

Abstract
Translation initiation factor eIF1A stimulates preinitiation complex (PIC) assembly and scanning, but the molecular mechanisms of its functions are not understood. We show that the F131A,F133A mutation in the C-terminal tail (CTT) of eIF1A impairs recruitment of the eIF2-GTP-Met-tRNAiMet ternary complex to 40S subunits, eliminating functional coupling with eIF1. Mutating residues 17–21 in the N-terminal tail (NTT) of eIF1A also reduces PIC assembly, but in a manner rescued by eIF1. Interestingly, the 131,133 CTT mutation enhances initiation at UUG codons (Sui− phenotype) and decreases leaky scanning at AUG, while the NTT mutation 17–21 suppresses the Sui− phenotypes of eIF5 and eIF2β mutations and increases leaky scanning. These findings and the opposite effects of the mutations on eIF1A binding to reconstituted PICs suggest that the NTT mutations promote an open, scanning-conducive conformation of the PIC, whereas the CTT mutations 131,133 have the reverse effect. We conclude that tight binding of eIF1A to the PIC is an important determinant of AUG selection and is modulated in opposite directions by residues in the NTT and CTT of eIF1A.
Keywords: eIF1A, GCN4 , initiation, Saccharomyces , translation
Introduction
Decoding the AUG start codon in mRNA by methionyl-initiator tRNA (Met-tRNAiMet) and the ribosome is stimulated by an array of eukaryotic translation initiation factors (eIFs). The Met-tRNAiMet is delivered to the 40S subunit in a ternary complex (TC) with the GTP-bound form of eIF2, forming the 43S PIC, in a process stimulated by eIF1, eIF1A, eIF3, and eIF5 (Hershey and Merrick, 2000; Hinnebusch, 2000; Asano et al, 2001b; Algire et al, 2002; Majumdar et al, 2003; Kolupaeva et al, 2005). In budding yeast, a web of interactions links eIFs 1, 3, 5, and the TC in a multifactor complex (MFC) whose formation promotes PIC assembly in vivo (Asano et al, 2001a; Valášek et al, 2002; Valasek et al, 2004; Singh et al, 2004; Jivotovskaya et al, 2006). The 40S binding of eIF1 also is stimulated by eIF1A (Maag and Lorsch, 2003; Majumdar et al, 2003) and the C -termini of these two factors are in close proximity in the 43S PIC (Maag et al, 2005), with eIF1A likely occupying the ‘A' site (Battiste et al, 2000) and eIF1 bound near the ‘P' site on the 40S subunit (Lomakin et al, 2003), the site of decoding during initiation.
The 43S PIC is recruited to the 5′ end of the mRNA and the AUG codon is selected as the PIC scans the leader, with the anticodon of Met-tRNAiMet inspecting successive triplets in the P-site (Hershey and Merrick, 2000). In a reconstituted mammalian system, eIF1 and eIF1A promote scanning and formation of a 48S initiation complex (IC) at the start codon (Pestova et al, 1998), and the absence of eIF1 allows selection of non-AUG triplets (Pestova and Kolupaeva, 2002). This fits with genetic studies in yeast that identified mutations in eIF1 (encoded by SUI1) that increase initiation at a UUG at the 5′ end of the HIS4 gene, suppressing mutations in the AUG codon (Sui− phenotype) (Donahue, 2000). It is thought that eIF1 promotes an open conformation of the PIC conducive to scanning and restricts base pairing of Met-tRNAiMet with non-AUGs in the P-site (Lomakin et al, 2003).
Our analysis of a reconstituted yeast system revealed that GTP bound to eIF2 is partially hydrolyzed in 48S PICs, dependent on eIF5, but the Pi is not released from eIF2-GDP-Pi when a non-AUG triplet occupies the P-site. Base pairing of Met-tRNAiMet with AUG triggers a conformational change wherein eIF1 and eIF1A move apart and eIF1 is released from its 40S binding site in the initiation complex. It appears that dissociation of eIF1 permits release of Pi from eIF2-GDP-Pi to finalize selection of the start codon (Algire et al, 2005; Maag et al, 2005). By contrast, AUG recognition elicits tighter binding of eIF1A to the complex, dependent on eIF5. A dominant Sui− mutation in eIF5 (SUI5) strengthens eIF1A binding with UUG in the P-site, correlating with increased initiation at UUG codons in SUI5 cells in vivo. A similar finding was made for an eIF1A mutant lacking the ∼50-residue unstructured CTT of eIF1A (tif11-ΔC, or just ΔC), which also confers a Sui− phenotype. It was proposed that the eIF1A CTT promotes formation of an open, scanning-conducive conformation of the PIC, and on AUG recognition, eIF5 antagonizes the eIF1A CTT, strengthening eIF1A's interactions and promoting a more closed conformation (Maag et al, 2006).
Translation of GCN4 mRNA is a sensitive indicator of defects in TC recruitment in vivo. GCN4 is repressed in amino acid-replete cells by upstream open reading frames (uORFs) in the 5′UTR. After translating uORF1, 40S subunits resume scanning and reinitiate translation at uORFs 2, 3 or 4, after which they dissociate from the mRNA and leave the GCN4 ORF untranslated. GCN4 translation is derepressed in amino acid-starved cells by phosphorylation of the α-subunit of eIF2 by kinase GCN2, which inhibits recycling of eIF2-GDP to eIF2-GTP. The ensuing reduction in TC concentration allows 40S subunits that translate uORF1 and resume scanning to bypass uORFs 2–4 and reinitiate downstream at GCN4. In nonstarved wild-type (WT) or gcn2Δ cells, by contrast, the high level of TC ensures that all 40S subunits scanning downstream from uORF1 quickly rebind TC, reinitiate at uORFs 2, 3 or 4, and dissociate from the mRNA without translating GCN4 (Hinnebusch, 2005). We found previously that the ΔC truncation of the eIF1A CTT conferred constitutive derepression of GCN4 translation in gcn2Δ cells. This Gcd− phenotype suggests that ΔC reduces the rate of TC binding to 40S subunits scanning downstream from uORF1, allowing a fraction to bypass uORFs 2–4 and reinitiate at GCN4 (Olsen et al, 2003). Supporting this interpretation, ΔC reduces TC binding to 40S subunits in both native and reconstituted PICs (Fekete et al, 2005).
