFA in the absence of TMSOTf led to soluble cyclic architectures that could be further derivatized at their periphery. Accordingly, the Boc-protected amines in heterocyclic peptide 3b were deprotected and then reacted with 4′-substituted terpyridyl carboxylic acid12 to afford, after Dmb group removal, peptide 5 for potential use in the design of metallopeptide nanotubular assemblies. In another example, the side chains of heterocyclic peptides 2b and 3b were first selectively deprotected in the presence of TFA and then condensed by dimerization via macrolactamization to afford tricyclic peptide 6. Dmb removal yielded peptide 7, which represents a covalently captured minimal repeating motif of a self-assembling heterocyclic peptide nanotube.2h,13
Scheme 3.
Postsynthetic Modification of Heterocyclic Peptides
It should be noted that the approach of tandem dimerization–macrocyclization can also be generalized for the expedient synthesis of unsymmetrical cyclic peptide sequences by taking advantage of the statistical product distribution arising from the reaction of two different linear azido–alkynes. As an example, subjecting an equimolar mixture of peptides 1a and 3a to the standard reaction conditions gave a 1:1 mixture of homo- and heterodimeric products from which 8 was readily isolated in 31% yield. This approach could provide a rapid and convergent route for the preparation of combinatorial libraries of heterocyclic peptide structures from readily available starting materials. In summary, the approach described here should provide a convenient entry for the design and synthesis of a variety of heterocyclic pseudo-hexapeptide structures with potential utility in biological and materials settings.
Supplementary Material
Figure 1.
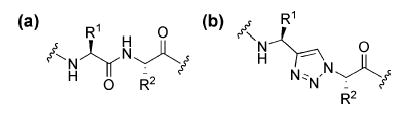
Structures of (a) a dipeptide and (b) a triazole ε-amino acid dipeptide surrogate.
Scheme 4.
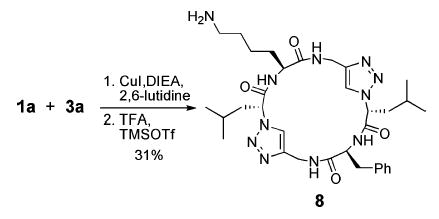
Synthesis of a Nonsymmetric Triazole Cyclic Peptide
Acknowledgments
We thank the National Institutes of Health (GM52190) and Office of Naval Research (N00014-03-1-0370) for financial support, The Netherlands Organization for Scientific Research for a travel grant to J.v.M., the National Science Foundation for a Predoctoral Fellowship to W.S.H., and John M. Beierle for synthesis and characterization of 8.
Footnotes
Supporting Information Available: Scheme for the synthesis of linear peptides 1a–4a, experimental methods, and compound characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Peptide nanotubes based on cyclic D,L-α -peptides: Ghadiri MR, Granja JR, Milligan RA, McRee DE, Khazanovich N. Nature. 1993;366:324–327. doi: 10.1038/366324a0.Hartgerink JD, Granja JR, Milligan RA, Ghadiri MR. J Am Chem Soc. 1996;118:43–50.Rosenthal-Aizman K, Svensson G, Unden A. J Am Chem Soc. 2004;126:3372–3373. doi: 10.1021/ja0372659. For a review, see: Bong DT, Clark TD, Granja JR, Ghadiri MR. Angew Chem, Int Ed. 2001;40:988–1011.
- 2.Peptide nanotubes based on other cyclic peptide architectures: Karle IL, Handa BK, Hassall CH. Acta Crystallogr, Sect B. 1975;31:555–560.Seebach D, Matthews JL, Meden A, Wessels T, Baerlocher C, McCusker LB. Helv Chim Acta. 1997;80:173–182.Clark TD, Buehler LK, Ghadiri MR. J Am Chem Soc. 1998;120:651–656.Ranganathan D, Lakshmi C, Karle IL. J Am Chem Soc. 1999;121:6103–6107.Gauthier D, Baillargeon P, Drouin M, Dory YL. Angew Chem, Int Ed. 2001;40:4635–4638. doi: 10.1002/1521-3773(20011217)40:24<4635::aid-anie4635>3.0.co;2-d.Semetey V, Didierjean C, Briand JP, Aubry A, Guichard G. Angew Chem, Int Ed. 2002;41:1895–1898. doi: 10.1002/1521-3773(20020603)41:11<1895::aid-anie1895>3.0.co;2-3.Amorin M, Castedo L, Granja JR. J Am Chem Soc. 2003;125:2844–2845. doi: 10.1021/ja0296273.Horne WS, Stout CD, Ghadiri MR. J Am Chem Soc. 2003;125:9372–9376. doi: 10.1021/ja034358h.
