

Summary
c-Jun, a major transcription factor in the activating protein 1 (AP-1) family of regulatory proteins, is activated by many physiologic and pathologic stimuli. However, whether c-jun is regulated by epigenetic modification of chromatin structure is not clear. We showed here that c-jun was transcriptionally repressed in response to osmotic stress via a truncated HDAC3 generated by caspase-7–dependent cleavage at aspartic acid 391. The activation of caspase-7, which is independent of cytochrome c release and activation of caspase-9 and caspase-12, depends on activation of caspase-8, which in turn requires MEK2 activity and secretion of FAS ligand. The cell apoptosis induced by the truncated HDAC3 or enhanced by c-Jun deficiency during osmotic stress was suppressed by exogenous expression of c-Jun, indicating that the downregulation of c-Jun by HDAC3-dependent transcriptional repression plays a role in regulating cell survival and apoptosis.
Introduction
The proto-oncogene c-jun is the cellular homologue of v-jun, the transforming oncogene in the genome of avian sarcoma virus 17 (Vogt, 2001). Jun family proteins are a major component of activating protein 1 (AP-1) transcription factors. AP-1 transcription factors are composed of homodimers and heterodimers of basic region-leucine zipper proteins that belong to the Jun (c-Jun, JunB, and JunD), Fos (c-Fos, FosB, Fra-1, and Fra-2), Maf (c-Maf, MafB, MafA, MafG/F/K, and Nrl), and ATF (ATF2, LRF1/ATF3, B-ATF, JDP1, and JDP2) subfamilies (Shaulian and Karin, 2002). AP-1 transcription factors are activated by various physiologic and pathologic stimuli, including phorbol ester, growth factors, oncoproteins such as Src and Ras, proinflammatory cytokines, chemotherapeutic drugs, osmotic stress, and ultraviolet radiation (Angel and Karin, 1991; Leppa and Bohmann, 1999; McCabe and Burrell, 2001; Sharp et al., 1991). c-Jun activation is instrumental in cell growth and differentiation, apoptosis, cell transformation, tissue morphogenesis, and inflammatory responses (Ip and Davis, 1998). Mouse embryos lacking c-Jun die at mid- to late-gestation and exhibit impaired hepatogenesis, altered fetal liver erythropoiesis, and generalized edema (Hilberg et al., 1993; Johnson et al., 1993). Liver-specific inactivation of c-Jun demonstrates that c-Jun is required at the early stage of chemically induced hepatocellular carcinomas in mice (Eferl et al., 2003).
The activity of individual AP-1 components can be regulated at multiple levels, including transcriptionally regulated protein expression, interaction with other proteins, and posttranslational modifications, such as phosphorylation, ubiquitination, and sumoylation (Muller et al., 2000; Musti et al., 1997). c-Jun can engage in a positive autoregulatory loop by binding to AP-1 binding sites of the c-jun enhancer (Vogt, 2001). c-Jun activity can be posttranslationally regulated by a mitogen-activated protein (MAP) kinase signaling pathway, which is composed of a cascade of three kinases: a MAP kinase; a MAP kinase/ERK kinase (MEK), a MAP kinase kinase (MKK), or MAP2K; and a MEK kinase (MEKK), MAP kinase kinase kinase (MKKK), or MAP3K (Leppa and Bohmann, 1999). Three subfamilies of MAP kinases are well characterized: extracellular signal-regulated protein kinases (ERKs), c-Jun N-terminal kinases (JNKs), and p38 MAP kinases (Leppa and Bohmann, 1999). In response to stress stimuli, c-Jun is activated by JNK phosphorylation at the serine (Ser) 63 and Ser73 residues in the transactivation domain near its N terminus (Vogt, 2001). c-Jun activity can be downregulated by ubiquitin/proteasome-mediated degradation via distinct E3 ubiquitin ligases or by sumoylation, depending on the cell and tissue type involved and the cellular regulatory influences that the cells are receiving (Bossis et al., 2005; Gao et al., 2004; Muller et al., 2000; Musti et al., 1997; Schmidt and Muller, 2002; Treier et al., 1994; Wei et al., 2005; Wertz et al., 2004).
In addition to the posttranslational modifications that regulate protein activity and stability, highly conserved histone acetyl transferases (HATs) and histone deacetylases (HDACs) dynamically regulate chromatin structure and transcription. HDACs repress gene expression via deacetylation of ε-amino groups on conserved lysine residues in the N-terminal tail domains of histones. HDACs are divided into three categories in humans. Class I HDACs are ubiquitously expressed in tissues and organs and are composed of HDACs 1, 2, 3, and 8, which are yeast Rpd3 homologues and have related catalytic domains. Class II HDACs include HDACs 4, 5, 6, 9, and 10, which have a domain similar to the deacetylase domain of yeast Hda1. Class III HDACs are related to yeast Sir2 protein and are involved in gene silencing (Marks et al., 2003). HDACs are also involved in the reversible acetylation of non-histone proteins (Marks et al., 2003).
Although the regulation of the activity and stability of c-Jun by posttranslational modifications has been intensively studied, whether c-jun is regulated by epigenetic modification of chromatin structure is not known. In this report, we demonstrate that c-Jun is downregulated in response to osmotic stress by transcriptional repression via caspase-7–dependent cleavage of HDAC3, which involves FAS ligand and MEK2-dependent activation of caspase-8. The downregulation of c-Jun promotes osmotic stress-induced apoptosis.
Results
Osmotic Stress Induces c-Jun Downregulation by Transcriptional Repression
To examine the effect of extracellular stimuli on c-Jun expression, we stimulated 3Y1 rat fibroblasts with tumor necrosis factor (TNF)-α, ultraviolet B (UVB) irradiation, or sorbitol as an osmotic stressor. As expected and consistent with previous findings (Dixit et al., 1989; Shaulian et al., 2000), treatment with TNF-α or UVB irradiation increased c-Jun protein levels (Figure 1A). However, c-Jun (Figure 1A), but not c-Fos (Figure 1B), was downregulated at 6 h of sorbitol stimulation. Similar data were also observed in NIH3T3 mouse fibroblasts (data not shown). All tested extracellular stimuli enhanced c-Jun phosphorylation at Ser63 (Figure 1A). Nevertheless, compared with 1 h of treatment, prolonged sorbitol treatment reduced the amount of phosphorylated c-Jun to an extent corresponding to the decrease in c-Jun protein level. These results demonstrate that the level of c-Jun protein is regulated differentially, depending on the extracellular stimuli and cellular milieu involved. Mannitol treatment also reduced the c-Jun protein level (Figure 1C), indicating that c-Jun downregulation is a general response to hyperosmotic stress. Real-time quantitative reverse-transcriptase polymerase chain reaction (RT-PCR) analysis showed a reduced level of c-jun mRNA upon sorbitol treatment (Figure 1D). Accordingly, c-jun promoter activity in NIH3T3 cells stably expressing pc-Jun-Luc containing the full-length c-jun promoter was suppressed in a time-dependent manner by sorbitol (Figure 1E). These data indicate that the transcriptional repression of c-jun is involved in sorbitol-induced c-Jun downregulation.
Figure 1. Osmotic Stress Induces c-Jun Downregulation by Transcriptional Repression.
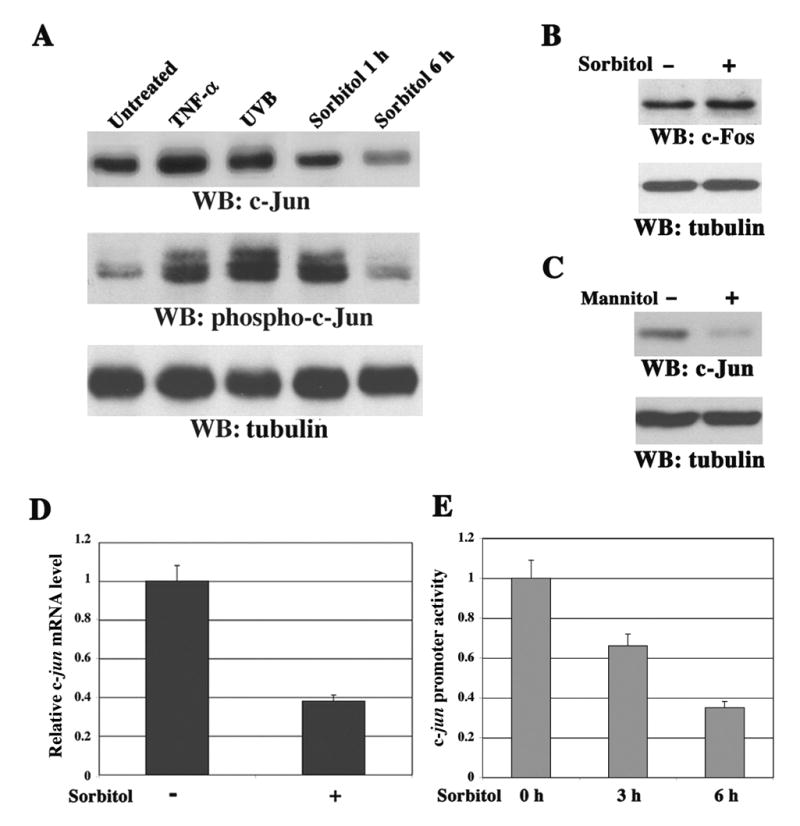
(A–C) Immunoblotting analyses with the indicated antibodies.