In this report, we provide new evidence supporting the dual role of the eIF1A CTT in TC recruitment and AUG selection, and pinpoint specific residues, Phe131 and Phe133, involved in both functions. We show that residues in the unstructured eIF1A NTT also affect PIC assembly in vivo, but through a distinct mechanism. Remarkably, mutations in the eIF1A NTT disrupt AUG selection in a manner opposite to that of the ΔC and SUI5 mutations, exhibiting a hyperaccurate phenotype. A concordance of genetic and biochemical data indicates that interaction of eIF1A with the initiation complex is a determinant of start codon selection that is modulated by the unstructured tails of eIF1A.
Results
Mutation of F131 and F133 in the CTT impairs PIC assembly
Previously, we showed that the ΔC truncation of eIF1A residues 108–153 confers a Gcd− phenotype and reduces native 43S PIC levels in vivo. Ala substitutions of CTT residues 128–132 (128DVNFE132) and 133–137 (133FGNAD137) also produce Gcd− phenotypes, as does the mutation F131A, F133A (henceforth 131,133) of the two Phe residues in these intervals. As substitutions of surrounding CTT residues 118–122, 123–127, 138–142, 143–148, and 149–153 did not confer Gcd− phenotypes (Fekete et al, 2005), it appeared that F131 and F133 play key roles in TC recruitment; hence, we set out to characterize the effects of the 131,133 mutation on 43S PIC assembly.
This and other eIF1A mutations described below were made in the TIF11-FL allele (tagged at the 5′ end with FLAG epitope) and introduced into a tif11Δ gcn2Δ strain by plasmid shuffling (Fekete et al, 2005). The 131,133 mutation produces a slow-growth (Slg−) phenotype on complete (SC) medium (Figure 1A) and decreased polysome content (Figure 1B), without reducing eIF1A expression (Fekete et al, 2005). The 131,133 mutation also restores growth on medium containing the inhibitor of histidine biosynthesis 3-aminotriazole (3AT), and this 3AT-resistant (3ATR) phenotype was diminished by a high-copy (hc) plasmid encoding the three subunits of eIF2 and tRNAiMet, comprising the TC (hc-TC) (Figure 1A). Gcd− mutations that derepress GCN4 translation independent of eIF2α phosphorylation by GCN2 derepress histidine biosynthetic genes under GCN4 control and overcome the 3-AT-sensitive (3-ATS) phenotype of gcn2Δ cells. Suppression of the 3ATR/Gcd− phenotype of the 131,133 mutant by hc-TC suggests that it results, at least partly, from a reduced rate of TC loading on 40S subunits scanning downstream from uORF1, allowing a fraction to bypass uORFs 2–4 and reinitiate at GCN4. Increasing the TC concentration with hc-TC is thought to boost the rate of TC loading by mass action.
Figure 1.
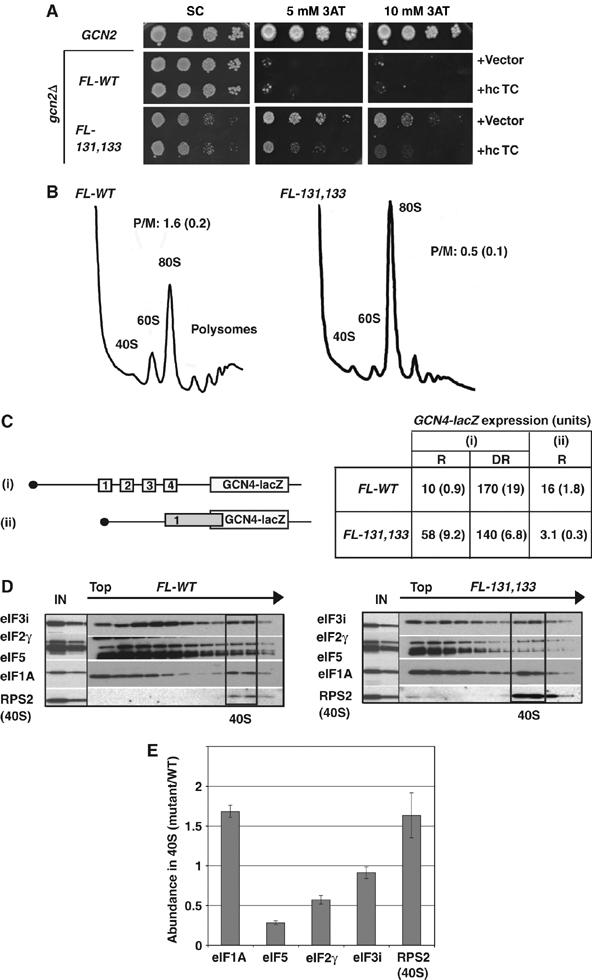
eIF1A CTT mutation 131,133 impairs GCN4 translational control, translation initiation and PIC assembly in vivo. (A) Dilutions of gcn2Δ strains containing the FLAG-tagged wild-type (FL-WT) (strain H2999) or 131,133 mutant (FL-131,133) allele of TIF11 (strain CFY615), plus empty vector or p1780-IMT (hc TC), together with an isogenic GCN2 strain, were grown at 30°C on SC lacking uracil and leucine (SC-UL) for 3 days and SC-UL lacking histidine (SC-ULH) and containing 3-AT for 7 days. (B) Polysome profiles of H2999 and CFY615 cultured in SC-L and crosslinked with HCHO. WCEs were resolved by sedimentation through sucrose gradients and the gradients scanned at A254. Polysome/monosome ratios (P/M, mean±s.e., n=3) are indicated. (C) GCN2+ strains H3583 (FL-WT) and CFY217 (FL-131,133) harboring GCN4-lacZ reporter plasmids p180 or pM226, depicted in (i) and (ii), respectively, were grown in repressing (R) medium (SC-U) or derepressing (DR) medium (SC-U lacking Ile and Val and containing 0.5 μg/ml sulfometuron (SM) to inhibit Ile/Val biosynthesis) for 6 h. Units of β-galactosidase activity (nmol of o-nitrophenyl-β-D-galactopyranoside cleaved per minper mg) were assayed in WCEs of three replicate cultures and reported as means±s.e. (n=6). (D) Native PICs were measured in the strains described in (A) after crosslinking cells with HCHO and resolving WCEs by sedimentation through sucrose gradients. Fractions were subjected to Western analysis using the indicated antibodies, analyzing 1 and 0.2% aliquots of each input WCE (IN) in parallel. Fractions containing free 40S subunits are boxed. (E) Initiation factor binding to 40S subunits in three replicates of the experiment in (D) was quantified by calculating the ratios of the 40S signals in the mutant relative to the WT. Results are means±s.e. (n=3).
