

Abstract
The COP9 signalosome (CSN) is a conserved protein complex that regulates assembly and activity of cullin-RING ubiquitin ligases (CRLs). Ubiquitin-dependent degradation of the NF-κB inhibitor IκBα preceeds nuclear translocation of NF-κB. For the first time, we show here an inducible interaction of the CSN with IκBα and that the CSN controls IκBα and NF-κB activity. Strikingly, disruption of the CSN by a small interfering RNA-mediated knockdown of single CSN subunits results in a reduced re-accumulation of IκBα and prolonged nuclear translocation of NF-κB in TNFα-stimulated cells. The control of IκBα by the CSN is regulated by deubiquitinylation of IκBα conferred by the CSN-associated deubiquitinylase USP15. Protein expression levels of cullin1 and the CRL substrate adapter β-TrCP are reduced in nonstimulated cells with a disrupted function of the CSN, which might account for an impaired basal turnover of IκBα. We propose that the CSN controls both CRL activity and stability of the CRL substrate IκBα. In consequence, basal and signal-induced CRL-dependent turnover of IκBα is precisely adapted to specific cellular needs.
Keywords: COP9 signalosome, cullin-RING ubiquitin-ligase, Nedd8, ubiquitin, USP15
Introduction
The COP9 signalosome (CSN), an eight-subunit (CSN1–8) protein complex, is evolutionarily conserved in the genomes of eukaryotes (Schwechheimer and Deng, 2001). It has been implicated to be functionally involved in pleiotropic biological processes as diverse as DNA replication and repair, cell-cycle progression and development (Cope and Deshaies, 2003; Nielsen, 2003). Furthermore, a multitude of proteins associates with CSN subunits (Bech-Otschir et al, 2002). All CSN subunits can be aligned to a corresponding paralogue in the lid subcomplex of the 19S regulatory particle of the 26S proteasome, which has encouraged proposals that the CSN participates in the ubiquitin proteolysis pathway (UPP) (Schwechheimer and Deng, 2001). The biochemical activities of the CSN, for example, detachment of the ubiquitin-like protein Nedd8 (deneddylation), deubiquitinylation and phosphorylation, are focused upon the control and dynamic regulation of multisubunit cullin-RING ubiquitin-ligases (CRLs) that share a cullin/zinc-binding RING-H2 protein (ROC-1 or ROC-2) core catalytic module (Petroski and Deshaies, 2005).
Cullin 1 (Cul1) and all other cullins identified so far (Lyapina et al, 2001; Schwechheimer et al, 2001; Zhou et al, 2001; Groisman et al, 2003; Liu et al, 2003; Pintard et al, 2003) represent the best-characterised substrates for deneddylase activity of the CSN. This intrinsic deneddylase activity has been pinpointed to a Jab1/Mpn domain metalloenzyme (JAMM) submotif located in the MPN domain of CSN5 (Cope et al, 2002). Whereas the mechanism of Nedd8 modification resembles the protein ubiquitinylation pathway, Nedd8 attachment does not target proteins for degradation. Rather neddylation enhances ubiquitin polymerisation and substrate-specific ubiquitinylation activity of the CRL Skp1/Cul1/F-box protein (SCF) in vitro by facilitating the recruitment of a ubiquitin-loaded E2 enzyme to the Cul1/ROC1 core catalytic module (Pan et al, 2004). Substrate specificity is mediated by association of Cul1 with the linker protein Skp1, which in turn recruits F-box-containing substrate adapter proteins (Petroski and Deshaies, 2005). Although in vitro studies supported the assumption that CSN due to its deneddylase activity is a negative regulator of the UPP, accumulating genetic data indicate the opposite. This led to the proposal that cycles of cullin neddylation and deneddylation might be required to promote efficient protein degradation via the UPP (Cope and Deshaies, 2003; Wee et al, 2005).
A conserved ubiquitin-depolymerising activity associated with the CSN has been identified as Ubp12/USP15, a conventional deubiquitinylase (DUB) of the cysteine protease family in yeast and mammalian cells, respectively (Zhou et al, 2003; Hetfeld et al, 2005). It has been proposed that CSN5 and Ubp12/USP15 cooperate to inactivate CRL core complexes between their cycles of operation to prevent spurious autoubiquitinylation and degradation of labile ligase components, so that the ligase activity is focused upon legitimate substrates and substrate-specific ubiquitinylation and UPP targeting is sustained (Zhou et al, 2003; Hetfeld et al, 2005; Wee et al, 2005). It is still unknown whether other substrates apart from cullins exist, that can be deubiquitinylated by the CSN-associated DUB.
Although it has been established in some cases that CSN either stabilises (Naumann et al, 1999) or sensitises proteins towards the UPP (Tomoda et al, 1999; Bech-Otschir et al, 2001), the impact of the CSN on UPP-dependent turnover of IκBα is unknown. IκBα binds to NF-κB transcription factors, thereby retaining them inactive in the cytosol. In response to various exogenous stimuli, IκBα is phosphorylated on Ser32/Ser36 residues by IκB kinase β (IKKβ), which is part of an IKK complex containing two catalytic subunits (IKKα and IKKβ) and the regulatory subunit IKKγ. IKKβ-phosphorylated IκBα is rapidly degraded via the UPP. Consequently, functional NF-κB molecules are free to enter the nucleus. Ubiquitinylation of IκBα is promoted by the CRL SCFβTrCP containing the F-box protein β-TrCP as substrate adapter, which specifically recognises and binds to Ser32/Ser36-phosphorylated IκBα (Karin and Ben-Neriah, 2000). Our current study was undertaken to illustrate the underlying mechanism by which the CSN regulates the degradation of IκBα and its inducible re-accumulation.
Results
CSN associates with IκBα
To elucidate whether the CSN affects UPP-dependent degradation of IκBα, we first analysed a putative interaction between the CSN and IκBα. For this purpose, cells were transiently transfected with FlagCSN2 to enhance CSN assembly. As shown in Figure 1A, transient association of IκBα with the CSN was observed in an immunoprecipitation performed with an IκBα-specific antibody after 20 and 60 min of TNFα stimulation. CSN associated with IκBα was detected with an anti-Flag antibody recognising the ectopically expressed FlagCSN2 subunit within the CSN. To some extent, the increased amount of FlagCSN2 co-immunoprecipitated in cells pretreated with the 26S proteasome inhibitor could be due to an enhanced level of endogenous IκBα. Immunoprecipitated endogenous IκBα and its TNFα-induced degradation and re-accumulation were detected with an IκBα-specific antibody. As expected, ectopic overexpression of FlagCSN2 coincided with an increased expression of endogenous CSN7 (Figure 1B), consistent with enhanced assembly of the CSN.
Figure 1.
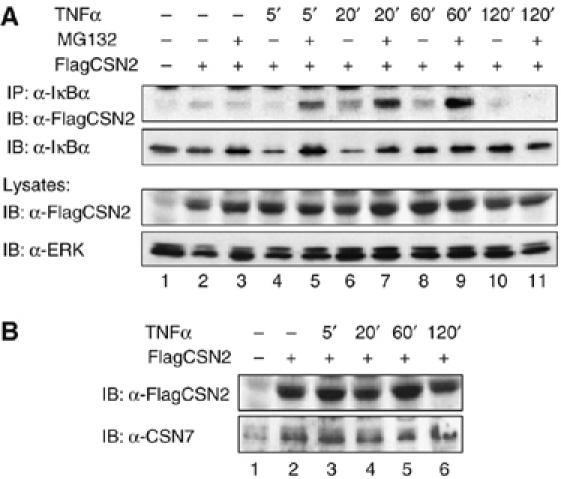
CSN associates with IκBα. (A) Cells were transiently transfected with a FlagCSN2 expression plasmid (lanes 2–11) to enhance CSN assembly. After 20 h, cells were stimulated with TNFα for the indicated periods of time (lanes 4–11) or left untreated (lanes 1–3). At 1 h before TNFα stimulation, cells were treated with either MG132 (lanes 3, 5, 7, 9 and 11) or vehicle (DMSO, lanes 1, 2, 4, 6, 8 and 10). After immunoprecipitation of endogenous IκBα from total lysates, interaction of the CSN with IκBα was detected with an anti-Flag antibody that recognises FlagCSN2 within the CSN (upper panel). The stripped immunoblot was developed with an anti-IκBα antibody to verify TNFα-induced turnover of IκBα, which is inhibited by MG132 (second panel). Ectopic expression of FlagCSN2 was detected in cell lysates using an anti-Flag antibody. Immunodetection of ERK was performed as a protein load control. (B) As a supplement for the experiment shown in (A), only samples obtained from cells not treated with MG132 were analysed. Ectopic expression of FlagCSN2 was detected using an anti-Flag antibody. Overexpression of FlagCSN2 enhanced CSN assembly, as shown in an immunoblot using an antibody against endogenous CSN7.