(A) 3Y1 cells were treated with TNF-α (10 ng/ml) for 6 h, exposed to UVB irradiation (180 mJ/cm2) 6 h before harvesting, or stimulated with sorbitol (500 mM) for 1 or 6 h.
(B–C) 3Y1 cells were treated for 6 h with or without sorbitol (500 mM) (B) or mannitol (500 mM) (C).
(D) The c-jun mRNA expression in NIH3T3 cells treated with or without sorbitol for 6 h was measured by real-time quantitative RT-PCR analyses. GAPDH mRNAs from the same cDNA library were amplified as a control. The relative levels of c-jun mRNA were normalized to the levels of untreated cells and of GAPDH mRNAs. Data represent the means ± SD of three independent experiments.
(E) The luciferase activity in NIH3T3 cells stably expressing pc-Jun-Luc was determined after cells were treated with or without sorbitol for 3 h and 6 h. The relative levels of luciferase activity were normalized to the levels of untreated cells and to the levels of luciferase activity of the Renilla control plasmid. Data represent the means ± SD of three independent experiments.
MEK2-Dependent and MEK1- or JNK1/2-Independent Transcriptional Downregulation of c-jun in Response to Sorbitol Treatment
Sorbitol treatment is known to activate both the JNK1/2 and MEK1/2-ERK pathways (Yan et al., 1994). Pretreatment with MEK1/2 inhibitor U0126, which inhibited MEK1/2 activation as detected by abrogated ERK1/2 phosphorylation, significantly reversed the sorbitol-induced inhibition of c-jun promoter activity (Figure 2A), reduction of mRNA level (Figure 2B), and downregulation of protein expression (Figure 2C). However, JNK inhibitor SP600125, which inhibited JNK1/2 activity and c-Jun phosphorylation, did not inhibit the effects of sorbitol on c-Jun (Figures 2A–C). The lack of requirement for JNK1/2 activity for c-Jun downregulation was further shown using JNK1/2+/+ and JNK1/2−/− fibroblasts (Ventura et al., 2004), which displayed a lack of sorbitol-induced phosphorylation of c-Jun at Ser63 but still downregulated c-Jun protein expression (Figure 2D) and the c-jun mRNA level in JNK1/2−/− cells (data not shown). The differential roles of MEK1 and MEK2 in c-Jun downregulation were revealed in MEK1−/− and MEK2−/− fibroblasts, in which a deficiency of MEK2, but not MEK1, partially blocked the downregulation of c-Jun protein expression (Figure 2E) and mRNA level (Figure 2F) upon sorbitol treatment for 3 h (data not shown) or 6 h, although ERK1/2 phosphorylation in MEK2−/− cells was not impaired (Figure 2E).
Figure 2. MEK2-Dependent and MEK1- or JNK1/2-Independent Transcriptional Downregulation of c-jun in Response to Sorbitol Treatment.
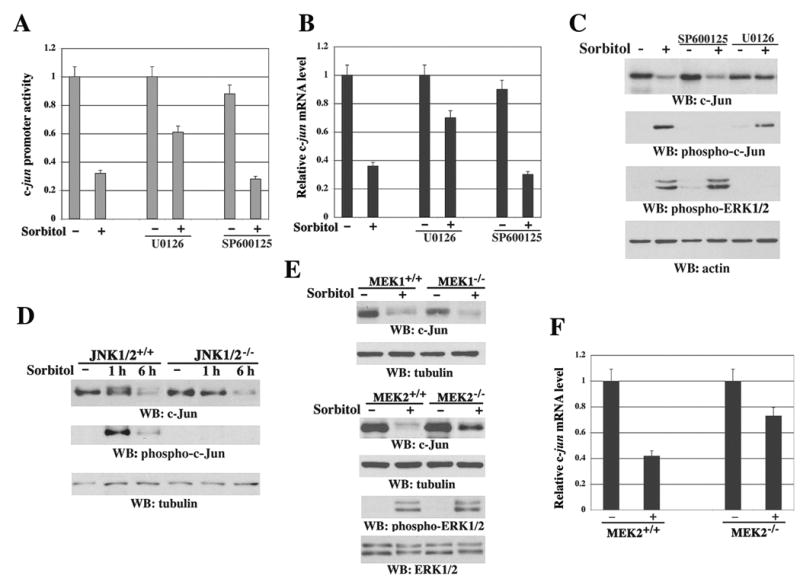
(A) The luciferase activity in NIH3T3 cells stably expressing pc-Jun-Luc was determined after cells were treated with or without U0126 (25 μM) or SP600125 (25 μM) for 30 min followed by sorbitol stimulation for 6 h. The relative levels of luciferase activity were normalized to the levels of untreated cells and to the levels of luciferase activity of the Renilla control plasmid. Data represent the means ± SD of three independent experiments.
(B) The c-jun mRNA expression in NIH3T3 cells was measured by real-time quantitative RT-PCR with or without pretreatment for 30 min with the indicated inhibitors and sorbitol stimulation for 6 h. The relative levels of c-jun mRNA were normalized to the levels of untreated cells and of GAPDH mRNA. Data represent the means ± SD of three independent experiments.
(C–E) Immunoblotting analyses with the indicated antibodies. 3Y1 cells were treated with or without the indicated inhibitors for 30 min followed by sorbitol stimulation for 6 h (C). JNK1/2+/+ and JNK1/2−/− cells were treated with or without sorbitol for 1 or 6 h (D). MEK1+/+, MEK1−/−, MEK2+/+, and MEK2−/− fibroblasts were treated with or without sorbitol for 6 h (E).
(F) MEK2+/+ and MEK2−/− fibroblasts were treated with or without sorbitol for 6 h. c-jun mRNA expression was measured by real-time quantitative RT-PCR. The relative levels of c-jun mRNA were normalized to the levels of untreated cells and of GAPDH mRNA. Data represent the means ± SD of three independent experiments.
HDAC Activity and Caspase-Dependent C-terminal Cleavage of HDAC3 Are Required for Sorbitol-Induced c-Jun Downregulation
Repression of gene transcription can be mediated by histone deacetylation through localized HDAC activity. Pretreatment with trichostatin A (TSA) and sodium butyrate (NaB), which are specific inhibitors of HDACs, dose-dependently inhibited sorbitol-induced c-Jun downregulation (Figure 3A). Differential effects of TSA on c-Jun expression have been observed in different experimental settings (Hazzalin and Mahadevan, 2005; Sng et al., 2005). Although the reason for enhanced c-Jun expression following TSA treatment alone (Figure S1, quantified by scanning densitometry) is not yet clear, since NaB, which by itself did not increase c-Jun expression level, also largely blocked the effect of sorbitol on c-Jun expression, this suggests that sorbitol-regulated HDAC activity plays a role in c-jun repression. NaB is an inhibitor of HDAC classes I and II (Canettieri et al., 2003). Because class II HDAC expression is mostly tissue specific (Marks et al., 2003), and expression of HDAC4–6 was hardly detectable in NIH3T3 cells (data not shown), we concentrated on class I HDACs, which are ubiquitously expressed (Marks et al., 2003). Sorbitol treatment resulted in a truncated protein, which was about 4 kDa smaller than the full-length HDAC3 and recognized by an antibody against the N terminus of HDAC3 (Figure 3B). The appearance of truncated HDAC3 correlated kinetically with a lower level of c-Jun expression. In contrast, truncated forms of HDAC1, HDAC2, or HDAC8 were not detected. The truncated HDAC3 fragment was not detected by an antibody against the extreme C terminus of HDAC3 (amino acids [aa] 411–428), but it was recognized by an antibody against HDAC3 (aa 330–428) (Figure 3C). This finding suggests that HDAC3 is truncated near the C terminus between aa 330 and aa 411.
Figure 3. HDAC Activity and Caspase-Dependent C-terminal Cleavage of HDAC3 Are Required for Sorbitol-Induced c-Jun Downregulation.
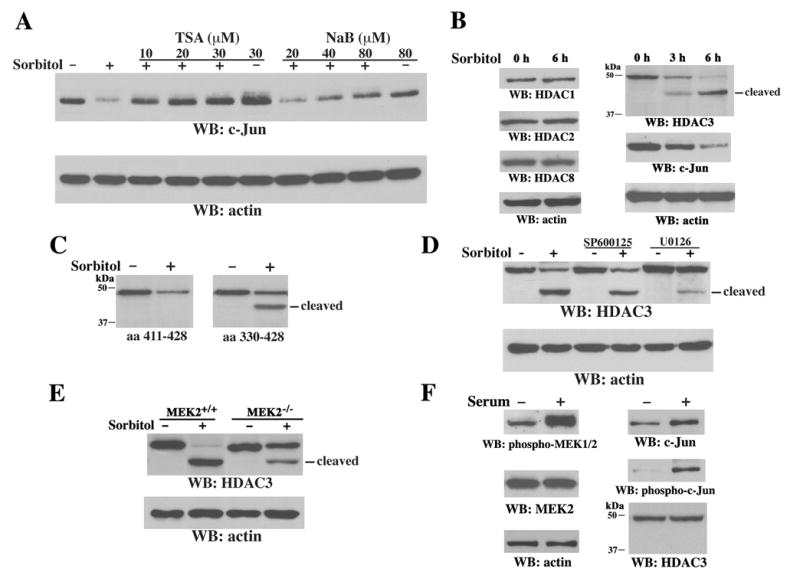
(A–F) Immunoblotting analyses with the indicated antibodies.