In a GCN2+ strain, the 131,133 mutation led to ∼six-fold derepression of a GCN4-lacZ reporter containing all four uORFs under non-starvation conditions compared with the ∼15-fold induction of this reporter on starvation of the TIF11+ strain for isoleucine and valine (Figure 1C). This indicates that 131,133 confers partial derepression of GCN4 translation, confirming its Gcd− phenotype.
Biochemical evidence that the 131,133 mutation impairs TC recruitment came first from our finding that it reduces eIF2 binding to 40S subunits in native PICs stabilized by formaldehyde crosslinking of living cells. The 40S binding of eIF5 was also reduced, whereas the mutant eIF1A protein showed higher than WT levels in the 40S fractions commensurate with the increased level of 40S protein RPS2 (Figure 1D–E). (As previously observed (Valasek et al, 2004; Fekete et al, 2005), the 40S binding defect of eIF2 and eIF5 in the mutant is associated with increased degradation of these factors during centrifugation.) Considering that the amount of free 40S subunits was elevated in the mutant, the approximately equal amounts of eIF3 in the mutant and WT 40S fractions suggest that eIF3 recruitment also is reduced by 131,133 (Figure 1D–E). Thus, 131,133 impairs 40S binding of three different MFC components in vivo. (No reduction in 40S-bound eIF1 was apparent; however, eIF1 expression is elevated in this mutant (Supplementary Figure S1), making it difficult to assess the efficiency of eIF1 binding.)
We further demonstrated that 131,133 impairs the rate of TC loading on 40S subunits in vitro in reactions containing eIF1 and mRNA. Interestingly, the 131,133 mutation reduced the rate of TC binding in the presence of eIF1 by >six-fold, but had no effect in the absence of eIF1 (Figure 2A–B), eliminating the stimulatory effect of eIF1 on TC loading (Figure 2B). The 131,133 mutation also reduces the rate of TC loading ∼four-fold in reactions lacking mRNA (0.009±0.003 versus 0.042±0.012 min−1; data not shown). To determine whether the defect in TC loading could arise from defective 40S binding of the 131,133 protein, we measured the Kd of eIF1A for 40S subunits in the presence and absence of eIF1 (Figure 2C–E), and new in the presence of both eIF1 and TC (Figure 2E). The 131,133 mutation does not weaken interaction of eIF1A with 40S subunits under any of these circumstances. Hence, 131,133 impairs the function of eIF1A bound to 40S subunits in accelerating TC loading.
Figure 2.
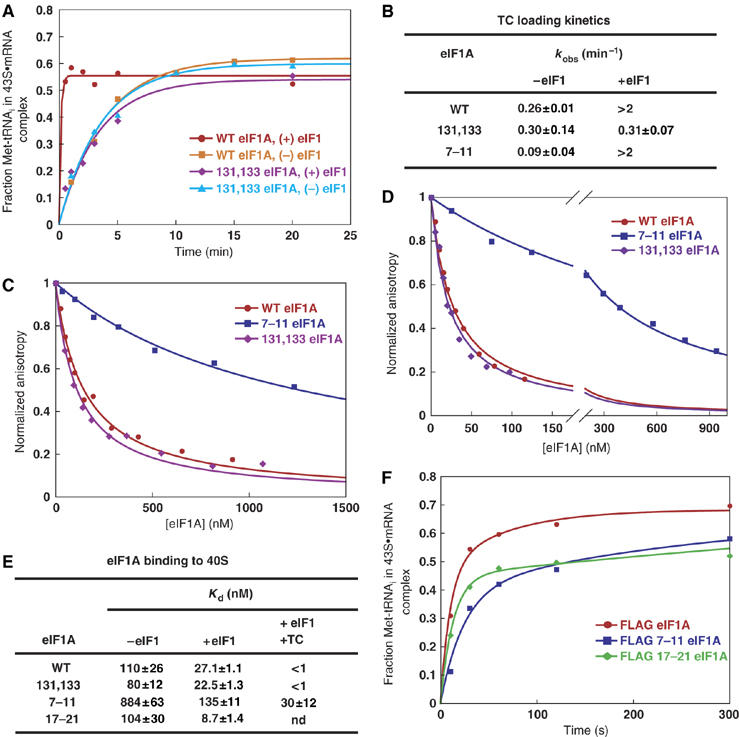
Effects of eIF1A mutations on kinetics of TC loading and 40S binding of eIF1A in vitro. (A) Preformed TC containing [35S]-Met-tRNAiMet was incubated with 40S subunits, eIF1, a model mRNA, and mutant or WT eIF1A, and the fraction of labeled [35S]-Met-tRNAiMet bound to 40S subunits was measured using a native gel assay. eIF1A was saturating (1 μM) in all cases. (B) Rate constants of 43S·mRNA complex formation measured as in (A). (C) Tetramethylrhodamine (TAMRA)-labeled WT eIF1A was prebound to 40S subunits and unlabeled mutant or WT eIF1A was added to compete with binding of labeled eIF1A. The decrease in fluorescence anisotropy of the labeled WT protein as a function of the concentration of unlabeled proteins yielded their Kd values. (D) Experiments as in (C) but with saturating eIF1 (1 μM). (E) Kd values for eIF1A binding to 40S subunits calculated from data in (C) and (D) in columns 2 and 3 and from experiments done with saturating preformed TC (0.9 μM) and eIF1 (1 μM) in column 4. (F) Mutations 7–11 and 17–21 reduce the rate of 43S·mRNA formation, measured in the presence of saturating eIF1, as in (A), when combined with the N-terminal FLAG tag. The 7–11 mutation reduces the rate constant to 1.3 min−1 from 3.5 min−1 for WT FL-eIF1A. Except in (F), eIF1A proteins lacked the FL tag.