Knockdown of CSN2 affects the basal protein level of IκBα
To show a functional role of the CSN in the turnover of IκBα, we transiently silenced csn2 gene expression by small interfering RNA (siRNA) technology. Very efficient knockdowns were obtained with two different CSN2-specific siRNA oligonucleotides, resulting in almost complete suppression of CSN2 expression (Figure 2A). This effect could not be attributed to technical manipulations required to introduce siRNA into cells, because a nontargeting siRNA (mock) was without effect. Knockdown of CSN2 suppressed CSN3 expression as well, which supports our previous data that the CSN2 subunit affects assembly of the CSN (Naumann et al, 1999). Thus, removal of a single CSN subunit is sufficient to destabilise the CSN complex and reduce the levels of other unrelated CSN subunits. Similar data have been described in mammalian cells by knockdown of CSN4 (Denti et al, 2006).
Figure 2.
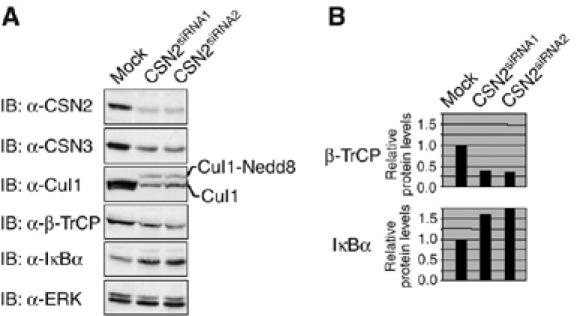
Knockdown of CSN2 affects the basal protein level of IκBα. Cells were transiently transfected with one of two different CSN2-specific siRNAs or transfected with a nontargeting siRNA (mock). After 48 h, they were harvested by RIPA lysis. (A) Cell lysates from mock-transfected and CSN2-knockdown cells were analysed by SDS–PAGE and Western blotting. Immunodetection of CSN subunits, Cul1, IκBα, β-TrCP and ERK (see indicated panels), was performed using specific antibodies. (B) Quantification of the results shown for β-TrCP and IκBα in (A). Densitometric values for protein bands were normalised to ERK content in each sample and depicted relative to the values of untreated mock-transfected cells. The data are representative for at least three independent experiments.
To confirm disruption of CSN function owing to knockdown of CSN2, we investigated the neddylation of Cul1. Neddylation mediates a characteristic mobility retardation of cullins in SDS–PAGE gels. Functionally, it promotes CRL catalytic activity, which is counteracted by the intrinsic deneddylase activity of the CSN. Thus, Cul1 should become hyperneddylated in cells that lack a functional CSN. As shown in Figure 2A, a molecular weight shift of Cul1 was observed in CSN2 knockdown compared to mock-transfected cells, the slower migrating band likely representing neddylated Cul1. Furthermore, in line with data obtained in Neurospora (He et al, 2005) and Drosophila (Wu et al, 2005), we observed a decrease in Cul1 expression in CSN2-knockdown cells, indicating that the CSN might regulate the stability of Cul1. The same seems to be true for the substrate adapter protein β-TrCP, the amount of which was also reduced in CSN2-knockdown cells (Figure 2A and B). Notably, the protein amount of the CRL substrate IκBα was increased in cells exerting a knockdown of CSN2 (Figure 2A and B). These results show that loss of function of the CSN is tightly connected with an impaired function of CRL.
CSN controls the inducible turnover of IκBα and nuclear translocation of NF-κB
Next, an impact of CSN on the inducible turnover of IκBα and on NF-κB activity was investigated. The IκBα protein was lost in mock-transfected cells within 10 min of TNFα stimulation owing to proteasomal degradation and was completely restored within 60 min of stimulation owing to de novo synthesis of IκBα. In contrast to that, reduced re-accumulation of IκBα was observed in CSN2-knockdown cells (Figure 3A and B). As the latter did not result from decreased mRNA expression (data not shown), post-transcriptional regulation of protein levels was suggested. Simultaneously, TNFα-induced Ser32/Ser36 phosphorylation of IκBα was markedly reduced in CSN2-knockdown cells (Figure 3A). Interestingly, however, the phospho-specific IκBα antibody prominently recognised high-molecular-weight forms of phosphorylated IκBα in CSN2-knockdown cells (Figure 3A), which could be enriched by immunoprecipitation of IκBα (Figure 3C). The appearance of these high-molecular-weight forms of phosphorylated IκBα strongly suggests enhancement of an additional post-translational modification of IκBα, most likely ubiquitinylation. Regarding NF-κB, TNFα stimulation resulted in Ser536 phosphorylation of p65 and in nuclear translocation of the p65 and p50 subunits (Figure 3D and E). Nuclear p65 was detected in mock-transfected cells as early as 10 min after stimulation, peaked at 20 min and declined within 90 min, whereas in CSN2-knockdown cells p65 stayed at the peak level in the nucleus up to 90 min. Similar results were obtained for the NF-κB p50 subunit, which transiently translocated to the nucleus in mock-transfected cells, but stayed at the peak level in CSN2-transfected cells up to 90 min. At the same time, transient accumulation and subsequent decline of Ser536-phosphorylated p65 were retarded in CSN2-knockdown cells (Figure 3D and E). These results suggest that the CSN exerts a functional role in the regulation of NF-κB activity by supporting the stability and re-accumulation of IκBα post induction. The observed differences between CSN2-knockdown and mock-transfected cells in the nuclear translocation of NF-κB were mirrored by differences in the NF-κB DNA-binding activity showing prolonged NF-κB DNA-binding activity in CSN2-knockdown cells (Figure 3F).
Figure 3.