A) 3Y1 cells were pretreated for 30 min with or without TSA or NaB at the concentrations shown before sorbitol treatment for 6 h.
(B) NIH3T3 cells were treated with sorbitol for the indicated time.
(C) Treatment of 3Y1 cells with or without sorbitol for 6 h was followed by immunoblotting with antibodies recognizing aa 411–428 (left) or 330–428 (right) of HDAC3.
(D) 3Y1 cells were pretreated for 30 min with or without SP600125 (25 μM) or U0126 (25 μM) before sorbitol treatment for 6 h
(E) MEK2+/+ and MEK2−/− cells were treated with or without sorbitol for 6 h.
(F) Serum-starved 3Y1 cells were treated with or without 20% serum for 6 h.
In line with the involvement of MEK in repressing c-jun transcription, pretreatment with U0126, but not SP600125, significantly inhibited sorbitol-induced HDAC3 cleavage (Figure 3D). Furthermore, in contrast to MEK2+/+ cells, MEK2−/− cells displayed reduced HDAC3 cleavage after sorbitol treatment (Figure 3E). These data indicate that sorbitol-induced activation of MEK2, but not MEK1 or JNK1/2, participates in caspase activation-dependent HDAC3 cleavage, which correlates with c-Jun downregulation. Nevertheless, serum stimulation, which activates MEK2 (Zheng and Guan, 1993) as detected by an anti-phospho-MEK1/2 antibody, increased both the expression and phosphorylation of c-Jun; whereas HDAC3 expression remained unchanged and no cleavage product was detected (Figure 3F). These results imply that MEK2 activation is necessary but not sufficient for sorbitol-induced HDAC3 cleavage and c-Jun downregulation.
Caspase-7 Cleaves HDAC3 at Aspartic Acid 391
Because sorbitol treatment can induce cells to undergo apoptosis (Matthews and Feldman, 1996), we examined the possible involvement of active caspases in HDAC3 cleavage. Pretreatment with the general caspase inhibitor Z-VAD-FMK blocked the sorbitol-induced truncation of HDAC3 and largely inhibited the downregulation of c-Jun (Figure 4A). A sequence analysis of HDAC3 aa 330–411 revealed that aa 388–391 (DRTD) are closely related to the consensus cleavage sequence of caspase-3 and caspase-7, both of which have a strong predilection for aspartic acid (Asp) at the P4 and P1 positions (Talanian et al., 1997). In vitro caspase cleavage reactions showed that purified human His-HDAC3 protein was cleaved by purified active His-caspase-7 but not by active His-caspase-1, −2, −3, or −4 (Figure 4B), whereas caspase-3 was still able to cleave poly(ADP-ribose) polymerase (PARP), a known substrate of caspase-3 (Koh et al., 2005). These results indicate that HDAC3 is a direct substrate of caspase-7.
Figure 4. Caspase-7 Cleaves HDAC3 at Asp391.
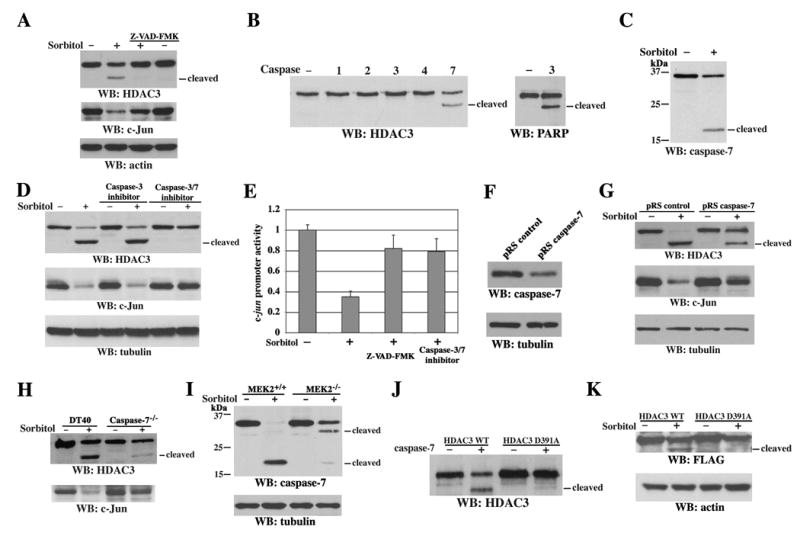
(A–D and F–K) Immunoblotting analyses with the indicated antibodies.
(A) 3Y1 cells were pretreated for 30 min with or without Z-VAD-FMK (75 μM) before sorbitol treatment for 6 h.
(B) In vitro caspase cleavage assay with mixed purified active His-caspase-1, −2, −3, −4, or −7 and purified His-HDAC3 or immunoprecipitated PARP from 293T cell lysate.
(C) 3Y1 cells were treated with or without sorbitol for 6 h.
(D) 3Y1 cells were pretreated with caspase-3 inhibitor V (25 μM) or caspase-3/7 inhibitor I (20 μM) for 30 min before sorbitol stimulation for 6 h.
(E) 293T cells stably expressing pc-Jun-Luc were pretreated with or without Z-VAD-FMK (75 μM) or caspase-3/7 inhibitor I (20 μM) before sorbitol stimulation for 6 h, and luciferase activity was determined. The relative levels of luciferase activity were normalized to the levels of untreated cells and to the levels of luciferase activity of the Renilla control plasmid. Data represent the means ± SD of three independent experiments.
(F) Cell lysates were prepared from 3Y1 cells expressing pRS control shRNA or pRS caspase-7 shRNA.
(G–I) 3Y1 cells expressing the indicated shRNA (G), DT40 cells and DT40 caspase-7−/− cells (H), and MEK2+/+ and MEK2−/− cells (I) were treated with or without sorbitol for 6 h.
(J) In vitro caspase cleavage assay with mixed purified His-caspase-7 and WT His-HDAC3 or the D391A mutant.
(K) 293T cells transiently expressing WT FLAG-HDAC3 or the D391A mutant were treated with or without sorbitol for 6 h.
Consistent with the in vitro data, caspase-7 was activated after sorbitol treatment, as shown by the appearance of the ~20-kDa active subunit (Figure 4C). Furthermore, caspase-3/7 inhibitor I, but not caspase-3 inhibitor V (Z-DQMD-FMK), blocked sorbitol-induced HDAC3 cleavage and c-Jun downregulation (Figure 4D). In line with these results, pretreatment with the general caspase inhibitor Z-VAD-FMK or caspase-3/7 inhibitor I significantly blocked sorbitol-induced suppression of c-jun promoter activity (Figure 4E), suggesting that the transcriptional repression of c-jun upon sorbitol treatment involves caspase-7–dependent cleavage of HDAC3. Expression of caspase-7 small hairpin RNA (shRNA), which reduced the endogenous caspase-7 protein level in 3Y1 cells (Figure 4F), reduced HDAC3 cleavage and c-Jun downregulation in response to sorbitol treatment (Figure 4G). Furthermore, DT40 caspase-7−/− chicken B cells were largely resistant to sorbitol-induced HDAC3 cleavage and c-Jun downregulation, in contrast to wild-type (WT) DT40 cells (Figure 4H). These results demonstrate the involvement of caspase-7 in sorbitol-induced cleavage of HDAC3 in vivo and the existence of a weak compensatory effect from other caspases when caspase-7 is absent. MEK2−/− cells, which showed reduced HDAC3 cleavage and decreased c-Jun downregulation upon sorbitol treatment, displayed reduced sorbitol-induced caspase-7 activation in contrast to their WT counterpart cells (Figure 4I). The cleaved ~32 kD fragment of caspase-7 in MEK2−/− cells is likely an intermediate product of caspase-7, since it showed up before the appearance of the active ~20 kD fragment of caspase-7 and became weaker as the active ~20 kD fragment of caspase-7 became more prominent (data not shown). Since MEK2 deficiency did not block the activation of caspase-7 in response to chemotherapeutic drug cisplatin (CDDP), which causes DNA damage (Figure S2), these results support the specific involvement of MEK2 activation in caspase-7–mediated HDAC3 cleavage in response to osmotic stress.
To identify the exact caspase-7 cleavage site in HDAC3, Asp391 of HDAC3 was mutated to alanine. Purified His-HDAC3 D391A mutant protein was resistant to cleavage by caspase-7, as shown in an in vitro caspase reaction (Figure 4J). Moreover, an N-terminally FLAG-tagged HDAC3 D391A mutant transiently expressed in 293T cells was resistant to sorbitol-induced cleavage, in contrast to WT FLAG-HDAC3 (Figure 4K). These data indicate that HDAC3 is cleaved in vivo at D391 by caspase-7 upon sorbitol treatment.