Mutations in the eIF1A NTT impair PIC assembly in a manner rescued by eIF1
To address the function of the NTT, we analyzed the phenotypes of Ala substitutions made in consecutive blocks of five amino acids of the TIF11-FL allele. Mutations of residues 7–11 and 12–16 were lethal and the 17–21 substitution conferred a Slg− phenotype, all without lowering the level of FL-eIF1A (Fekete et al, 2005). Interestingly, these phenotypes were suppressed by overexpressing WT eIF1 from a hc SUI1 plasmid, whereas the Slg− phenotype of the CTT mutation 131,133 was exacerbated by hc SUI1 (Figure 3A–B). The diminished polysome content of the 17–21 mutant was also partially suppressed by hc SUI1 (Figure 3C), and 17–21 lowered the levels of eIF1, eIF5, eIF2, and eIF3 in the 40S fractions in a manner rescued by eIF1 overexpression (Figure 3D–E). The 40S binding of eIF1A itself was not diminished by the 17–21 mutation, and eIF1 overexpression led to a higher than WT level of 40S-bound 17–21 protein (Figure 3E). Thus, the FL-17–21 mutation impairs PIC assembly by 40S-bound eIF1A in a manner rescued by increasing the eIF1 level in the cell.
Figure 3.
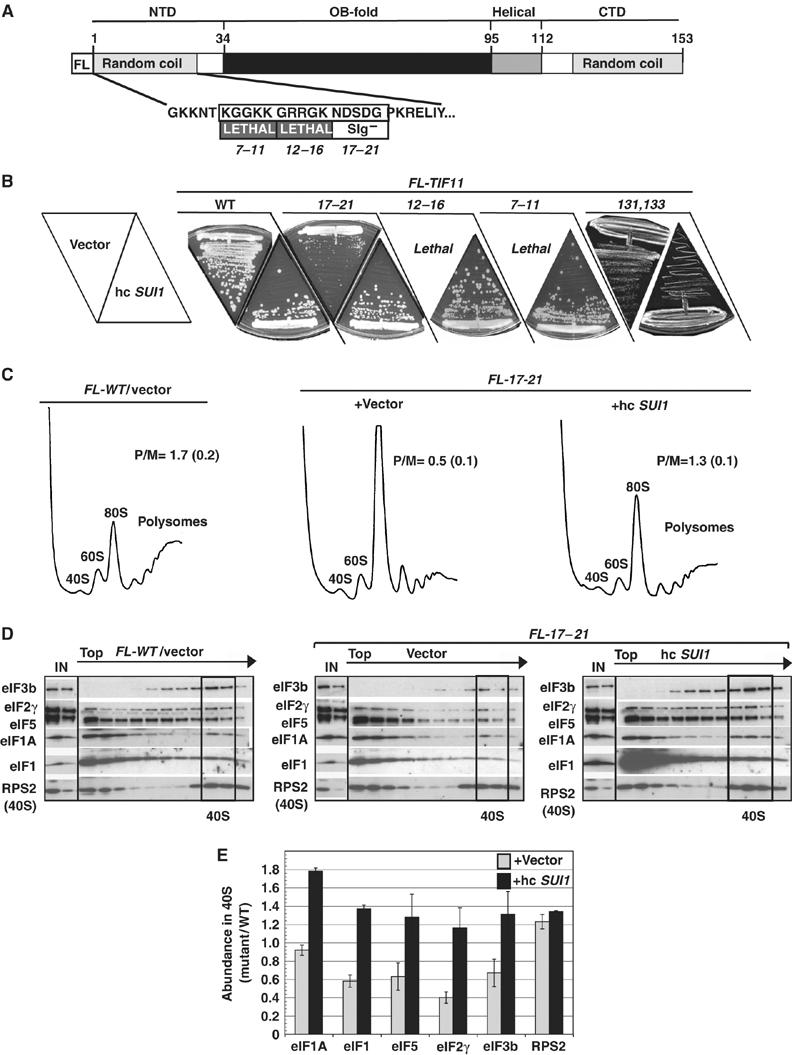
eIF1A NTT mutations reduce translation initiation and PIC levels in a manner suppressed by eIF1 overexpression. (A) Schematic of N-terminally FLAG-tagged eIF1A showing alanine-substituted NTT residues and corresponding growth phenotypes of three mutants. (B) gcn2Δ strains with the indicated FL-TIF11 alleles and harboring hc SUI1 plasmid YEp-SUI1-U (lower sectors) or empty vector (upper sectors) were grown on SC-UL medium at 30°C. (C) Polysome profiles of strains from (B) harboring FL-WT (H2999) or FL-17–21 (CFY177) alleles of TIF11 and empty vector or hc SUI1, measured as described in Figure 1B. (D) Native PICs in strains described in (C) measured as described in Figure 1D. (E) eIF binding to 40S subunits in three replicates of the experiment in (D) quantified by calculating the ratio of the 40S signal in the FL-17–21 mutant, harboring empty vector or hc SUI1 plasmid, relative to the WT strain. Results are means±s.e. (n=3).