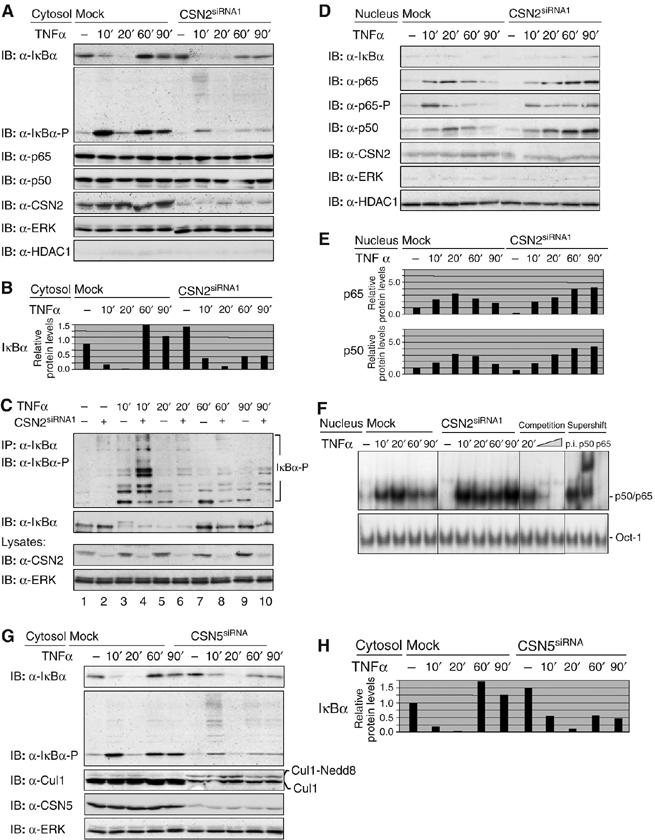
Knockdown of the CSN affects the inducible turnover of IκBα and nuclear translocation of NF-κB. Cells were transiently transfected with CSN2siRNA1 or CSN5siRNA. After 48 h, cells were stimulated for different periods of time, as indicated, with TNFα. The cells were harvested in hypotonic lysis buffer, followed by cytosolic/nuclear fractionation. Efficient knockdown of CSN2 or CSN5 (indicated panels) was analysed in cell lysates by immunodetection. In order to control the purity of cytosolic and nuclear preparations, ERK was immunodetected as a cytosolic and HDAC1 as a nuclear marker protein. (A) Cytosolic fractions of CSN2-knockdown cells were analysed by SDS–PAGE and Western blotting. To confirm TNFα-induced phosphorylation and turnover of IκBα, immunodetection of IκBα and its Ser32/Ser36-phosphorylated form was performed using specific antibodies. The p65 and p50 subunits (see the indicated panels) were immunodetected to show their predominant cytosolic localisation. (B) Quantification of the results shown for IκBα in (A) was performed as described in Figure 2B. (C) Endogenous IκBα was immunoprecipitated from cytosolic fractions of HeLa cells, transfected with CSN2siRNA1 or a nontargeting siRNA. Immunoprecipitates were analysed by SDS–PAGE and Western blotting. The IκBα protein level and its multiple high-molecular-weight forms of phosphorylated IκBα were immunodetected using IκBα- and phospho-IκBα-specific antibodies, respectively. (D) Nuclear fractions were analysed by SDS–PAGE and Western blotting. Translocated p65, Ser536-phosphorylated p65, as well as p50 (indicated panels) were immunodetected using specific antibodies. (E) Quantification of the results shown for p50 and p65 in (D). Densitometric values for protein bands were normalised to HDAC1 content in each sample and depicted relative to the values of untreated mock-transfected cells. (F) Nuclear fractions were analysed by Electrophoretic mobility shift assay (EMSA). The shifted DNA-bound NF-κB p50/p65 heterodimer and the shifted DNA-bound Oct-1 transcription factor (used as a load control) are indicated. To test the specificity, competition (using 10 and 20 ng of the nonlabelled double-stranded Igκ oligonucleotide) and antibody supershifting using anti-p50 and anti-p65 antibodies as well as preimmune serum were performed. (G) Cytosolic fractions of CSN5-knockdown cells were analysed by SDS–PAGE and Western blotting. To confirm TNFα-induced phosphorylation and turnover of IκBα, immunodetection of IκBα and its Ser32/Ser36-phosphorylated form was performed using specific antibodies. Neddylated Cul1 (Cul1-Nedd8) was immunodetected, using a Cul1-specific antibody. (H) Quantification of the results shown for IκBα in (G) was performed as described in Figure 2B.
To further support a role of CSN in the fine-tuning of the IκB/NF-κB system, we set out to confirm our results by knockdown of CSN5, the subunit exerting deneddylase activity when integrated in the CSN complex. As illustrated in Figure 3G, hyperneddylation and reduced expression of Cul1 were evident in CSN5-knockdown cells to a similar extent as shown for CSN2-knockdown cells (Figure 2A). Most importantly, knockdown of CSN5 resulted in a reduced re-accumulation of IκBα after its TNFα-induced degradation. Furthermore, as in CSN2-knockdown cells, high-molecular-weight forms of phosphorylated IκBα molecules prominently accumulated in CSN5-knockdown cells in response to TNFα stimulation (Figure 3G and H). Collectively, these data thus clearly indicate that the observed effects result from impaired function of the CSN.
CSN regulates CRL assembly
We investigated whether the re-accumulation of IκBα in TNFα-stimulated cells correlates with Cul1 deneddylation activity of the CSN. To determine the inducible association of β-TrCP with Cul1, we immunoprecipitated β-TrCP from TNFα-stimulated CSN2-knockdown and mock-transfected cells (Figure 4A). Interaction of Cul1 with β-TrCP increased in mock-transfected cells within 20 min of TNFα treatment (Figure 4A). Notably, neddylated Cul1 associated with β-TrCP could be detected with an anti-Nedd8 antibody. This result is consistent with published data (Petroski and Deshaies, 2005) showing that the substrate adapter β-TrCP is specifically recruited into the activated/neddylated CRL SCFβ−TrCP responsible for ubiquitinylation of IκBα. In CSN2-knockdown cells, constitutive interaction of neddylated Cul1 with β-TrCP was observed and this interaction was not further enhanced by TNFα stimulation (Figure 4A). This indicates that CSN2-knockdown cells exert constitutive CRL activity and that the reduced amount of β-TrCP (Figure 4A) efficiently integrates into active CRL complexes. However, as shown before (Figure 2), hyperneddylation of Cul1 coincides with an accumulation and thus putatively decreased basal turnover of the IκBα protein in nonstimulated CSN2-knockdown cells. Thus, in the absence of stimulation, spurious autoubiquitinylation of Cul1 and β-TrCP might be facilitated, explaining their reduced protein levels and a decreased basal CRL activity. Regarding the reduced re-accumulation of de novo synthesised IκBα in response to TNFα stimulation in CSN2-knockdown cells (Figure 4A), one might speculate about a causal role of constitutively active CRL complexes, which could rapidly degrade re-synthesised IκBα. However, the fact that IκBα does efficiently re-accumulate in mock-transfected cells within 60 min of TNFα stimulation in the presence of active CRL complexes (Figure 4A) strongly indicates that the CSN supports the re-accumulation of IκBα by an additional mechanism, which is not functional in CSN2-knockdown cells. Constitutive association of neddylated Cul1 with β-TrCP in CSN2-knockdown cells was confirmed in a co-immunoprecipitation using a Nedd8-specific antibody (Figure 4B).
Figure 4.
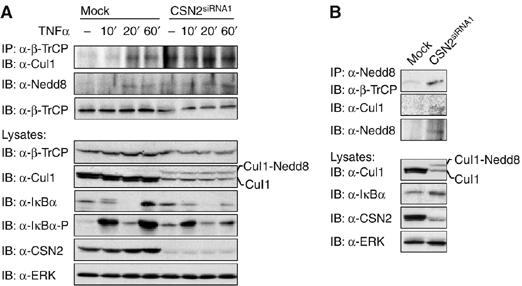
CSN regulates CRL assembly. (A) Cells were transiently transfected with CSN2siRNA1 to functionally disrupt the CSN. At 48 h after transfection, mock-transfected and CSN2-knockdown cells were stimulated for different periods of time with TNFα or left untreated, as indicated. The cells were harvested in hypotonic lysis buffer followed by cytosolic/nuclear fractionation. Endogenous β-TrCP was immunoprecipitated from the cytosolic fractions and the immunoprecipitates analysed by SDS–PAGE and Western blotting. Cul1 associated with β-TrCP as well as neddylated Cul1 (Cul1-Nedd8) was immunodetected, using Cul1- or Nedd8-specific antibodies, as indicated. Reduced expression of immunodetected β-TrCP and Cul1 in lysates of CSN2siRNA1-treated cells was shown by use of protein-specific antibodies. IκBα and phosphorylated IκBα were immunodetected using specific antibodies. Immunodetection of CSN2 was shown as a control for efficient knockdown. Equal amounts of protein in the lysates were controlled by immunodetection of ERK (indicated panels). (B) Endogenous Nedd8 was immunoprecipitated from cytosolic fractions of HeLa cells, transfected with CSN2siRNA1 or a nontargeting siRNA. Immunoprecipitates from mock-transfected and CSN2siRNA1-treated cells were analysed by SDS–PAGE and Western blotting. Associated β-TrCP as well as Cul1 was detected using β-TrCP- and Cul1-specific antibodies, respectively. The neddylation state of Cul1 was immunodetected in the total cell lysate using a Cul1-specific antibody. To verify the functional impairment of CSN, hyperneddylation and a decrease in Cul1 expression, as well as increased expression of IκBα in CSN2-knockdown versus mock-transfected cells was immunodetected in the cell lysates using Cul1- or IκBα-specific antibodies, as indicated. ERK was immunodetected to show equal amounts of protein in the cell lysates.