MEK2- and FAS Ligand-Dependent Caspase-8 Activation, but not Cytochrome c Release or Activation of Caspase-9 and Caspase-12, Is Necessary for Sorbitol-induced Caspase-7 Activation
Caspase-7 can be activated by upstream initiator caspase-9 and caspase-8, which are activated by cytochrome c release from mitochondria and engagement of death receptors, respectively (Nunez et al., 1998). Caspase-12 is another major initiator caspase and is involved in endoplasmic reticulum (ER)-stress-mediated apoptosis (Nakagawa et al., 2000). To investigate the mechanism of caspase-7 activation in response to osmotic stress, caspase-9−/− and caspase-12−/− with their WT counterpart cells, and 3Y1 cells pretreated with the caspase-8 inhibitor Ac-IETD-CHO, were treated with sorbitol. Immunoblotting analyses showed that inhibition of caspase-8, but not caspase-9 or caspase-12 deficiency, largely blocked sorbitol-induced caspase-7 activation (Figure 5A). Consistently, caspase-7 activation was also abrogated in caspase-8−/−but not in caspase-8+/+ mouse fibroblasts (Figure 5A). Furthermore, the kinetics of caspase-8 activation, as reflected by the cleavage of pro-caspase-8, correlated with the kinetics of caspase-7 activation and HDAC3 cleavage and showed early activation at 30 min of sorbitol treatment (Figure 5B). In contrast, cytochrome c release from mitochondria into the cytosol was first detected at 8 h of treatment and became more pronounced at 14 h, which kinetically correlated with the reduced cytochrome c level in the membrane fraction (Figure 5C). These results indicate that osmotic stress-induced caspase-7 activation is dependent on caspase-8 activation but not on cytochrome c release or activation of caspase-9 or caspase-12. Consistent with the involvement of MEK2 in caspase-7 activation, MEK2−/− cells showed a partial inhibition of caspase-8 activation in contrast to MEK2+/+ cells (Figure 5D), implicating a role for MEK2 in caspase-8 activation in response to osmotic stress. The lack of detectable caspase-8 cleavage in MEK2−/−cells upon sorbitol treatment is due to the remaining abundant full-length caspase-8. Caspase-8 is best known to be activated by ligand binding-induced trimerization of death receptors, such as FAS (also called APO-1 or CD95), TNF receptor, DR3, DR4 (TRAIL-R1), DR5 (TRAIL-R2), and R6 (Kruidering and Evan, 2000). To test the possible involvement of death receptors, in conjunction with MEK2, for caspase-8 activation, serum-free media were collected after sorbitol treatment of 3Y1 cells. Immunoblotting with anti-FAS ligand antibodies showed an increased secretion of FAS ligand from 3Y1 cells (Figure 5E) upon prolonged osmotic stress, whereas the lack of detectable tubulin in these media indicates that they are not contaminated with broken cells. Incubation with a neutralizing FAS ligand antibody before sorbitol treatment reduced sorbitol-induced activation of caspase-8 and caspase-7, HDAC3 cleavage, and c-Jun downregulation, but not MEK1/2 phosphorylation (Figure 5F), indicating that secreted FAS ligand is another factor contributing to caspase-8-induced transcriptional repression of c-Jun. Consistently, a reduction in sorbitol-induced caspase-8 activation was observed by pre-treatment of MEK2-deficient cells with a neutralizing FAS ligand antibody (Figure 5G), supporting the idea that there are combinatorial effects of MEK2 activation and secretion of FAS ligand on caspase-8 activation. Blocking the function of FAS did not completely rescue sorbitol-induced reduction of full-length caspase-8, implying that additional factors may also contribute to the activation of caspase-8.
Figure 5. MEK2- and FAS Ligand-Dependent Caspase-8 Activation Is Necessary for Sorbitol-Induced Caspase-7 Activation.
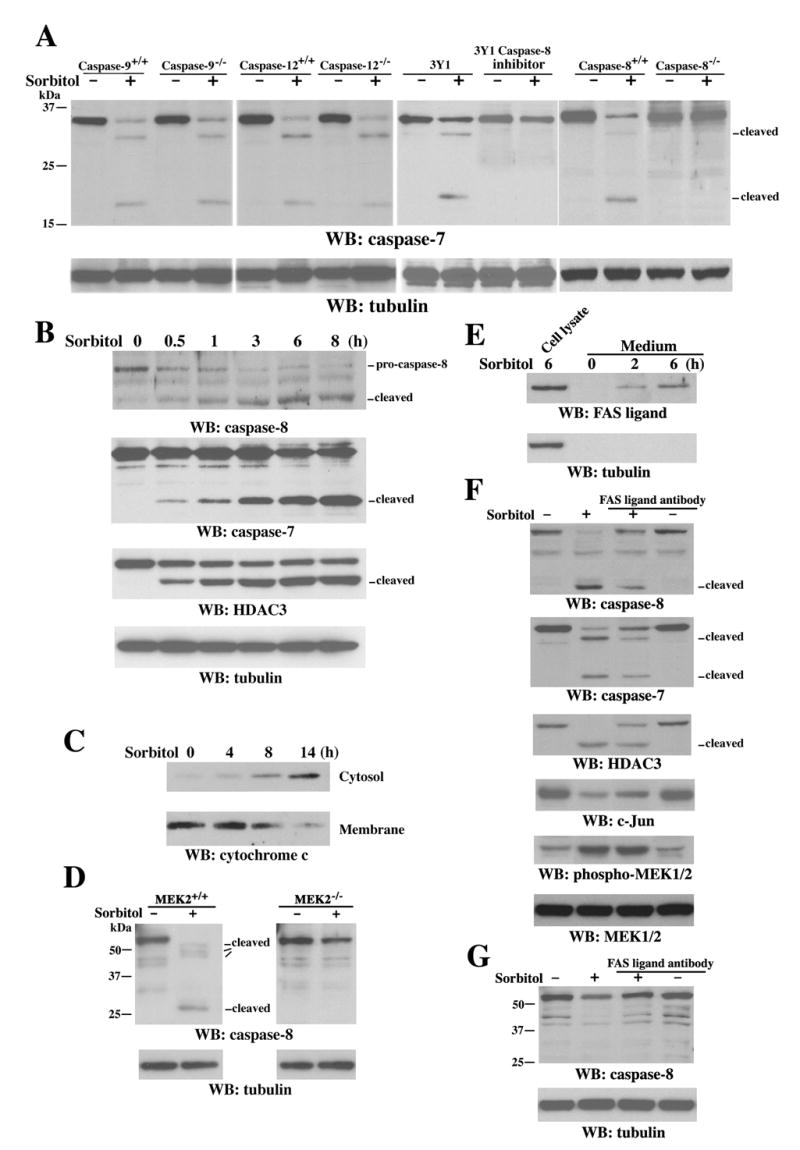
(A–G) Immunoblotting analyses with the indicated antibodies.
(A) Caspase-9−/−, caspase-12−/−, caspase-8−/− 3T3 fibroblast, and their WT counterpart cells were treated with or without sorbitol for 6 h. 3Y1 cells were pretreated with or without caspase-8 inhibitor Ac-IETD-CHO (10 μM) for 30 min before sorbitol treatment for 6 h.
(B–C) 3Y1 cells were treated with or without sorbitol for the indicated time. Cytosol and membrane fractions of the cells were prepared (C).
(D) MEK2+/+ and MEK2−/− cells were treated with or without sorbitol for 6 h.
(E) 3Y1 cells were treated with or without sorbitol for the indicated time. Serum-free media were collected for protein precipitation.
(F–G) Incubation with or without a neutralizing FAS ligand antibody (4 μg/ml) before sorbitol treatment of 3Y1 cells (F) or MEK2−/− cells (G) for 6 h was followed by cell lysate preparation.
Truncated HDAC3 (aa 1–391) Is Recruited to the c-jun Promoter Region and Is Responsible for Sorbitol-Induced Transcriptional Repression of c-jun
The blockage of sorbitol-induced downregulation of c-Jun by HDAC inhibitors (Figure 3A) indicates that HDAC activity is involved in transcriptional repression. However, sorbitol treatment did not significantly alter HDAC3 activity, as determined by in vitro analyses of immunoprecipitated HDAC3 from untreated or treated cells (data not shown). In addition, truncated HDAC3 (aa 1–391) and WT HDAC3 have similar cytoplasmic and nuclear distributions (Figure S3A), and sorbitol treatment did not result in an obvious nuclear translocation of truncated HDAC3 (Figure S3B). To examine whether sorbitol treatment induces recruitment of HDAC3 to the c-jun promoter, which thereby causes repression of c-jun transcription, NIH3T3 cells were treated with sorbitol, and chromatin immunoprecipitation (ChIP) assays were carried out with an antibody that recognizes C-terminally-truncated HDAC3. PCR with mouse c-jun promoter-specific primers detected the recruitment of much more HDAC3 to the promoter after sorbitol treatment (Figure 6A, left panel). Consistently, more truncated FLAG-tagged HDAC3 (aa 1–391) than full-length HDAC3 bound to the c-jun promoter region (Figure 6A, right panel). Furthermore, a lower level of acetylated histone H3 was detected in the c-jun promoter region in cells expressing HDAC3 (aa 1–391) than in cells expressing WT HDAC3 (Figure 6B). These data imply that sorbitol treatment results in local chromatin modification by recruiting truncated HDAC3 (aa 1–391) to the c-jun promoter, thus leading to histone deacetylation and repression of c-jun transcription.
Figure 6. Truncated HDAC3 (aa 1–391) Is Recruited to the c-jun Promoter Region and Is Responsible for Sorbitol-Induced Transcriptional Repression of c-jun.