The FL tag contributes to the phenotypes of the NTT mutations. In untagged TIF11 or TIF11-HA (with a C-terminal HA3 tag instead), the 17–21 mutation has no effect on growth; and 7–11 produces Slg−, rather than lethality (that is not suppressed by hc SUI1) and does not reduce 40S binding of MFC components (data not shown). Thus, it appears that the NTT mutations strongly impair PIC assembly (in a manner rescued by eIF1) only when combined with the FL tag.
This last conclusion is consistent with results from in vitro analysis. Untagged 7–11 protein shows defects in 40S binding in the presence or absence of eIF1 and TC (Figure 2C–E). It also reduces the rate of TC loading in the absence of eIF1 when the mutant is supplied at a concentration high enough to compensate for its 40S binding defect (Figure 2B); however, there is no detectable defect in TC loading in the presence of eIF1 under these conditions (Figure 2B). Attaching FL to WT eIF1A reduces the rate of TC loading in the presence of eIF1 by ∼30% (data not shown), and introducing the 17–21 or 7–11 mutations into FL-eIF1A reduces the rate further, with the 7–11 mutation having the greater effect (Figure 2F). eIF1 strongly stimulates the reaction rates for both of these eIF1A NTT mutants (data not shown).
These in vitro results coincide with our in vivo findings that combining FL with the 7–11 and 17–21 mutations produces additive defects in PIC assembly and growth that are suppressed by eIF1 overexpression. In addition to increasing the rate of TC loading, eIF1 overexpression also enhances 40S binding of the mutant eIF1As (Figures 2E and 3E) and other MFC components (Figure 3E), which probably contributes to suppression of the translation initiation defects in NTT mutants by excess eIF1 (Figure 3B and C). The residual Slg− phenotype of untagged 7–11 (which is not rescued by eIF1 overexpression) implies that this NTT mutation also disrupts a step downstream of PIC assembly, which fits with results below indicating that it impairs AUG recognition.
The 131,133 CTT mutation increases initiation at UUG codons in vivo
We have shown previously that the ΔC mutation in eIF1A confers increased initiation at a UUG triplet in HIS4, reducing the histidine auxotrophy (His− phenotype) produced by the absence of the AUG start codon in the his4-303 allele (Sui− phenotype) (Fekete et al, 2005). We found that the 131,133 mutation also perturbs CTT function in start codon selection. First, 131,133 confers a marked increase in the expression of a HIS4-lacZ reporter containing a UUG start codon relative to that produced by a matched reporter containing an AUG (Figure 4A). Although we did not observe a His+ phenotype in 131,133 cells harboring his4-303, this might be explained by the strong Slg− phenotype of this mutant. Supporting this interpretation, Western analysis of WT and 131,133 cells harboring an myc-tagged version of chromosomal his4-303 revealed a ∼nine-fold increase in the expression of myc-tagged his4-303 protein, with little change in his4-303-myc mRNA in the mutant cells (Figure 4B).
Figure 4.
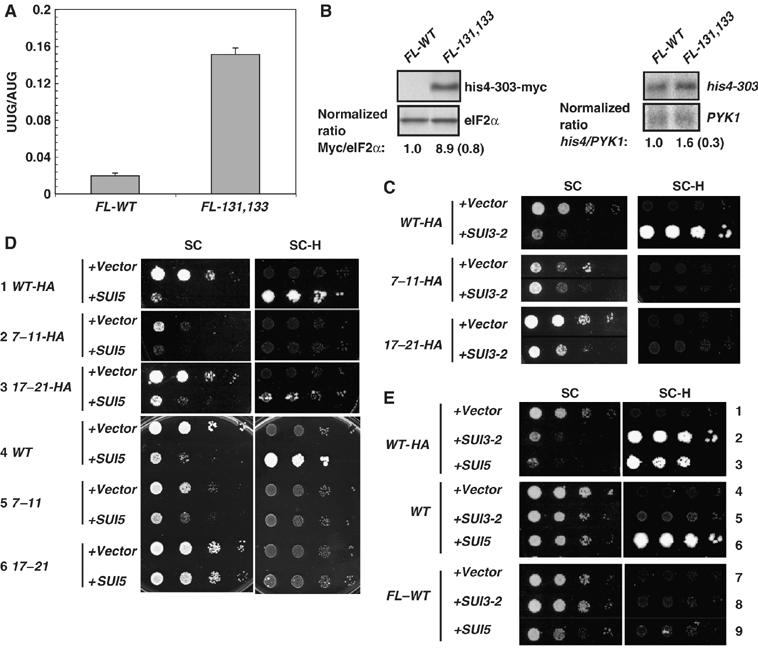
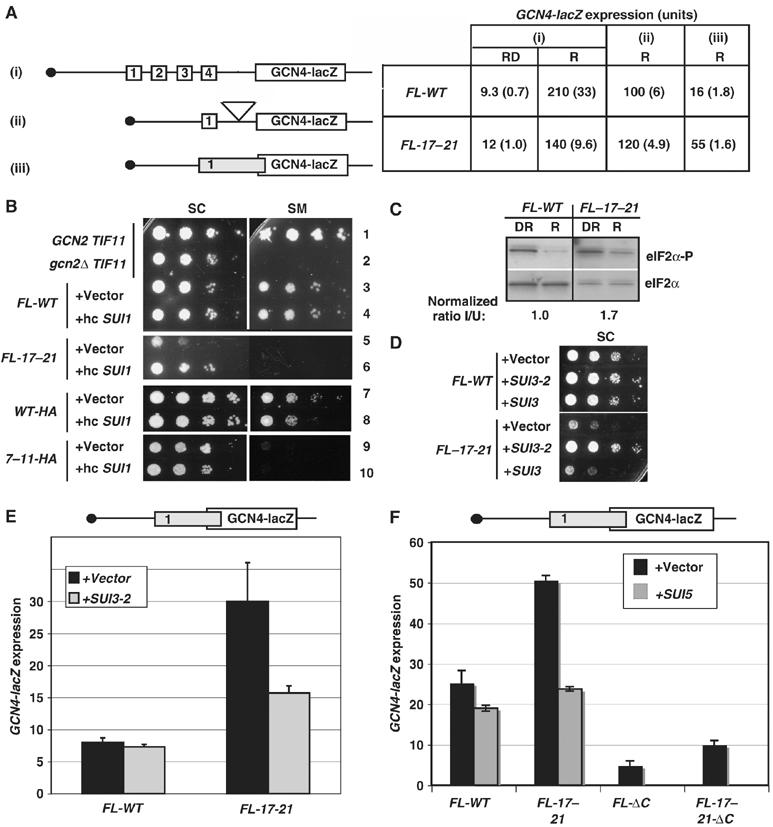