CSN promotes deubiquitinylation of IκBα
To analyse if the CSN might stabilise IκBα by deubiquitinylation, endogenous IκBα was immunoprecipitated from CSN2-knockdown and mock-transfected cells and its ubiquitinylation during the time course of TNFα stimulation was determined. Stimulation of the cells with TNFα resulted in rapid degradation of IκBα and its re-accumulation within 60 min (Figure 5A, second panel). TNFα-induced ubiquitinylation of immunoprecipitated IκBα was detected in the presence of MG132, and clearly enhanced in CSN2 knockdown compared to mock-transfected cells (Figure 5A). Notably, in CSN2-knockdown cells but not in mock-transfected cells, ubiquitinylation of IκBα was detectable after 10 min of TNFα stimulation already in the absence of MG132. This correlated well with the accumulation of high-molecular-weight forms of phosphorylated IκBα molecules, which was most prominent at this time point (Figure 3C). Thus, the observed high-molecular-weight forms of phosphorylated IκBα molecules represent ubiquitinylated and phosphorylated IκBα, which accumulates in CSN-depleted cells.
Figure 5.
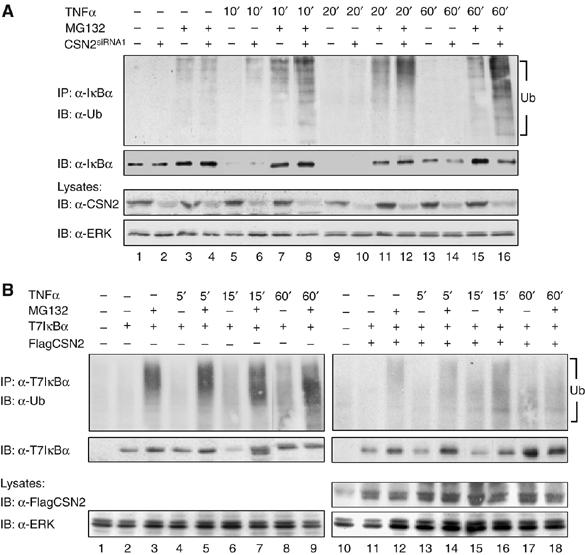
CSN promotes deubiquitinylation of IκBα. (A) Cells were either transiently transfected with CSN2siRNA1 (lanes 2, 4, 6, 8, 10, 12, 14 and 16) or mock transfectd (lanes 1, 3, 5, 7, 9, 11, 13 and 15). Then, cells were stimulated for different periods of time with TNFα or left untreated as indicated. At 1 h before stimulation, MG132 (lanes 3, 4, 7, 8, 11, 12, 15 and 16) or vehicle (DMSO, lanes 1, 2, 5, 6, 9, 10, 13 and 14), was added to the culture medium. After harvest of the cells by RIPA lysis, endogenous IκBα was immunoprecipitated, using an anti-IκBα antibody. Ubiquitinylated IκBα was immunodetected with an anti-ubiquitin antibody. The blot was stripped and developed with an anti-IκBα antibody to show TNFα-induced degradation and re-accumulation of IκBα. In addition, CSN2 was immunodetected in the cell lysates, using an anti-CSN2 antibody, to verify efficient knockdown of the CSN2 protein. Immunodetection of ERK in the cell lysates was performed as protein load control. (B) T7IκBα was expressed either alone (lanes 2–9) or in combination with FlagCSN2 (lanes 11–18) by transient transfection of equal amounts of cDNA in HeLa cells. Before cell lysis, the cells were stimulated with TNFα (lanes 4–9 and 13–18) for the indicated periods of time or left untreated (lanes 1–3 and 10–12). Additionally, the cells were treated for 1 h with either MG132 (lanes 3, 5, 7 9, 12, 14, 16 and 18) or vehicle (DMSO, lanes 1, 2, 4, 6, 8, 10, 11, 13, 15 and 17) before stimulation. Ectopically expressed T7IκBα was immunoprecipitated from cell lysates using an anti-T7 antibody and the protein complexes were subjected to SDS–PAGE and Western blotting. T7IκBα ubiquitinylation in TNFα-stimulated cells was determined by immunostaining with an anti-ubiquitin antibody (upper panels). The stripped immunoblots were developed with an anti-IκBα antibody to detect the TNFα-induced proteolytic turnover of T7IκBα, which was inhibited by MG132 (second panels). Ectopic expression of FlagCSN2, which mediates enhanced expression of CSN, was recognised by immunodetection with an anti-Flag antibody in the total cell lysate. Immunodetection of ERK was performed as protein load control.
To further investigate the impact of enhanced assembly of the CSN on the deubiquitinylation of IκBα, we cotransfected cDNAs encoding T7IκBα and FlagCSN2 or transfected T7IκBα alone (Figure 5B). As overexpression of IκBα can be toxic for cells, ectopic expression of the protein was kept below the expression level of endogenous IκBα (data not shown). Ectopically expressed T7IκBα was immunoprecipitated and its ubiquitinylation during the time course of TNFα stimulation was determined. Stimulation of the cells with TNFα resulted in degradation of ectopically expressed T7IκBα within 15 min, which re-accumulated within 60 min (Figure 5B, second panels). The observed increase in T7IκBα accumulation compared to T7IκBα in nonstimulated cells is consistent with the data reported by Place et al (2001) and can be explained by a deregulated synthesis of the ectopically expressed protein, which occurs under the control of the strong CMV promoter. TNFα-induced ubiquitinylation of T7IκBα was observed in MG132-treated cells (Figure 5B, upper panel). Only very weak ubiquitinylation of ectopically expressed T7IκBα was detectable during the time course of TNFα stimulation under the condition of enhanced CSN assembly induced by ectopic coexpression of FlagCSN2 (Figure 5B, upper panel). Overall, these data strongly suggest that deubiquitinylation of IκBα by the CSN plays a causal role in CSN-mediated stabilisation of IκBα.
CSN-associated USP15 deubiquitinylates IκBα
One candidate factor for IκBα deubiquitinylation is represented by USP15, the DUB known to be associated with the CSN. USP15 belongs to the large group of ubiquitin-specific processing proteases (USPs) responsible for processing of inactive ubiquitin precursors, removing ubiquitin from cellular adducts and ubiquitinylated proteins, and keeping the 26S proteasome free of inhibitory ubiquitin chains (Hetfeld et al, 2005). In order to analyse if deubiquitinylation of IκBα by the CSN might be executed by USP15, we investigated the impact of this DUB on the inducible turnover of IκBα. As illustrated in Figure 6A and B, reduced re-accumulation of IκBα after its TNFα-induced degradation was observed in cells showing efficient suppression of USP15 expression. As in CSN2-knockdown cells (Figure 3F), this reduction in IκBα re-accumulation in response to TNFα stimulation coincided with a decelerated decline in NF-κB DNA-binding activity (Figure 6C). However, this effect was less pronounced than in CSN2-depleted cells (Figure 3F). Additionally, TNFα-induced Ser32/Ser36 phosphorylation of IκBα was reduced in USP15-knockdown cells (Figure 6A). The analysis of IκBα immunoprecipitates from USP15-knockdown cells clearly demonstrated that high-molecular-weight forms of phosphorylated IκBα molecules accumulated in these cells in response to TNFα stimulation and could be stabilised by pretreatment of the cells with MG132 (Figure 6D, upper panel). Immunodetection with an ubiquitin-specific antibody revealed that the presence and abundance of high-molecular-weight forms of phosphorylated IκBα molecules correlate with the presence and abundance of ubiquitinylated IκBα molecules (Figure 6D, second panel).
Figure 6.