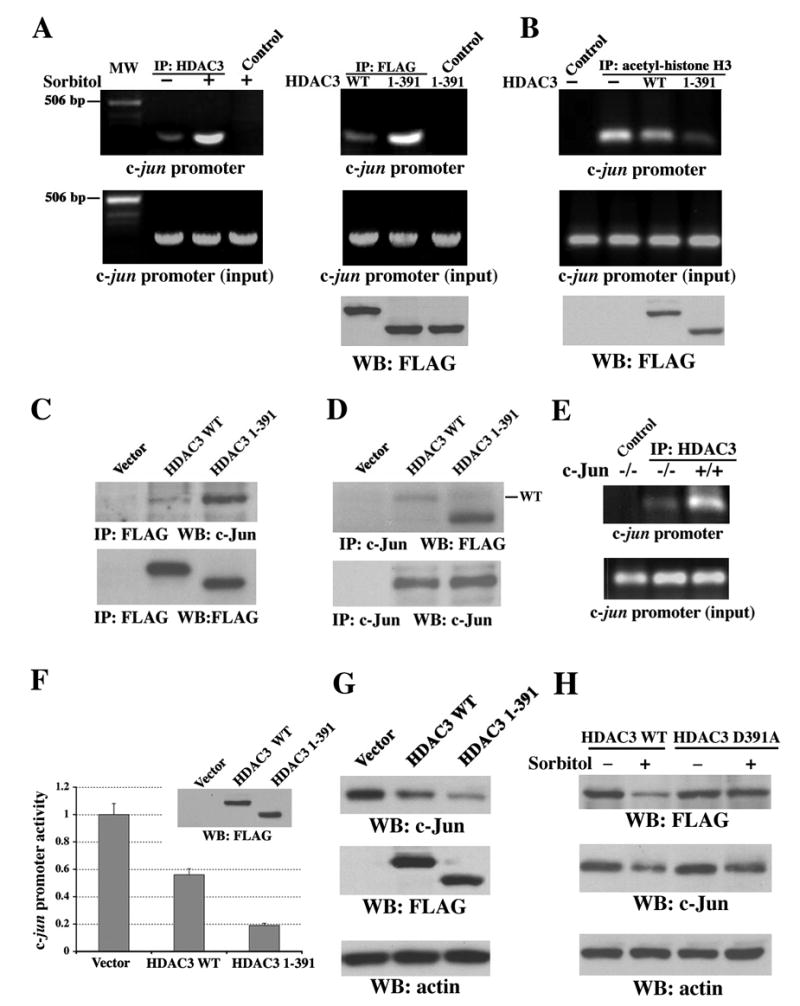
(A–D and F–H) Immunoblotting analyses with the indicated antibodies.
(A) Chromatin was prepared from NIH3T3 cells (left) treated with or without sorbitol for 3 h or from 293T cells (right) transiently expressing WT FLAG-HDAC3 or FLAG-HDAC3 (aa 1–391). ChIP analysis was carried out with primers for mouse (left) or human (right) c-jun promoter. A rabbit anti-HDAC3 antibody (H-99), mouse anti-FLAG antibody, or normal rabbit or mouse IgG as a control was used for immunoprecipitation. MW, molecular weight.
(B) 293T cells transiently expressing with or without WT FLAG-HDAC3 or FLAG-HDAC3 (aa 1–391) were used for ChIP analysis with an acetyl-histone H3 antibody for immunoprecipitation.
(C–D) 293T cells transiently expressing with or without WT FLAG-HDAC3 or FLAG-HDAC3 (aa 1–391) were used for immunoprecipitation with anti-FLAG (C) or anti-c-Jun (D) antibodies.
(E) Chromatin prepared from c-Jun−/− and c-Jun+/+ cells, which were treated with sorbitol for 3 h, were used for ChIP analysis with a rabbit anti-HDAC3 antibody (H-99) or normal rabbit IgG as a control for immunoprecipitation.
(F) pFLAG (vector), pFLAG-HDAC3 WT, or pFLAG-HDAC3 (aa 1–391) was transiently transfected into 293T cells expressing pc-Jun-Luc and subjected to luciferase-reporter and immunoblotting analyses with an anti-FLAG antibody. The relative levels of luciferase activity were normalized to the levels of pFLAG-transfected cells and to the levels of luciferase activity of the Renilla control plasmid. Data represent the means ± SD of three independent experiments.
(G) 293T cells were transiently transfected with pFLAG, pFLAG-HDAC3 WT, or pFLAG-HDAC3 (aa 1–391).
(H) NIH 3T3 cells transiently transfected with pFLAG-HDAC3 WT or pFLAG-HDAC3 D391A and were treated with or without sorbitol for 6 h.
HDAC3 has been shown to interact with the N terminus of c-Jun and function as a suppressing component of c-Jun transcriptional activity (Weiss et al., 2003). Immunoblotting of immunoprecipitated WT-HDAC3 or HDAC3 (aa 1–391) with an anti-c-Jun antibody revealed an enhanced association between truncated HDAC3 and c-Jun (Figure 6C), which was further confirmed by reciprocal immunoprecipitation (Figure 6D). To test the dependence of c-Jun on recruitment of the truncated HDAC3 to the c-jun promoter region, sorbitol-treated c-Jun−/− cells and their WT counterpart cells were analyzed by ChIP assay with an antibody that recognizes full-length and truncated HDAC3. As shown in Figure 6E, the absence of c-Jun reduced the binding of HDAC3 to the c-jun promoter after sorbitol treatment, which indicates a requirement for c-Jun and involvement of other transcriptional regulators for recruiting HDAC3 to the c-jun promoter.
To examine the effects of truncated HDAC3 on c-Jun expression, WT FLAG-HDAC3 or truncated FLAG-HDAC3 (aa 1–391) was transfected into 293T cells stably expressing pc-Jun-Luc. HDAC3 (aa 1–391) suppressed c-jun promoter activity significantly more than WT HDAC3 did (Figure 6F). Similar results were also observed in NIH3T3 cells (data not shown). Consistent with these data, the HDAC3 (aa 1–391) expressed in 293T cells reduced c-Jun expression much more than WT HDAC3 did (Figure 6G), whereas the expression of HDAC3 D391A, which is resistant to caspase-7-dependent cleavage, reduced sorbitol-induced c-Jun downregulation in contrast to the expression of WT HDAC3 (Figure 6H). These results demonstrate that sorbitol treatment results in caspase-7–dependent cleavage of HDAC3 at Asp391, which is responsible for sorbitol-induced transcriptional repression of c-jun.
Anti-Apoptotic Function of c-Jun in Sorbitol-Induced Cell Apoptosis
Prolonged stress stimuli, such as sorbitol treatment, can cause cell apoptosis (Matthews and Feldman, 1996). Given that c-Jun is downregulated by HDAC3-dependent transcriptional repression, we next examined the role of HDAC3 cleavage, which subsequently results in c-Jun downregulation, in apoptosis. V5-tagged HDAC3 (aa 1–391) transiently expressed in NIH3T3 cells showed both cytosolic and nuclear localization (Figure 7A), as did its WT counterpart with or without sorbitol treatment (Figure S3A). NIH3T3 cells expressing truncated HDAC3 (aa 1–391) displayed condensed and fragmented nuclei, which is a phenotypic change typical of cells undergoing apoptosis, and this was largely prevented by the simultaneous overexpression of c-Jun (Figure 7A). These data indicate that cleavage of HDAC3 at Asp391 is sufficient to induce c-Jun downregulation-dependent apoptosis. To examine the role of c-Jun in sorbitol-induced apoptosis, c-Jun+/+ and c-Jun−/− 3T3 cells were treated with or without sorbitol for 12 h. Trypan blue exclusion analyses showed that many more c-Jun−/− cells than c-Jun+/+ cells underwent apoptosis (Figure 7B). c-Jun−/− cells transiently expressing WT His-c-Jun, but not its DNA binding mutant c-Jun Δ270–272 (Figure 7C) or Δ256–258 (data not shown) (Brown et al., 1996), showed an increased resistance to sorbitol-induced apoptosis, in contrast to untransfected cells. These results indicate that c-Jun has anti-apoptotic effects, which are dependent upon its DNA-binding ability, and that a deficiency in c-Jun promotes sorbitol-induced apoptosis.
Figure 7. Anti-apoptotic Function of c-Jun in Sorbitol-Induced Apoptosis and the Mechanism of Osmotic Stress-Induced Downregulation of c-Jun.
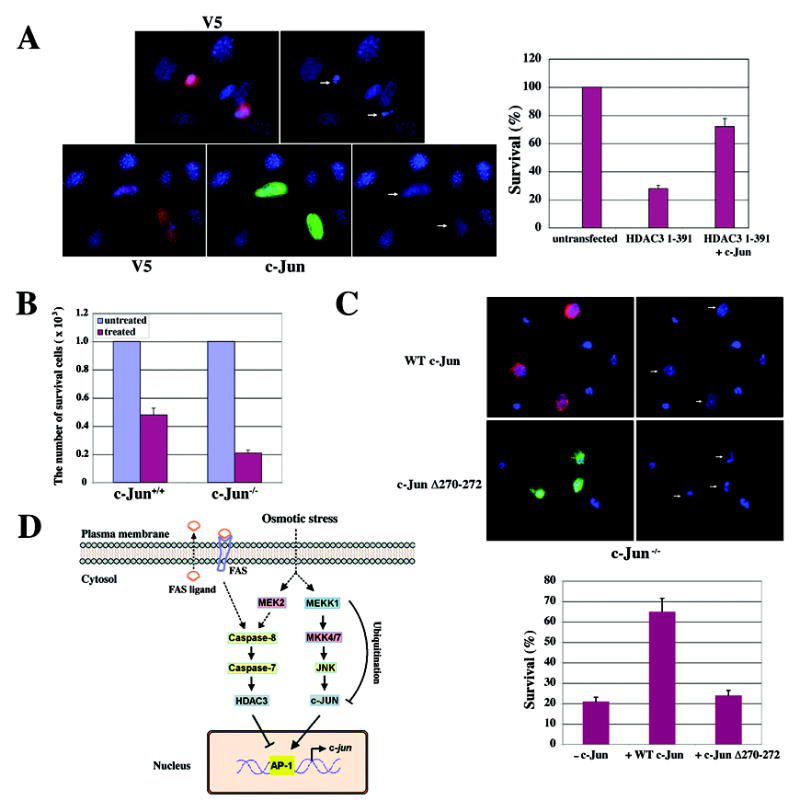
(A) NIH3T3 cells were transfected with pCDNA6/V5-His-HDAC3 (aa 1–391) or co-transfected with a pMT35-His-c-Jun construct for 48 h. Cells were stained with an anti-V5 antibody (V5; red) (top) or with both the anti-V5 antibody and an anti-c-Jun antibody (c-Jun; green) (bottom). Nuclei were stained with Hoechst 33342 (blue). The white arrows point to transfected cells (left). One hundred cells with or without expression of V5-His-HDAC3 (aa 1–391) and with or without co-expression of V5-His-HDAC3 (aa 1–391) and His-c-Jun were counted. The relative levels of cell survival were normalized to the levels of untransfected cells. Data represent the means ± SD of three independent experiments (right).