Opposite effects of eIF1A CTT and NTT mutations on initiation at UUG codons in vivo. (A) his4-303 strains containing the FL-WT (H3583) or FL-131,133 (CFY217) TIF11 alleles and harboring HIS4-lacZ reporter plasmids with an AUG (p367) or UUG (p391) start codon grown in SC-U and assayed for β-galactosidase activity. Specific activities for three transformants were used to calculate the mean±s.e. ratios (n=3) for the UUG versus AUG reporter. (B) Cultures of his4-303-myc strains harboring FL-WT (CFY636) or FL-131,133 (CFY798) alleles of TIF11 were grown in SC-H. (Left panel) Western analysis of WCEs with antibodies against Myc epitope or eIF2α. Normalized ratios of Myc:eIF2α signals from three experiments are given as means±s.e. (n=3). Right panel: Northern analysis of HIS4 and PYK1 mRNAs. Normalized ratios of the his4-303:PYK1 mRNA signals from three independent experiments are given as means±s.e. (n=3). (C) TIF11 NTT mutations suppress the Slg− and Sui− phenotypes of SUI3-2. Serial dilutions of his4-303 strains containing HA-tagged wild-type (WT-HA) (CFY840), 7–11-HA (CFY842), or 17–21-HA (CFY844) TIF11 alleles harboring vector or SUI3-2 plasmid pRSSUI3-S264Y-U were grown at 30°C on SC-U for 2 days and SC-UH for 7 days. (D) Growth on SC-U (2 days) and SC-UH (10 days) of his4-303 strains containing WT-HA, 7–11-HA, 17–21-HA, or untagged wild-type (WT), 7–11 or 17–21 alleles of TIF11, harboring vector or SUI5 plasmid YCpTIF5-G31R-U. (E) Growth on SC-U (2 days) and SC-UH (14 days) of HA-tagged (WT-HA), untagged (WT), or FLAG-tagged (FL-WT) wild-type alleles of TIF11 carrying vector, SUI3-2 plasmid pRSSUI3–264Y-U or SUI5 plasmid YCpTIF5-G31R-U.
We also observed a decrease in leaky scanning of AUG start codons in the 131,133 mutant using a GCN4-lacZ reporter containing only an elongated version of uORF1 that overlaps the GCN4 ORF. In WT cells, expression of this construct is very low because most ribosomes translate the elongated uORF1 and fail to reinitiate upstream at GCN4 (Figure 1C, WT, construct ii). Interestingly, 131,133 conferred five-fold lower expression of this construct, suggesting diminished leaky scanning past the uORF1 AUG codon (Figure 1C, 131,133, ii). It could be argued instead that 131,133 allows increased initiation at UUGs upstream of uORF1, but because translation of uORF1 is required for derepression, this should equally impair derepression of the WT GCN4-lacZ construct with all four uORFs, which was not observed (Figure 1C, 131,133, i). A third indication that 131,133 alters AUG selection is that the mutation is synthetically lethal with the Sui− SUI5 allele (data not shown), as observed previously on combining the tif11-ΔC mutation with SUI5 (Fekete et al, 2005). We suggested previously that this synthetic lethality results from an additive effect of the mutations in elevating UUG selection, which reaches an intolerable level in the double mutant.
The eIF1A NTT mutations suppress utilization of UUG start codons in Sui− mutants
Remarkably, the eIF1A NTT mutations affect initiation at UUG triplets in a manner opposite to that of Sui− CTT mutations, suppressing the Sui− phenotypes conferred by SUI5 and the eIF2β mutation SUI3-2 (SUI3-S264Y). As expected (Huang et al, 1997), introducing plasmid-borne SUI3-2 into the TIF11-HA his4-303 strain confers a dominant His+/Sui− phenotype and impairs growth on SC medium (Figure 4C, top two rows). The His+ phenotype of SUI3-2 was eliminated in strains containing the 7–11 or 17–21 mutations even though they grew better on SC medium than the corresponding WT TIF11-HA strain with SUI3-2 (Figure 4C). Similarly, 17–21 and 7–11 suppressed the dominant His+/Sui− phenotype of SUI5 in TIF11-HA strains (Figure 4D, strains 1–3), and also when present in untagged TIF11 (strains 4–6). The untagged 17–21 mutation also suppressed the Slg− phenotype of SUI5 (Figure 4D, strains 4 and 6). Hence, the NTT mutations have Ssu− (Suppressor of Sui−) phenotypes indicating reduced initiation at UUG codons in Sui− mutants.