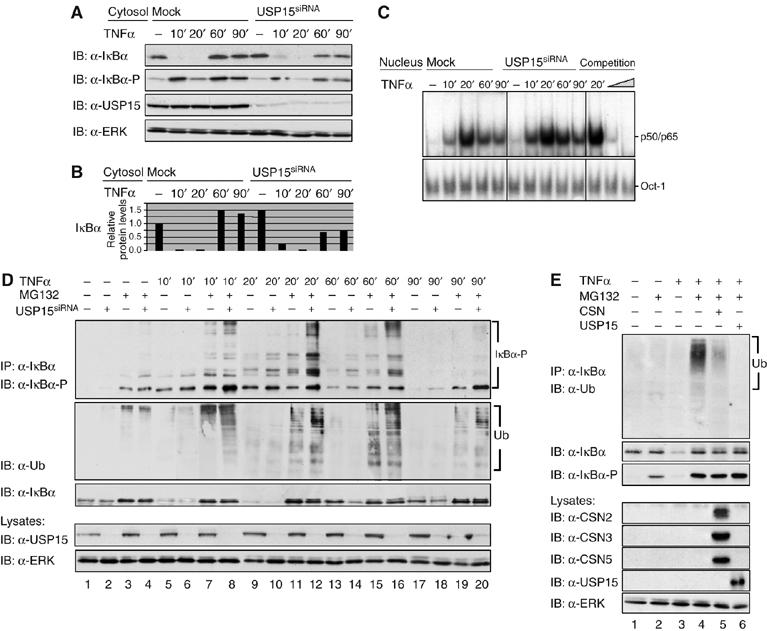
CSN-associated USP15 deubiquitinylates IκBα. (A) Cells were either transiently transfected with USP15siRNA or mock transfected. After 48 h, cells were stimulated for different periods of time, as indicated, with TNFα. Cytosolic fractions of USP15-knockdown cells were analysed by SDS–PAGE and Western blotting. To confirm TNFα-induced phosphorylation and turnover of IκBα, immunodetection of IκBα and its Ser32/Ser36-phosphorylated form was performed using specific antibodies. USP15 was immunodetected to confirm knockdown efficiency. Immunodetection of ERK in the cell lysates was performed as protein load control. (B) Quantification of the results shown for IκBα in (A) was performed as described in Figure 2B. (C) Nuclear fractions were analysed by EMSA as described in Figure 3F. (D) Cells were either transiently transfected with USP15siRNA (lanes 2, 4, 6, 8, 10, 12, 14, 16, 18 and 20) or mock-transfected (lanes 1, 3, 5, 7, 9, 11, 13, 15, 17 and 19). Then, cells were stimulated for different periods of time with TNFα (lanes 5–20) or left untreated (lanes 1–4). At 1 h before stimulation, MG132 (lanes 3, 4, 7, 8, 11, 12, 15, 16, 19 and 20) or vehicle (DMSO, lanes 1, 2, 5, 6, 9, 10, 13, 14, 17 and 18) was added to the culture medium. After harvest of the cells, endogenous IκBα was immunoprecipitated from cytosolic fractions of HeLa cells using an anti-IκBα antibody. Immunoprecipitates were analysed by SDS–PAGE and Western blotting. The IκBα protein and its multiple high-molecular-weight forms of phosphorylated IκBα were immunodetected using IκBα- and phospho-IκBα-specific antibodies. Ubiquitinylated IκBα was immunodetected with an anti-ubiquitin antibody. USP15 was immunodetected in the cell lysates, using an anti-USP15 antibody, to verify efficient knockdown of the USP15 protein. Immunodetection of ERK in the cell lysates was performed as protein load control. (E) Cells were preincubated for 1 h (lanes 2 and 4–6) with MG132 or treated with vehicle (DMSO, lanes 1 and 3) and subsequently stimulated (lanes 3–6) or not (lanes 1 and 2) with TNFα for 15 min. IκBα immunoprecipitates were incubated for 4 h at 37°C in the presence of an ATP regenerating system. In addition, purified CSN (lane 5) or recombinant USP15 (lane 6) was added to catalyse deubiquitinylation of IκBα. Aliquots of the reactions were analysed by SDS–PAGE and Western blotting. Immunodetection of ubiquitin, IκBα and its Ser32/Ser36-phosphorylated form, CSN subunits and USP15 (see indicated panels) was performed using specific antibodies. Immunodetection of ERK in the cell lysates was used as a control for equal amounts of protein applied to each immunoprecipitation.
To directly prove the ability of CSN and USP15 to deubiquitinylate IκBα, we performed an in vitro DUB assay in which we used IκBα as a substrate, which was immunoprecipitated from TNFα-stimulated cells treated with MG132. Immunodetection with an anti-ubiquitin antibody showed the ubiquitinylation of the substrate (Figure 6E, upper panel, lane 4). Purified CSN (Figure 5E, lane 5, upper panel) and, more efficiently, purified USP15 (Figure 5E, lane 6, upper panel) deubiquitinylated IκBα. Similarly, the CSN and USP15 efficiently cleaved the Lys48 isopeptide bonds of Ub4 as a substrate (data not shown).
Collectively, these results show that USP15 has a previously unknown impact on the stability of IκBα in TNFα-stimulated cells and could be, at least in part, responsible for the CSN deubiquitinylation activity towards IκBα.
Discussion
The CSN is a conserved protein complex in eukaryotic cells, involved in the regulation of ubiquitin-dependent proteolysis. As major findings of the present study, we report here that (i) the CSN interacts with IκBα in an inducible and transient manner, (ii) the CSN controls NF-κB activity by supporting the re-accumulation of IκBα after its signal-induced degradation, (iii) apart from CSN-mediated deneddylation, which controls CRL activity, stabilisation of de novo-synthesised IκBα does primarily depend on CSN-associated DUB activity, represented by USP15 (Figure 7).
Figure 7.
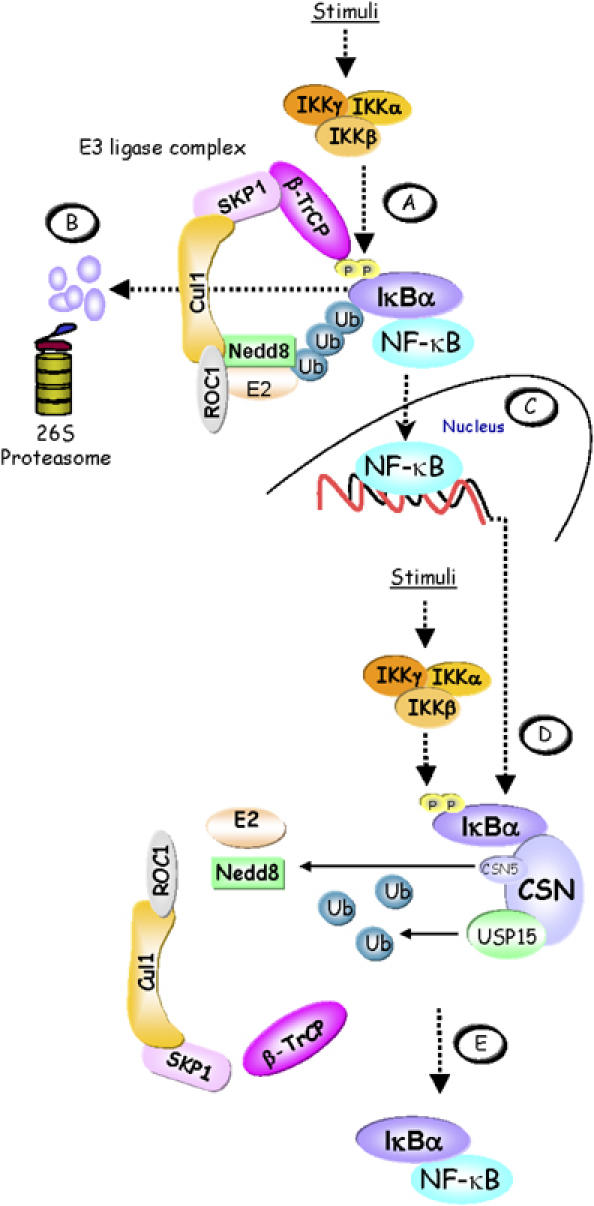
Model of CSN-mediated IκBα control. (A) In stimulated cells, the activated IKK complex phosphorylates Ser32 and Ser36 in the N-terminus of IκBα. When phosphorylated, these serine residues are part of a ‘degron' that is specifically recognised by β-TrCP, the CRL substrate adapter for IκBα. CRL activity is promoted by neddylation of Cul1. The latter facilitates the recruitment of a ubiquitin-loaded ubiquitin-conjugating enzyme (E2) to the ROC1 subunit of the CRL, which then cooperates with the CRL to ubiquitinylate IκBα. (B) Ubiquitinylated IκBα is degraded via the 26S proteasome releasing the previously bound and inactivated NF-κB. (C) Released NF-κB translocates to the nucleus to activate target genes, including the IκBα gene. As part of a negative feedback loop, re-accumulating IκBα can dissociate DNA-bound NF-κB and re-transport it to the cytosol. (D) During persistent stimulation, re-accumulated IκBα becomes phosphorylated in the cytosol by the IKK complex, thus initiating the cycle again. However, the CSN can enhance the stability of IκBα and thereby contribute to shut down NF-κB activity. This might be brought about in part by cycles of CSN5-mediated deneddylation of Cul1, which controls CRL activity. Most importantly, however, IκBα is deubiquitinylated by the CSN-associated DUB USP15. (E) Re-accumulated IκBα firmly associates with NF-κB, keeping it inactive in the cytosol.