(B) c-Jun+/+ and c-Jun−/− cells were treated with or without sorbitol for 12 h, and a trypan blue exclusion assay was carried out. The relative survival levels of treated cell were normalized to the survival levels of 1×103 cells without sorbitol treatment. Data represent the means ± SD of three independent experiments.
(C) Two days after transfection of c-Jun−/− cells with pMT35-His-c-Jun or pFLAG-c-Jun Δ270–272, the cells were treated with sorbitol for 12 h. Cells expressing His-c-Jun (red) or FLAG-c-Jun Δ270–272 (Green) were detected with an anti-His antibody or anti-FLAG antibody. Nuclei were stained with Hoechst 33342 (blue). The white arrows point to transfected cells (top). One hundred cells with or without expression of His-c-Jun or FLAG-c-Jun Δ270–272 and with or without sorbitol treatment were counted. The relative levels of cell survival were normalized to the levels of cells expressing His-c-Jun or FLAG-c-Jun Δ270–272 without sorbitol treatment. Data represent the means ± SD of three independent experiments (Bottom).
(D) The mechanism of osmotic stress-induced downregulation of c-Jun. c-Jun is phosphorylated and activated by the MEKK1–MEK4/7–JNK kinase cascade at an early stage of osmotic stress. Prolonged osmotic stress downregulates c-Jun protein via transcriptional repression and MEKK1-mediated degradation. Activation of the FAS/MEK2–dependent caspase-8–caspase-7–HDAC3 cleavage cascade leads to deacetylation of histone at the c-jun promoter region and the subsequent transcriptional repression of c-jun. Downregulation of c-Jun promotes osmotic stress-induced cell apoptosis. (The arrow with the dashed line indicates an indirect effect.).
Discussion
c-Jun, functioning as a major AP-1 transcription factor, is phosphorylated and activated in response to diverse extracellular stimuli and regulates gene expression. Stimulation with TNF-α and UVB irradiation induced c-Jun phosphorylation and increased its expression. In contrast, sorbitol-induced c-Jun phosphorylation diminished after prolonged treatment and was accompanied by reduced c-Jun expression. These results indicate that c-Jun activity is dynamically regulated via the dual mechanisms of phosphorylation and protein expression and that the activation of c-Jun is either transient or sustained, depending on the nature of the extracellular stimuli to which the cells are exposed.
Transcriptional repression is a mechanism underlying the downregulation of c-Jun. That inhibition of HDAC rescued the sorbitol-induced transcriptional downregulation of c-Jun underscores the importance of functionally regulated HDAC activity in c-Jun downregulation, although the possible contribution of reduced HAT activity to histone deacetylation cannot be excluded. Intriguingly, HDAC3 is cleaved at Asp391 near its C terminus in response to sorbitol. It has been shown that a non-selective deletion mutant of HDAC3 (aa 1–401) immunoprecipitated from HeLa cells has reduced catalytic activity in vitro (Yang et al., 2002). However, we did not observe any dramatically altered activity of HDAC3 (aa 1–391) expressed in NIH3T3 cells, which is 37 aa shorter than WT HDAC3. These disparities could result from differences in cellular and experimental systems, as well as from possible structural changes of the protein by deletions. Truncated HDAC3 (aa 1–391) and WT HDAC3 have similar cytoplasmic and nuclear distributions, and sorbitol treatment did not cause nuclear translocation of the truncated HDAC3. Nevertheless, HDAC3 (aa 1–391), in contrast to WT HDAC3, has an increased association with c-Jun, enhanced recruitment to c-jun promoter regions in a c-Jun dependent manner, and increased repression of c-jun promoter activity and reduction of c-Jun expression. Acetylated histone H3, an HDAC3 substrate (Marks et al., 2003), was deacetylated more in the c-jun promoter region in cells expressing HDAC3 (aa 1–391) than in cells expressing WT HDAC3. This result, together with the finding that the HDAC inhibitors TSA and NaB abrogated sorbitol-induced c-Jun downregulation, provides strong evidence that the transcriptional downregulation of c-jun is regulated by HDAC3-mediated histone deacetylation. HDAC3 has been shown as an important component of N-CoR (nuclear receptor corepressor)–SMRT (silencing mediator for retinoid and thyroid receptors) corepressor complexes that also include GPS2, TBL1, and TBLR1 (Yoon et al., 2003; Zhang et al., 2002), and ectopically expressed GPS2 inhibits JNK activity in an HDAC activity-dependent manner (Zhang et al., 2002). Further investigation is needed to ascertain whether the truncation of HDAC3 alters the formation and/or composition, and subsequently the function, of the corepressor complexes, which in combination with recruitment c-Jun to HDAC3, result in targeted gene repression. Similar to the repression of c-jun by localized HDAC3 activity, HDAC4 has been shown to be cleaved in a caspase-3–dependent manner (Liu et al., 2004; Paroni et al., 2004), with the N-terminal fragment of HDAC4 translocating into the nucleus and repressing myocyte enhancer factor-2c (MEF2c) transcription (Paroni et al., 2004). Caspase-dependent HDAC3 degradation, which is involved in the regulation of E2F-1 transcription, has also been observed in neuronal apoptosis (Panteleeva et al., 2004).
Inhibition of HDAC3 cleavage and c-Jun downregulation by a caspase-3/7 inhibitor, but not a caspase-3 inhibitor, implies involvement of caspase-7 activity. This supposition is supported by the reduction in HDAC3 cleavage observed in DT40 caspase-7−/− cells and in the presence of an shRNA against caspase-7. Inhibition of caspase-7 by either means partially blocked the sorbitol-induced cell apoptosis (data not shown). Since caspase-7 deficiency did not completely abolish the sorbitol-induced HDAC3 cleavage, other caspases may compensate. The in vitro cleavage of HDAC3 by caspase-7, but not caspase-1, −2, −3, or −4, indicates that HDAC3 is a direct substrate of caspase-7. Caspases-3, −6, and −7 are effector caspases and can be activated by initiator caspases, such as caspase-2, −8, −9, or −10 (Nunez et al., 1998). Caspase-12 is an initiator caspase that regulates ER stress-mediated apoptosis. Inhibition or deficiency of caspase-8, but not caspase-9, −12, or −2 (data not shown), blocked caspase-7 activation, indicating a requirement for caspase-8 in caspase-7 activation in response to osmotic stress. Consistent with caspase-7 activation upon sorbitol treatment being independent of caspase-9, cytochrome c release from mitochondria occurred at a late stage of osmotic stress, in contrast to an early activation of caspases-8 and −7 and cleavage of HDAC3. These findings strongly suggest that caspase-7–dependent HDAC3 cleavage is not regulated by cytochrome c release.
MEK1/2 inhibitor U0126, but not JNK1/2 inhibitor SP600125, and MEK2 deficiency, but not MEK1 or JNK1/2 deficiency, partially blocked sorbitol-induced activation of caspases-8 and −7 (data not shown for MEK1−/− or JNK1/2−/− cells), HDAC3 cleavage, and c-Jun downregulation. These findings point to the involvement of MEK2 in caspase-8– and caspase-7–dependent HDAC3 cleavage. Since ERK1/2 activation was not affected in MEK2−/− cells, it seems that ERK1/2 activity is not involved in the regulation of HDAC3 cleavage in response to sorbitol treatment. Serum stimulation, which activates MEK1/2 and ERK1/2, did not induce either HDAC3 cleavage or c-Jun downregulation. These results imply that activated MEK2 induced by osmotic stress, in contrast to that stimulated by serum, mediates a different signal transduction process that leads to the functional regulation of different substrates.
Inhibition or deficiency of caspase-8 completely blocked activation of caspase 7, whereas MEK2 deficiency only partially blocked caspase-8 activation, HDAC3 cleavage, and c-Jun downregulation. These results indicate the involvement of other mechanisms in activation of caspase-8. Caspase-8 is best known to be activated by ligand binding-induced trimerization of death receptors (Kruidering and Evan, 2000). Sorbitol treatment induced a secretion of FAS ligand, which was detected at the early stages of osmotic stress. Blocking the binding of FAS ligand to FAS by a neutralizing FAS ligand antibody, which did not affect MEK1/2-ERK1/2 activation, partially blocked caspase-8 activation. A more complete inhibition of caspase-8 activation was observed by pre-treatment of MEK2-deficient cells with a neutralizing FAS ligand antibody. These results indicate that, in addition to MEK2, the activation of FAS death receptors mediated by osmotic stress-induced secretion of FAS ligand is another regulatory mechanism for caspase-8 activation.