Interestingly, attaching the FL tag to the N-terminus of eIF1A confers an Ssu− phenotype on its own, suppressing the strong Sui− phenotype of SUI5 (Figure 4E, rows 6 and 9), whereas the C-HA3 tag exacerbates the Sui− and Slg− phenotypes of SUI3-2 (Figure 4E, rows 2 and 5) and the Slg− phenotype of SUI5 (Figure 4E, rows 3 and 6). These opposite effects of the C-terminal and N-terminal tags reinforce the notion that altering the eIF1A CTT favors, whereas altering the NTT decreases, UUG selection in Sui− mutants.
The FL-17–21 NTT mutation also affects leaky scanning in a manner opposite to that of CTT mutation 131,133, producing 3.4-fold higher expression of the GCN4-lacZ reporter with elongated uORF1 (Figure 5A, iii). Leaky scanning of uORF1 in a construct containing all four WT uORFs should impair induction of GCN4 because reinitiation after translation of uORF1 is required to bypass uORFs 3–4. In accordance with this possibility, FL-17–21 decreases induction of the WT GCN4-lacZ reporter, reducing the induction ratio from 23 (observed in WT) to 12 (Figure 5A, i). Consistently, FL-17–21 confers sensitivity to SM (SMS) (Figure 5B, cf. rows 3 and 5), indicating a Gcn− phenotype, as also observed for gcn2Δ (Figure 5B, rows 1–2). Unlike gcn2Δ, however, the FL-17–21 mutation does not decrease eIF2α phosphorylation in SM-induced cells (Figure 5C). The impaired derepression of GCN4 cannot be attributed to reduced reinitiation following uORF1 translation because expression of a construct containing WT uORF1 as the sole uORF was unaffected by the FL-17–21 mutation (Figure 5A, ii). The 7–11 mutation in the TIF11-HA background (where it is viable) also confers a strong SMS phenotype (Figure 5B, rows 7 and 9). These data suggest that eIF1A NTT mutations impair GCN4 translational control, at least partly, by allowing a fraction of PICs scanning from the cap to bypass the uORF1 AUG.
Figure 5.
eIF1A NTT mutation FL-17–21 confers leaky scanning and slow-growth phenotypes suppressed by SUI3–2. (A) FL-17–21 impairs derepression of GCN4 and increases leaky scanning of uORF1. GCN2 strains containing FL-WT (H3583) or FL-17–21 (CFY245) alleles of TIF11 and GCN4-lacZ reporter plasmids p180, pM199, or pM226, depicted in (i), (ii), and (iii), respectively, were assayed in repressing and derepressing conditions as in Figure 1C. Units of specific activity are means±s.e. (n=6). (B) Growth on SC-UL or SC-ULIV+0.5 μg/ml SM of GCN2 strains containing FL-WT, FL-17–21, WT-HA, or 7–11-HA alleles of TIF11 and either vector or hc SUI1 plasmid YEplac195-SUI1. Isogenic GCN2 (H1642) and Δgcn2 (H1895) strains were analyzed as controls (top rows). (C) Western analysis of WCEs of GCN2 strains containing FL-WT or FL-17–21 alleles of TIF11 described in (A) grown under repressing and derepressing conditions with antibodies specific for eIF2α phosphorylated on Ser-51 (eIF2α-P; upper panel) or total eIF2α (lower panel). Mean ratios of eIF2α-P/eIF2α were calculated and normalized to the WT ratio in each medium from two independent experiments. (D) Growth on SC-UL of FL-WT and FL-17–21 strains described in (A) carrying vector, pRSSUI3-S264Y-U (SUI3-2), or p291 (SUI3). (E) GCN2 strains containing FL-WT or FL-17–21 alleles of TIF11 and GCN4-lacZ reporter pM226 described in (A), and either vector or YCpSUI3-S264Y-W (SUI3-2), were assayed under repressing conditions as described in Figure 1C. Units of specific activity are means±s.e. (n=6). (F) Same as (E) except that FL-WT and FL-17–21 strains harboring plasmid-borne SUI5 and strains containing FL-ΔC or FL-17–21-ΔC alleles of TIF11 and empty vector were analyzed.
Whereas eIF1 overexpression suppresses the Slg− phenotype of the FL-17–21 mutant, it does not suppress the SMs/Gcn− phenotype of FL-17–21 cells (Figure 5B, rows 5–6). These results fit with the idea that eIF1 overexpression overcomes the defect in PIC assembly (and attendant Slg− phenotype), but not the defect in start codon selection that contributes to the SMS/Gcn− phenotype in FL-17–21 cells.
Interestingly, SUI3–2 eliminates the Slg− phenotype of FL-17–21 (Figure 5D), reduces the increased leaky scanning conferred by FL-17–21 (Figure 5E), and partially suppresses the SMS/Gcn− phenotype in FL-17–21 cells (data not shown). Thus, FL-17–21 and SUI3-2 mutually suppress their opposite effects on start codon selection. It is remarkable that SUI5, or introducing ΔC into the CTT, also suppresses the leaky scanning produced by FL-17–21 (Figure 5F). Thus, the Sui− mutations SUI3-2, SUI5, and ΔC, which increase UUG initiation at his4-303, also increase recognition of the uORF1 AUG in the FL-17–21 background, whereas FL-17–21 reduces both initiation events. These data also support the notion that NTT (FL-17–21) and CTT (ΔC) mutations in eIF1A have opposite effects on AUG selection.
eIF1A mutations in the NTT and CTT have opposite effects on the stability of eIF1A interactions in reconstituted PICs
We considered the possibility that both the Ssu− and leaky scanning phenotypes of the eIF1A NTT mutations could arise from stabilization of an open, scanning conformation of the PIC, reducing the probability of initiation at either UUG or AUG codons. Our previous work indicates that eIF1A dissociates more slowly at AUG versus non-AUG codons, suggesting that tighter binding of eIF1A is a hallmark of the closed, scanning-arrested IC formed at AUG (Maag et al, 2006). (Note that eIF1A dissociation in all cases is too slow to be physiologically relevant, but is a sensitive probe of the interactions between eIF1A and the PIC.) Accordingly, we asked whether the FL-17–21 mutation increases the rate of eIF1A dissociation at both UUG and AUG complexes.