NF-κB activity is subject to negative feedback regulation through NF-κB-dependent induction of iκbα gene transcription (Sun et al, 1993), and Place et al (2001) noted that the rebound of IκBα in stimulated cells shows a surprisingly fast kinetics even in the presence of N-terminal phosphorylation. They concluded that in addition to the induction of iκbα gene transcription, other mechanisms, such as protection of IκBα from degradation, must take effect to allow this rapid replenishment. Our observation that the CSN inducibly associates with IκBα and might selectively support the re-accumulation of IκBα (Figure 1) led us to examine if deneddylase and DUB activity of the CSN affect the re-accumulation of IκBα after its TNFα-induced proteasomal degradation. To disrupt CSN function, we transiently silenced the expression of the CSN2 and CSN5 subunits. Knockdown of the protein expression of CSN subunits and destabilisation of the CSN complex coincided with hyperneddylation of Cul1, as well as reduced expression of the CRL subunits β-TrCP and Cul1 (Figures 2 and 3), likely owing to autoubiquitinylation-induced accelerated proteasomal degradation. Simultaneously, the basal protein level of the CRL substrate IκBα was affected in nonstimulated cells (Figure 2). This indicated that knockdown of single CSN subunits strongly interfered with CSN function. However, in line with a previous publication (Cope and Deshaies, 2006), neddylation of Cul1 in CSN-depleted cells was not complete, indicating either residual CSN deneddylation activity or, more likely, the presence of other deneddylases, which could partially compensate the impaired deneddylase activity of the CSN. In addition, hyperneddylation of Cul1 in CSN2- or CSN5-knockdown cells was associated with reduced Cul1 expression in HeLa cells (Figures 2 and 3G) and Neurospora (He et al, 2005), whereas normal Cul1 expression levels have been observed in CSN5-depleted HEK293 cells (Cope and Deshaies, 2006). It is unclear at present why CSN5-knockdown HEK293 and HeLa cells behave differently.
During detailed characterisation of the CSN2- and CSN5-knockdown cells, a reduced ability to re-accumulate IκBα after its TNFα-induced degradation was observed (Figure 3). The reduced re-accumulation of IκBα coincided with a prolonged nuclear translocation of the p65 and p50 NF-κB subunits and NF-κB DNA-binding activity, which is consistent with a defect in the negative NF-κB feedback regulation. Apart from the function of the CSN to enhance the re-accumulation of IκBα, it seems that the CSN also affects the extent of degradation of IκBα (Figure 3A and G) as well as the resulting translocation of p50/p65 to the nucleus (Figure 3F) in TNFα-stimulated cells. To investigate these effects of the CSN in more detail, additional experiments applying lower doses of TNFα and the analysis of shorter periods of time are required. Overall, these data suggest an important role of the CSN in the fine-tuning of TNFα-induced NF-κB activity.
Signal-induced assembly of SCFβ−TrCP, the CRL responsible for ubiquitinylation of IκBα, requires the substrate adapter β-TrCP and the linker protein Skp1 to recruit Ser32/Ser36-phosphorylated IκBα to Cul1/Roc1 core complexes, which are kept in an inactive state in nonstimulated cells via their association with the regulatory protein CAND1 (Petroski and Deshaies, 2005). Inactivation of the Cul1/Roc1 core complexes is relieved through dissociation of CAND1 by an ATP-dependent mechanism and counteracted by neddylation of Cul1, which prevents re-association of the Cul1/Roc1 core complexes with CAND1 (Liu et al, 2002; Zheng et al, 2002; Goldenberg et al, 2004). Immunoprecipitation of β-TrCP thus allowed to selectively analyse the formation of activated CRL complexes during the time course of TNFα stimulation (Figure 4). We could show that the interaction of β-TrCP with Cul1 in its neddylated state, indicative for active CRL complexes, is inducible in mock-transfected cells, but constitutive in CSN2-knockdown cells. Most notably, however, these experiments clearly demonstrated that the reduced ability of CSN2-knockdown cells to re-accumulate IκBα cannot convincingly be explained solely by constitutively activated CRL complexes, because efficient re-accumulation of IκBα did occur in mock-transfected cells in the presence of active CRL complexes (Figure 4). Therefore, an additional mechanism, like deubiquitinylation of IκBα, was anticipated to be involved in the protection of de novo-synthesised IκBα from proteasomal degradation in stimulated cells.
It has been established that the CSN supports substrate-specific proteolysis via the UPP through its deneddylation and deubiquitinylation activities. To explain this mechanistically, it has been proposed that cycles of neddylation and deneddylation might be required to allow CRLs to rapidly adapt to changing substrate adapter requirements and to protect CRL subunits from autoubiquitinylation-dependent degradation (Wolf et al, 2003; Petroski and Deshaies, 2005).
No evidence had been obtained so far that the CSN has the capability to contribute in the regulation of CRL substrates, such as IκBα. In the present study, we show that ubiquitinylation of IκBα is increased in CSN2-knockdown cells, whereas enhanced assembly of the CSN by overexpression of CSN2 coincides with a sharply reduced ubiquitinylation of IκBα in stimulated cells (Figure 5A and B). To confirm our hypothesis that CSN-dependent deubiquitinylation of IκBα might stabilise de novo-synthesised IκBα and support its re-accumulation after TNFα-induced degradation, we performed a knockdown of USP15 by siRNA technology. Up to now USP15 represents the only DUB known to be associated with the CSN (Zhou et al 2003; Hetfeld et al, 2005). As illustrated in Figure 6, we successfully elucidated a functional role of USP15 in the control of IκBα at which USP15 actively supports the re-accumulation of IκBα post induction. Strikingly, as observed in CSN2-depleted cells (Figure 3), high-molecular-weight forms of phosphorylated IκBα accumulated in USP15-knockdown cells in response to TNFα stimulation, which could be stabilised by pretreatment of the cells with the proteasome inhibitor MG132. Furthermore, kinetics and abundance of the high-molecular-weight IκBα molecules correlated well with the kinetics and abundance of the high-molecular-weight IκBα molecules, immunodetected with a ubiquitin-specific antibody. Interestingly, consistent with USP15 acting as a polyubiquitin depolymerising enzyme, shorter trees of phosphorylated, most likely polyubiquitinylated, IκBα molecules, were observed in mock-transfected cells. In addition, the ability of the CSN-associated USP15 to efficiently deubiquitinylate IκBα could be formally proven in vitro (Figure 6E). Taken together, these data conclusively show the ability of CSN to efficiently deliver deubiquitinylation activity (USP15) towards the CRL substrate IκBα to scavenge it from degradation. This demonstrates a novel role of USP15 in the IκBα/NF-κB regulation. However, the milder phenotype of USP15-knockdown cells compared to cells depleted for CSN might indicate that other as yet not identified DUBs might be associated with the CSN, which could be involved also in the control of the signal-induced turnover of IκBα. This issue needs to be addressed in future studies.
In summary, our results provide first evidence that in addition to its well-established functions in the control upon CRL assembly and activity, the CSN does also control the stability of CRL substrates, which have been targeted to the UPP by ubiquitinylation.