Pretreatment with the general caspase inhibitor Z-VAD-FMK, which blocked the sorbitol-induced HDAC3 cleavage, did not completely reverse the downregulation of c-Jun (Figure 4A), implying an additional mechanism involved in c-Jun downregulation. Indeed, we found that osmotic stress also results in post-translational regulation, which involves MEKK1-mediated c-Jun ubiquitination and degradation (Xia et al., 2007), although this regulation plays a minor role in contrast to transcriptional repression. Thus, c-Jun downregulation in response to osmotic stress was regulated at more than one level.
c-Jun may have pro- and anti-apoptotic functions that depend on the cell or tissue type and the specific apoptotic stimuli involved (Jochum et al., 2001). The truncated HDAC3 (aa 1–391), which downregulates c-Jun, by itself is sufficient to induce apoptosis. The fact that the apoptotic effect of HDAC3 (aa 1–391) can be largely blocked by exogenous expression of c-Jun, but not its DNA-binding mutants, indicates that transcriptional repression of c-jun and subsequent c-jun–induced downstream gene expression by HDAC3 (aa 1–391) are indispensable for inducing cell apoptosis. That a genetic deficiency of c-Jun promotes sorbitol-induced apoptosis, which can be rescued by expression of c-Jun, provides additional evidence of the anti-apoptotic functions of c-Jun. These results are consonant with the fact that c-Jun−/− fibroblasts are much less capable than c-Jun+/+ cells of escaping UV-induced cell apoptosis (Wisdom et al., 1999). These results are also consistent with the massive level of apoptosis of hepatoblasts and erythroblasts resulting from c-Jun deficiency in the developing mouse liver in vivo (Eferl et al., 1999). Although more studies are needed to characterize the anti-apoptotic functions of c-Jun, it has been shown that c-Jun suppresses p53 transcription by directly binding to a variant AP-1 site in the p53 promoter (Schreiber et al., 1999). In addition, c-Jun protects hepatocytes from apoptosis by antagonizing p53 activity in liver-specific c-Jun conditional mutant mice (Eferl et al., 2003).
In summary, our results reveal a novel mechanism for the regulation of signal transduction in response to extracellular stimuli and provide a model for the dynamic regulation of c-Jun expression during stress responses in living cells (Figure 7D). c-Jun is phosphorylated and activated by the MEKK1-MKK4/7-JNK1/2 kinase cascade in response to brief treatment with sorbitol. Activated c-Jun counteracts extracellular stress by transcriptionally suppressing the expression of pro-apoptotic proteins or by activating the expression of anti-apoptotic proteins, which allows cells to recover from transient stress stimuli. When exposure to the stress is prolonged, c-Jun expression is downregulated through transcriptional repression and post-translational downregulation that involves MEKK1-mediated c-Jun ubiquitination. In combination with the effects of osmotic stress-induced secretion of FAS ligand and MEK2 activation, activation of caspase-8 leads to mitochondria-cytochrome c release-independent activation of caspase-7 and HDAC3 cleavage. The truncated HDAC3, which is generated by caspase-7 cleavage at the early phase of osmotic stress, binds to the c-jun promoter region in a c-Jun dependent manner, deacetylates histones, and thereby represses c-jun transcription. The depletion of c-Jun by MEKK1-mediated protein degradation and HDAC3-regulated transcriptional repression disrupts the positive feedback resulting from c-Jun binding to its own promoter, which further reduces c-jun transcription. The abrogated c-Jun expression, which downregulates c-Jun activation occurred during the early stage of exposure to stress, promotes osmotic stimulation-induced cell apoptosis. Our proposed model for the dynamic regulation of cell survival molecules, which are activated at an early stage of stress for counteracting apoptosis and downregulated at a late stage for promoting apoptosis, may represent an important general cellular mechanism.
Experimental Procedures
Cell Stimulation with Osmotic Stress
Cells were grown in media containing 0.5% serum for 16 h to induce the cultures to quiescence and then treated with sorbitol (500 mM) or mannitol (500 mM).
Subcellular Fractionation
Nuclei and cytosol were isolated by hypotonic lysis and isopycnic centrifugation on OptiPrepTM (Accurate Chemicals, Westbury, NY) iodixanol density gradient following the manufacturer’s protocol.
Real-Time Quantitative RT-PCR
First-strand cDNA was synthesized using the ProStar First Strand RT-PCR kit (Stratagene, Cedar Creek, TX). The c-jun primers 5′-GCCAACCTCAGCAACTTCAAC-3′ (forward) and 5′-GGAAGAGCCGCAGACCGT-3′ (reverse) were used in PCR. The primers for GAPDH RNA were 5′-ATGGGGAAGGTGAAGGTCGG-3′ (forward) and 5′-GACGGTGCCATGGAATTTGC-3′ (reverse). Real-time quantitative RT-PCR was performed with a kit (IQ SYBR Green Supermix kit; Bio-Rad Laboratories), according to the manufacturer's instructions.
ChIP Assay
ChIP was performed using an Upstate Biotechnology kit. Chromatin prepared from NIH3T3 cells, c-Jun−/− cells, c-Jun+/+ cells, or 293T cells (in a 10-cm dish) was used to determine total DNA input and for overnight incubation with the specific antibodies or with normal rabbit or mouse immunoglobulin G. The mouse c-jun promoter-specific primers used in PCR were 5′-CGGCAGCCTCCGTCACTAGACAG-3′ (forward) and 5′-AGATTCTTCTCTGGGCCCGCG-3′ (reverse). The human c-jun promoter-specific primers used in PCR were 5′-CGACTGTAGGAGGGCAGCGG-3′ (forward) and 5′-AGCCCTTATCCAGCCCGAGC-3′ (reverse).
In Vitro Caspase Cleavage Assay
Immunoprecipitated PARP from 293T cell lysate or 500 ng of purified WT His-HDAC3 or His-HDAC D391A protein was incubated with 2 units of caspases at 37°C for 4 h in a reaction solution (pH 7.2) containing 50 mM NaCl, 50 mM HEPES, 10 mM EDTA, 5% glycerol, and 0.1% CHAPS.
Cell Viability Assay
A total of 5 × 105 c-Jun−/− and c-Jun+/+ 3T3 mouse fibroblasts were plated. The cells were treated with or without sorbitol for 12 h after serum starvation for 16 h. Trypan blue exclusion assays were carried out, and cell viability was analyzed with a Vi-cell analyzer (Beckman Coulter, Fullerton, CA).
Immunofluorescence Analysis
Cells were fixed and incubated with primary antibodies, Alexa Fluor dye-conjugated secondary antibodies, and Hoechst 33342, according to standard protocols. Cells were examined using a deconvolutional microscope (Zeiss, Thornwood, NY) with a 60-Å oil immersion objective. Axio Vision software from Zeiss was used to deconvolve Z-series images.
Supplementary Material
Acknowledgments
We thank Dirk Bohmann (University of Rochester Medical Center) for the pMT35-His-c-Jun construct, Roger Davis (University of Massachusetts Medical School) for the JNK1/2−/− and JNK1/2+/+ fibroblasts, William Earnshaw (University of Edinburgh) for the DT40 and DT40 caspase-7−/− cells, Ann MacLaren (The Scripps Research Institute) for the c-Jun−/− and c-Jun+/+ 3T3 fibroblasts, Jean Charron (Centre de Recherche en Cancérologie de l'Université Laval, l'Hôtel-Dieu de Québec) for the MEK1−/−, MEK1+/+, MEK2−/−, and MEK2+/+ cells, Junying Yuan (Harvard Medical School) for the caspase-12−/− and caspase-12+/+ cells, Richard Flavell (Yale University School of Medicine) for the caspase-9−/− and caspase-9+/+ cells, Kazuhiro Sakamaki (Graduate School of Biostudies, Kyoto University) for the caspase-8−/− and caspase-8+/+ cells, and Wayne V. Vedeckis (Louisiana State University Medical Center) for the pc-Jun-Luc construct. We thank Dexing Fang for preparation of the graphs and Theresa Willis and Elizabeth Hess for critical reading of this manuscript.