- 3.(a) Ghadiri MR, Granja JR, Buehler LK. Nature. 1994;369:301–304. doi: 10.1038/369301a0. [DOI] [PubMed] [Google Scholar]; (b) Granja JR, Ghadiri MR. J Am Chem Soc. 1994;116:10785–10786. [Google Scholar]; (c) Motesharei K, Ghadiri MR. J Am Chem Soc. 1997;119:11306–11312. [Google Scholar]; (d) Fernandez-Lopez S, Kim HS, Choi EC, Delgado M, Granja JR, Khasanov A, Kraehenbuehl K, Long G, Weinberger DA, Wilcoxen KM, Ghadiri MR. Nature. 2001;412:452–456. doi: 10.1038/35086601. [DOI] [PubMed] [Google Scholar]; (e) Sanchez-Quesada J, Kim HS, Ghadiri MR. Angew Chem, Int Ed. 2001;40:2503–2506. [PubMed] [Google Scholar]; (f) Couet J, Jeyaprakash JD, Samuel S, Kopyshev A, Santer S, Biesalski M. Angew Chem, Int Ed. 2005;44:3297–3301. doi: 10.1002/anie.200462993. [DOI] [PubMed] [Google Scholar]; (g) Horne WS, Wiethoff CM, Cui C, Wilcoxen KM, Amorin M, Ghadiri MR, Nemerow GR. Bioorg Med Chem. 2005;13:5145–5153. doi: 10.1016/j.bmc.2005.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horne WS, Yadav MK, Stout CD, Ghadiri MR. J Am Chem Soc. 2004;126:15366–15367. doi: 10.1021/ja0450408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horne WS. Ph.D. Thesis. The Scripps Research Institute; La Jolla, CA: 2005. [Google Scholar]
- 6.(a) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem, Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; (b) Tornoe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 7.(a) Bodine KD, Gin DY, Gin MS. J Am Chem Soc. 2004;126:1638–1639. doi: 10.1021/ja039374t. [DOI] [PubMed] [Google Scholar]; (b) Billing JF, Nilsson UJ. J Org Chem. 2005;70:4847–4850. doi: 10.1021/jo050585l. [DOI] [PubMed] [Google Scholar]; (c) Punna S, Kuzelka J, Wang Q, Finn MG. Angew Chem, Int Ed. 2005;44:2215–2220. doi: 10.1002/anie.200461656. [DOI] [PubMed] [Google Scholar]
- 8.(a) Ghadiri MR, Kobayashi K, Granja JR, Chadha RK, McRee DE. Angew Chem, Int Ed Engl. 1995;34:93–95. [Google Scholar]; (b) Clark TD, Buriak JM, Kobayashi K, Isler MP, McRee DE, Ghadiri MR. J Am Chem Soc. 1998;120:8949–8962. [Google Scholar]
- 9.(a) Weygand F, Steglich W, Bjarnaso J, Akhtar R, Khan NM. Tetrahedron Lett. 1966:3483–3487. doi: 10.1016/s0040-4039(01)99959-9. [DOI] [PubMed] [Google Scholar]; (b) Blaakmeer J, Tijsseklasen T, Tesser GI. Int J Pept Protein Res. 1991;37:556–564. [PubMed] [Google Scholar]
- 10.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Org Lett. 2004;6:2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 11.Cacchi S, Fabrizi G, Goggiamani A. Org Lett. 2003;5:4269–4272. doi: 10.1021/ol0354371. [DOI] [PubMed] [Google Scholar]
- 12.Fallahpour RA. Synthesis. 2000:1138–1142. [Google Scholar]
- 13.(a) Clark TD, Ghadiri MR. J Am Chem Soc. 1995;117:12364–12365. [Google Scholar]; (b) Clark TD, Kobayashi K, Ghadiri MR. Chem–Eur J. 1999;5:782–792. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.