Materials and methods
Expression constructs and antibodies
The expression construct for CSN2 was generated by cloning the PCR-amplified full-length cDNA into the eukaryotic expression vector pcDNA3 (Invitrogen). In addition, a sequence encoding the Flag tag was included in the 5′ end PCR primers. The expression construct for IκBα was generated by cloning the PCR-amplified full-length cDNAs into the eukaryotic pcDNA3 expression vector. In addition, a sequence encoding the His6 tag and the T7 tag has been included in the 5′ end PCR primers for the generation of the construct. CSN and USP15 were purified as described previously (Seeger et al, 1998; Hetfeld et al, 2005).
The following antibodies were used in this study: Flag M2 (Sigma), T7 (Novagen), CSN2 (Abcam), CSN3 (Calbiochem), Cul1 (Abcam), CSN5, HDAC1, p50, p65, ERK, IκBα (Santa Cruz Biotechnology Inc.), CSN7 (Affiniti/Biomol), phospho-p65, phospho-IκBα (Cell Signaling), Nedd8 (Axxora), β-TrCP (Zytomed), USP15 (Abnova) and ubiquitin (Babco/HISS Diagnostics).
Cell culture and transfection
HeLa cells were cultured as described previously (Naumann et al, 1993). They were continously kept in a subconfluent state. Twenty-four hours before transient transfection or stimulation with TNFα, cells were seeded on 10 cm diameter Petri dishes and grown to 60–70% confluency. At least 4 h before stimulation with TNFα (20 ng/ml, Calbiochem), cells were transferred to serum-free medium. One hour before stimulation, MG132 (50 μM, Calbiochem) was added as indicated.
Transient transfections were carried out in serum-free medium using 1–5 μg cDNA per plate and kationic lipids (Effectene, Qiagen) according to the manufacturer's instructions. Six hours post-transfection, fresh medium supplemented with FCS was added and cell culture continued for 20 h.
For transient knockdown of CSN2, cells were seeded 24 h before siRNA transfection on six-well plates or 60-mm-diameter Petri dishes and grown to 50% confluency. Immediately before transfection, cells were washed twice and then resuspended in OptiMEM serum-free medium (Invitrogen). Transfection of siRNA duplexes (50 nM final concentration) with siLentFect Lipid Reagent (Bio-Rad) was performed according to the manufacturer's instructions, using 1 μl siLentFect Lipid Reagent/10pmol siRNA duplex. After 6 h to overnight culture of the cells in the presence of siRNA, the medium was removed and cell culture continued for 48 h. Afterwards, cells were harvested by radioimmunoprecipitation assay (RIPA) lysis or used for cytosolic/nuclear fractionations as described. siRNA oligonucleotide pairs with the following sequences were purchased from Eurogentec: hCSN2siRNA1, 5′-GCACUGAAACAAAUGAUUA-3′ (forward and reverse); hCSN2siRNA2, 5′-UGGAUUUACUGCAGGAA UU-3′ (forward and reverse); hCSN5siRNA, GCUCAGAGUAUCGAUG AAA (forward and reverse); hUSP15siRNA, GCACGUGAUUAUUCCU GUU (forward and reverse). A scrambled nontargeting siRNA was transfected as mock control. Before use, the oligonucleotide pairs were annealed following the protocol suggested by the manufacturer.
Immunoprecipitation
Cells were washed once in ice-cold PBS and then lysed in 400 μl ice-cold RIPA buffer (50 mM Tris (pH 7.5), 150 mM NaCl, 5 mM EDTA, 10 mM K2HPO4, 10% v/v glycerol, 1% v/v Triton X-100, 0.15% SDS, 1 mM Na3VO4, 1 mM sodium molybdate, 20 mM NaF, 10 mM sodium pyrophosphate, 0.1 mM PMSF, 20 mM glycero-2-phosphate and EDTA-free protease inhibitor cocktail (Roche)). Cells were disrupted by passing the cell suspensions through a 27-gauge needle. After clearance of the lysates by centrifugation (2500 g) immunoprecipitations (IPs) of the supernatants were performed.
For IPs of T7IκBα and β-TrCP, cell lysates (400 μl) were diluted (1:2) with IP buffer (20 mM Tris pH 7.4, 150 mM NaCl, 2 mM EDTA, 0,05% v/v Triton X-100, 20 mM NaF, 2 mM Na3VO4 and EDTA-free protease inhibitor cocktail (Roche)) and incubated on a permanent rotator (10 r.p.m.) at 4°C for 3 h to overnight with protein A or protein G sepharose beads (25 μg per reaction) preloaded with the respective antibodies (1–2 μg). Afterwards, the beads were washed four times with 800 μl ice-cold IP buffer. After complete removal of the buffer, 40 μl of 2 × SDS–PAGE sample buffer was added. The samples were incubated for 30 min at 56°C with continous shaking followed by boiling for 5 min. The IP samples were analysed by SDS–PAGE and blotted to PVDF membranes (Millipore). Western blots were developed using the enhanced chemiluminescence (ECL) method (Amersham Biosciences). If required, aliquots of the lysates were also separated by SDS–PAGE and analysed in Western blots. For IPs of IκBα and Nedd8, the antibodies (1–2 μg per reaction) were directly added to the cell lysates. The reactions were rotated overnight at 4°C. Immune complexes were then recovered by addition of 25 μg protein G sepharose beads per reaction.
Preparation of cytosolic and nuclear fractions
Cytosolic and nuclear fractions were prepared as described previously (Naumann and Scheidereit, 1994). Briefly, cells grown on Petri dishes (10 cm diameter) were trypsinised, washed twice in ice-cold PBS and harvested by centrifugation (500 g, 5 min, 4°C). The cell pellets were washed once in 1 ml of buffer A and then resuspended in 0.5 ml of buffer A (10 mM Tris (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 10 mM K2HPO4, 10% glycerol, 0.5 mM DTT, 0.5 mM PMSF, 1 mM NaVO4 and 10 mM NaF) and incubated for 10 min on ice, to allow the cells to swell. Cell lysis was initiated by adding 5 μl of 12.5% NP-40 (Sigma) per vial and allowed to proceed for 5 min on ice under vigorous shaking. Separation of the nuclei from the cytosolic supernatants was then carried out by centrifugation (2000 g, 10 min, 4°C). The nuclear pellets were washed once in buffer A (500 μl), then resuspended in 40 μl buffer C (20 mM Tris (pH 7.9), 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 10 mM K2HPO4, 10% glycerol, 0.5 mM DTT, 0.5 mM PMSF, 1 mM NaVO4 and 10 mM NaF) per sample and incubated for 30 min on ice under vigorous shaking. Finally, cytosolic and nuclear fractions were cleared by centrifugation (13 000 g, 10 min, 4°C).
Electrophoretic mobility shift assay
A gel retardation assay for the detection of NF-κB and Oct-1 was performed with Igκ-oligo and H2B oligo-probes as described previoulsy (Naumann et al, 1993; Naumann and Scheidereit, 1994). The oligonucleotides containing the NF-κB or the Oct-1 recognition sites were labelled using the T4 polynucleotide kinase in the presence of [γ-32P] dATP. The DNA-binding reactions were performed with 20 μl binding buffer (2 μg poly(dI-dC), 1 μg BSA, 5 mM DTT, 20 mM HEPES, pH 8.4, 60 mM KCl and 10% glycerol) for 20 min at 30°C. For competition, the cold Igκ oligonucleotide was used. Supershifts were performed with the indicated antibodies. The reaction products were analysed by electrophoresis in a 5% polyacrylamide gel using 12.5 mM Tris, 12.5 mM boric acid and 0.25 mM EDTA, pH 8.3. The gels were dried and exposed to Amersham TM film (Amersham) at −70°C using an intensifying screen.
In vitro DUB assay
IκBα was immunopreciptated from RIPA lysates of cells stimulated with TNFα in the presence or absence of MG132 as described above. The immunoprecipitates, resuspended to a final volume of 30 μl in DUB assay buffer (30 mM Tris (pH 7.8), 10 mM KCl, 5 mM MgCl2, 5% glycerol, 0.1 mM (for CSN) or 5 mM (for USP15) DTT and 2 mM ATP), were incubated for 4 h at 37°C with continous shaking in the presence of either recombinant USP15 (0.5 μg) or purified CSN (3 μg) and an ATP-regenerating system. Reactions were stopped on ice by the addition of 4 × SDS–PAGE sample buffer and boiling for 4 min. Aliquots were then analysed by SDS–PAGE and Western blotting.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft by a grant to MN (Na 292/7-2). We thank Nicole Wundrack for assistence. The authors declare that they have no financial interest related to this work.