This work was supported by the Pediatric Brain Tumor Foundation (Z.L.), the Charlotte Geyer Foundation (Z.L.), an institutional research grant from The University of Texas M. D. Anderson Cancer Center (Z.L.), and National Cancer Institute grants 1R01CA109035-01A1 (Z.L.), CA55418 (T.H.), and CA82683 (T.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- Bossis G, Malnou CE, Farras R, Andermarcher E, Hipskind R, Rodriguez M, Schmidt D, Muller S, Jariel-Encontre I, Piechaczyk M. Down-regulation of c-Fos/c-Jun AP-1 dimer activity by sumoylation. Mol Cell Biol. 2005;25:6964–6979. doi: 10.1128/MCB.25.16.6964-6979.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PH, Kim SH, Wise SC, Sabichi AL, Birrer MJ. Dominant-negative mutants of cJun inhibit AP-1 activity through multiple mechanisms and with different potencies. Cell Growth Differ. 1996;7:1013–1021. [PubMed] [Google Scholar]
- Canettieri G, Morantte I, Guzman E, Asahara H, Herzig S, Anderson SD, Yates JR, 3rd, Montminy M. Attenuation of a phosphorylation-dependent activator by an HDAC-PP1 complex. Nat Struct Biol. 2003;10:175–181. doi: 10.1038/nsb895. [DOI] [PubMed] [Google Scholar]
- Dixit VM, Marks RM, Sarma V, Prochownik EV. The antimitogenic action of tumor necrosis factor is associated with increased AP-1/c-jun proto-oncogene transcription. J Biol Chem. 1989;264:16905–16909. [PubMed] [Google Scholar]
- Eferl R, Ricci R, Kenner L, Zenz R, David JP, Rath M, Wagner EF. Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell. 2003;112:181–192. doi: 10.1016/s0092-8674(03)00042-4. [DOI] [PubMed] [Google Scholar]
- Eferl R, Sibilia M, Hilberg F, Fuchsbichler A, Kufferath I, Guertl B, Zenz R, Wagner EF, Zatloukal K. Functions of c-Jun in liver and heart development. J Cell Biol. 1999;145:1049–1061. doi: 10.1083/jcb.145.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Labuda T, Xia Y, Gallagher E, Fang D, Liu YC, Karin M. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science. 2004;306:271–275. doi: 10.1126/science.1099414. [DOI] [PubMed] [Google Scholar]
- Hazzalin CA, Mahadevan LC. Dynamic acetylation of all lysine 4-methylated histone H3 in the mouse nucleus: analysis at c-fos and c-jun. PLoS Biol. 2005;3:e393. doi: 10.1371/journal.pbio.0030393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilberg F, Aguzzi A, Howells N, Wagner EF. c-jun is essential for normal mouse development and hepatogenesis. Nature. 1993;365:179–181. doi: 10.1038/365179a0. [DOI] [PubMed] [Google Scholar]
- Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK)--from inflammation to development. Curr Opin Cell Biol. 1998;10:205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- Jochum W, Passegue E, Wagner EF. AP-1 in mouse development and tumorigenesis. Oncogene. 2001;20:2401–2412. doi: 10.1038/sj.onc.1204389. [DOI] [PubMed] [Google Scholar]
- Johnson RS, van Lingen B, Papaioannou VE, Spiegelman BM. A null mutation at the c-jun locus causes embryonic lethality and retarded cell growth in culture. Genes Dev. 1993;7:1309–1317. doi: 10.1101/gad.7.7b.1309. [DOI] [PubMed] [Google Scholar]
- Koh DW, Dawson TM, Dawson VL. Mediation of cell death by poly(ADP-ribose) polymerase-1. Pharmacol Res. 2005;52:5–14. doi: 10.1016/j.phrs.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Kruidering M, Evan GI. Caspase-8 in apoptosis: the beginning of "the end"? IUBMB Life. 2000;50:85–90. doi: 10.1080/713803693. [DOI] [PubMed] [Google Scholar]
- Leppa S, Bohmann D. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene. 1999;18:6158–6162. doi: 10.1038/sj.onc.1203173. [DOI] [PubMed] [Google Scholar]
- Liu F, Dowling M, Yang XJ, Kao GD. Caspase-mediated specific cleavage of human histone deacetylase 4. J Biol Chem. 2004;279:34537–34546. doi: 10.1074/jbc.M402475200. [DOI] [PubMed] [Google Scholar]
- Marks PA, Miller T, Richon VM. Histone deacetylases. Curr Opin Pharmacol. 2003;3:344–351. doi: 10.1016/s1471-4892(03)00084-5. [DOI] [PubMed] [Google Scholar]
- Matthews CC, Feldman EL. Insulin-like growth factor I rescues SH-SY5Y human neuroblastoma cells from hyperosmotic induced programmed cell death. J Cell Physiol. 1996;166:323–331. doi: 10.1002/(SICI)1097-4652(199602)166:2<323::AID-JCP10>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- McCabe JT, Burrell AS. Alterations of AP-1 and CREB protein DNA binding in rat supraoptic and paraventricular nuclei by acute and repeated hyperosmotic stress. Brain Res Bull. 2001;55:347–358. doi: 10.1016/s0361-9230(01)00520-2. [DOI] [PubMed] [Google Scholar]
- Muller S, Berger M, Lehembre F, Seeler JS, Haupt Y, Dejean A. c-Jun and p53 activity is modulated by SUMO-1 modification. J Biol Chem. 2000;275:13321–13329. doi: 10.1074/jbc.275.18.13321. [DOI] [PubMed] [Google Scholar]
- Musti AM, Treier M, Bohmann D. Reduced ubiquitin-dependent degradation of c-Jun after phosphorylation by MAP kinases. Science. 1997;275:400–402. doi: 10.1126/science.275.5298.400. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Nunez G, Benedict MA, Hu Y, Inohara N. Caspases: the proteases of the apoptotic pathway. Oncogene. 1998;17:3237–3245. doi: 10.1038/sj.onc.1202581. [DOI] [PubMed] [Google Scholar]
- Panteleeva I, Rouaux C, Larmet Y, Boutillier S, Loeffler JP, Boutillier AL. HDAC-3 participates in the repression of e2f-dependent gene transcription in primary differentiated neurons. Ann N Y Acad Sci. 2004;1030:656–660. doi: 10.1196/annals.1329.076. [DOI] [PubMed] [Google Scholar]
- Paroni G, Mizzau M, Henderson C, Del Sal G, Schneider C, Brancolini C. Caspase-dependent regulation of histone deacetylase 4 nuclear-cytoplasmic shuttling promotes apoptosis. Mol Biol Cell. 2004;15:2804–2818. doi: 10.1091/mbc.E03-08-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Muller S. Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc Natl Acad Sci U S A. 2002;99:2872–2877. doi: 10.1073/pnas.052559499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber M, Kolbus A, Piu F, Szabowski A, Mohle-Steinlein U, Tian J, Karin M, Angel P, Wagner EF. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999;13:607–619. doi: 10.1101/gad.13.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp FR, Sagar SM, Hicks K, Lowenstein D, Hisanaga K. c-fos mRNA, Fos, and Fos-related antigen induction by hypertonic saline and stress. J Neurosci. 1991;11:2321–2331. doi: 10.1523/JNEUROSCI.11-08-02321.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Schreiber M, Piu F, Beeche M, Wagner EF, Karin M. The mammalian UV response: c-Jun induction is required for exit from p53-imposed growth arrest. Cell. 2000;103:897–907. doi: 10.1016/s0092-8674(00)00193-8. [DOI] [PubMed] [Google Scholar]
- Sng JC, Taniura H, Yoneda Y. Inhibition of histone deacetylation by trichostatin A intensifies the transcriptions of neuronal c-fos and c-jun genes after kainate stimulation. Neurosci Lett. 2005;386:150–155. doi: 10.1016/j.neulet.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW. Substrate specificities of caspase family proteases. J Biol Chem. 1997;272:9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- Treier M, Staszewski LM, Bohmann D. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell. 1994;78:787–798. doi: 10.1016/s0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- Ventura JJ, Kennedy NJ, Flavell RA, Davis RJ. JNK regulates autocrine expression of TGF-beta1. Mol Cell. 2004;15:269–278. doi: 10.1016/j.molcel.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Vogt PK. Jun, the oncoprotein. Oncogene. 2001;20:2365–2377. doi: 10.1038/sj.onc.1204443. [DOI] [PubMed] [Google Scholar]
- Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Weiss C, Schneider S, Wagner EF, Zhang X, Seto E, Bohmann D. JNK phosphorylation relieves HDAC3-dependent suppression of the transcriptional activity of c-Jun. Embo J. 2003;22:3686–3695. doi: 10.1093/emboj/cdg364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz IE, O'Rourke KM, Zhang Z, Dornan D, Arnott D, Deshaies RJ, Dixit VM. Human De-etiolated-1 regulates c-Jun by assembling a CUL4A ubiquitin ligase. Science. 2004;303:1371–1374. doi: 10.1126/science.1093549. [DOI] [PubMed] [Google Scholar]
- Wisdom R, Johnson RS, Moore C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. Embo J. 1999;18:188–197. doi: 10.1093/emboj/18.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Wang J, Xu S, Johnson GL, Hunter T, Lu Z. MEKK1 Mediates the Ubiquitination and Degradation of c-Jun in Response to Osmotic Stress. Mol Cell Biol. 2007;27:510–517. doi: 10.1128/MCB.01355-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M, Dai T, Deak JC, Kyriakis JM, Zon LI, Woodgett JR, Templeton DJ. Activation of stress-activated protein kinase by MEKK1 phosphorylation of its activator SEK1. Nature. 1994;372:798–800. doi: 10.1038/372798a0. [DOI] [PubMed] [Google Scholar]
- Yang WM, Tsai SC, Wen YD, Fejer G, Seto E. Functional domains of histone deacetylase-3. J Biol Chem. 2002;277:9447–9454. doi: 10.1074/jbc.M105993200. [DOI] [PubMed] [Google Scholar]
- Yoon HG, Chan DW, Huang ZQ, Li J, Fondell JD, Qin J, Wong J. Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. Embo J. 2003;22:1336–1346. doi: 10.1093/emboj/cdg120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kalkum M, Chait BT, Roeder RG. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol Cell. 2002;9:611–623. doi: 10.1016/s1097-2765(02)00468-9. [DOI] [PubMed] [Google Scholar]
- Zheng CF, Guan KL. Cloning and characterization of two distinct human extracellular signal-regulated kinase activator kinases, MEK1 and MEK2. J Biol Chem. 1993;268:11435–11439. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.