References
- Bech-Otschir D, Kraft R, Huang X, Henklein P, Kapelari B, Pollmann C, Dubiel W (2001) COP9 signalosome-specific phosphorylation targets p53 to degradation by the ubiquitin system. EMBO J 20: 1630–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bech-Otschir D, Seeger M, Dubiel W (2002) The COP9 signalosome: at the interface between signal transduction and ubiquitin-dependent proteolysis. J Cell Sci 115: 467–473 [DOI] [PubMed] [Google Scholar]
- Cope GA, Suh GS, Aravind L, Schwarz SE, Zipursky SL, Koonin EV, Deshaies RJ (2002) Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science 298: 608–611 [DOI] [PubMed] [Google Scholar]
- Cope GA, Deshaies RJ (2003) COP9 signalosome: a multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell 114: 663–671 [DOI] [PubMed] [Google Scholar]
- Cope GA, Deshaies RJ (2006) Targeted silencing of Jab1/Csn5 in human cells downregulates SCF activity through reduction of F-box protein levels. BMC Biochem 7: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denti S, Fernandez-Sanchez ME, Rogge L, Bianchi E (2006) The COP9 signalosome regulates Skp2 levels and proliferation of human cells. J Biol Chem 281: 32188–32196 [DOI] [PubMed] [Google Scholar]
- Goldenberg SJ, Cascio TC, Shumway SD, Garbutt KC, Liu J, Xiong Y, Zheng N (2004) Structure of the Cand1–Cul1–Roc1 complex reveals regulatory mechanisms for the assembly of the multisubunit cullin-dependent ubiquitin ligase. Cell 119: 517–528 [DOI] [PubMed] [Google Scholar]
- Groisman R, Polanowska J, Kuraoka I, Sawada JL, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y (2003) The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in reponse to DNA damage. Cell 113: 357–367 [DOI] [PubMed] [Google Scholar]
- He Q, Cheng P, He Q, Liu Y (2005) The COP9 signalosome regulates the Neurospora circadian clock by controlling the stability of the SCFFWD-1 complex. Genes Dev 19: 1518–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetfeld BKJ, Helfrich A, Kapelari B, Scheel H, Hofmann K, Guterman M, Schade R, Kloetzel PM, Dubiel W (2005) The zinc finger of the CSN-associated deubiquitinating enzyme USP15 is essential to rescue the E3 ligase Rbx1. Curr Biol 15: 1217–1221 [DOI] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y (2000) Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol 18: 621–663 [DOI] [PubMed] [Google Scholar]
- Liu J, Furukawa M, Matsumoto T, Xiong Y (2002) Nedd8 modification of Cul1 dissociates p120CAND1, an inhibitor of Cul1-Skp1 binding and SCF ligases. Mol Cell 10: 1511–1518 [DOI] [PubMed] [Google Scholar]
- Liu C, Powell KA, Mundt K, Wu L, Carr AM, Caspari T (2003) Cop9/signalosome subunits and Pcu4 regulate ribonucleotide reductase by both checkpoint-dependent and -independent mechanisms. Genes Dev 17: 1130–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyapina S, Cope GA, Shevchenko A, Serino G, Tsuge T, Zhou C, Wolf DA, Wei N, Deshaies RJ (2001) Promotion of Nedd8–Cul1 conjugate cleavage by COP9 signalosome. Science 292: 1382–1385 [DOI] [PubMed] [Google Scholar]
- Naumann M, Wulczyn FG, Scheidereit C (1993) The NF-κB precursor p105 and the proto-oncogene product Bcl-3 are IκB molecules and control nuclear translocation of NF-κB. EMBO J 12: 213–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann M, Bech-Otschir D, Huang X, Ferrell K, Dubiel W (1999) COP9 signalosome-directed c-Jun activation/stabilization is independent of JNK. J Biol Chem 274: 35297–35300 [DOI] [PubMed] [Google Scholar]
- Naumann M, Scheidereit C (1994) Activation of NF-κB in vivo is regulated by multiple phosphorylations. EMBO J 13: 4597–4607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen O (2003) COP9 signalosome: a provider of DNA building blocks. Curr Biol 13: R565–R567 [DOI] [PubMed] [Google Scholar]
- Pan ZQ, Kentsis A, Dias DC, Yamoah K, Wu K (2004) Nedd8 on cullin: building an expressway to protein destruction. Oncogene 23: 1985–1997 [DOI] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ (2005) Function and regulation of cullin-RING ubiquitin-ligases. Nat Rev Mol Cell Biol 6: 9–20 [DOI] [PubMed] [Google Scholar]
- Pintard L, Kurz T, Glaser S, Willis JH, Peter M, Bowerman B (2003) Neddylation and deneddylation of Cul-3 is required to target MEI-1/katanin for degradation at the meiosis-to-mitosis transition in C. elegans. Curr Biol 13: 911–921 [DOI] [PubMed] [Google Scholar]
- Place RF, Haspeslagh D, Hubbard AK, Giardina C (2001) Cytokine-induced stabilization of newly synthesized IκBα. Biochem Biophys Res Commun 283: 813–820 [DOI] [PubMed] [Google Scholar]
- Schwechheimer C, Deng XW (2001) COP9 signalosome revisited: a novel mediator of protein degradation. Trends Cell Biol 11: 420–426 [DOI] [PubMed] [Google Scholar]
- Schwechheimer C, Serino G, Callis J, Crosby WL, Lyapina S, Deshaies RJ, Gray WM, Estelle M, Deng XW (2001) Interactions of the COP9 signalosome with the E3 ubiquitin ligase SCFTIR1 in mediating auxin response. Science 292: 1379–1381 [DOI] [PubMed] [Google Scholar]
- Seeger M, Kraft R, Ferrell K, Bech-Otschir D, Dumdey R, Schade R, Gordon C, Naumann M, Dubiel W (1998) A novel protein complex involved in signal transduction possessing similarities to 26S proteasome subunits. FASEB J 12: 469–478 [PubMed] [Google Scholar]
- Sun SC, Ganchi PA, Ballard DW, Greene WC (1993) NF-κB controls expression of inhibitor IκBα: evidence for an inducible autoregulatory pathway. Science 259: 1912–1915 [DOI] [PubMed] [Google Scholar]
- Tomoda K, Kubota Y, Kato JY (1999) Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature 398: 160–165 [DOI] [PubMed] [Google Scholar]
- Wee S, Geyer RK, Toda T, Wolf DA (2005) CSN facilitates cullin-RING ubiquitin ligase function by counteracting autocatalytic adapter instability. Nat Cell Biol 7: 387–391 [DOI] [PubMed] [Google Scholar]
- Wolf DA, Zhou C, Wee S (2003) The COP9 signalosome: an assembly and maintenance platform for cullin ubiquitin ligases. Nat Cell Biol 5: 1029–1033 [DOI] [PubMed] [Google Scholar]
- Wu JT, Lin HC, Hu YC, Chien CT (2005) Neddylation and deneddylation regulate Cul1 and Cul3 protein accumulation. Nat Cell Biol 7: 1014–1020 [DOI] [PubMed] [Google Scholar]
- Zheng J, Yang X, Harrell JM, Ryzhikov S, Shim EH, Lykke-Andersen K, Wei N, Sun H, Kobayashi R, Zhang H (2002) CAND1 binds to unneddylated Cul1 and regulates the formation of SCF ubiquitin E3 ligase complex. Mol Cell 10: 1519–1526 [DOI] [PubMed] [Google Scholar]
- Zhou C, Seibert V, Geyer R, Rhee E, Lyapina S, Cope G, Deshaies RJ, Wolf DA (2001) The fission yeast COP9/signalosome is involved in cullin modification by ubiquitin-related Ned8p. BMC Biochem 2: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Wee S, Rhee E, Naumann M, Dubiel W, Wolf DA (2003) Fission yeast COP9/signalosome suppresses cullin activity through recruitment of the deubiquitylating enzyme Ubp12p. Mol Cell 11: 927–938 [DOI] [PubMed] [Google Scholar]