43S·mRNA(AUG) or 43S·mRNA(UUG) complexes were assembled with eIF1A, tagged at its C-terminus with fluorescein, in the presence of eIF5, chased with excess non-labeled eIF1A, and eIF1A dissociation was measured over time as the decrease in fluorescence anisotropy. WT eIF1A dissociates with biphasic kinetics, with rate constants for the fast and slow phases designated k1 and k2, respectively. Kamp is the ratio of the amplitudes of the slow to fast kinetic phases; hence values of Kamp>1 indicate that the slow phase dominates the reaction. When 40S binding of eIF1A is saturated (as here), the anisotropy (r) reflects the restriction of rotational motion of the eIF1A CTT in the complex, with higher r values indicating less freedom (Maag et al, 2006).
In accordance with earlier findings (Maag et al, 2006), the kinetics of eIF1A dissociation from AUG complexes in the presence of WT eIF5 is dominated by the slow phase (Kamp of 5.5; Table I, row 3), whereas replacing AUG with UUG reduces Kamp to 4.0 and increases k1 and k2, indicating weaker eIF1A binding. The CTT is also relatively less restricted (smaller r value) at UUG (Table I, rows 1–4, UUG). The SUI5 mutation in eIF5 (G31R) decreases k2 and increases r at UUG (Table I, rows 1–8), indicating tighter eIF1A binding at this near-cognate codon. Similar results were reported for the Sui− ΔC mutation in eIF1A (Maag et al, 2006). These findings allowed us to propose that tighter eIF1A binding to UUG complexes contributes to the increased initiation at UUG in these Sui− mutants. It is interesting that SUI5/G31R and ΔC both weaken eIF1A binding and increase rotational freedom of the CTT for AUG complexes (Table I, AUG) (Maag et al, 2006), suggesting that these mutations disfavor AUG recognition while increasing selection of UUG codons.
Table 1.
Effects of eIF1A mutations on the kinetics of eIF1A dissociation from 43S.AUG and 43S.UUG PICsa
| eIF1A | eIF5 | AUG | UUG | ||
|---|---|---|---|---|---|
| 1 | WT | WT | k1 | 5.7±1.2 | 10.9±1.6 |
| 2 | k2 | 0.47±0.07 | 1.0±0.23 | ||
| 3 | Kamp | 5.5±0.7 | 4.0±0.88 | ||
| 4 | r bound | 0.205±0.004 | 0.185±0.007 | ||
| 5 | WT | G31R | k1 | 13.8±2.0 | 12±1.8 |
| 6 | k2 | 2.9±0.6 | 0.53±0.12 | ||
| 7 | Kamp | 1.3±0.6 | 5.8±2.8 | ||
| 8 | r bound | 0.187±0.005 | 0.206±0.007 | ||
| 9 | FL-17–21 | WT | k1 | 8.8±1.3 | 13.5±0.5b |
| 10 | k2 | 0.95±0.03 | |||
| 11 | Kamp | 0.16±0.04 | |||
| 12 | r bound | 0.184±0.003 | 0.171±0.0005 | ||
| 13 | FL-17–21 | G31R | k1 | 34±1b | 6.5±0b |
| 14 | k2 | ||||
| 15 | Kamp | ||||
| 16 | r bound | 0.1530±0.0007 | 0.1726±0.0016 | ||
| 17 | 7–11 | WT | k1 | 17±2.5b | 73±1b |
| 18 | k2 | ||||
| 19 | Kamp | ||||
| 20 | r bound | 0.19±0.01 | 0.1808±0.0009 | ||
| 21 | 7–11 | G31R | k1 | 216±0b | 28±2b |
| 22 | k2 | ||||
| 23 | Kamp | ||||
| 24 | r bound | 0.1686±0.0002 | 0.1819±0.0039 | ||
| 25 | 131,133 | WT | k1 | 8.1±1.5 | 2.1±0.005b |
| 26 | k2 | 0.315±0.015 | |||
| 27 | Kamp | 4.7±0.2 | |||
| 28 | r bound | 0.216±0.001 | 0.221±0.0001 | ||
| 29 | 131,133 | G31R | k1 | 10±0.65 | 7.8±1.2 |
| 30 | k2 | 2.3±0.65 | 0.49±0.01 | ||
| 31 | Kamp | 0.27±0.065 | 6.5±0.8 | ||
| 32 | r bound | 0.210±0.001 | 0.218±0.0025 | ||
| All rates are 10−3 s−1. | |||||
| ar bound is the fluorescence anisotropy of the labeled eIF1A in the 43S·mRNA complex. | |||||
| bSingle exponential. | |||||
Dissociation of FL-17–21 from AUG complexes containing WT eIF5 showed a dramatic reduction in Kamp (Table I, AUG, rows 3 and 11) so that the rapid phase now dominates the reaction (see also kinetic data in Figure 6A). For the corresponding UUG complexes, the reaction is monophasic with a rate constant even higher than k1 for WT complexes at UUG (Table I, UUG, rows 1 and 9). The CTT is also less restricted (smaller r) for both AUG and UUG complexes with FL-17–21. Similar conclusions hold for the double-mutant complexes formed with SUI5/G31R and FL-17–21 proteins, which exhibit rapid, monophasic dissociation and reduced r values for both AUG and UUG complexes, relative to those formed with SUI5/G31R and WT eIF1A (Table I, rows 13–16, and Figure 6A). The untagged 7–11 mutant also produced monophasic dissociation from AUG and UUG complexes at even higher rates and decreased r values (Table I, rows 17–24). Thus, the FL-17–21 and 7–11 NTT mutations weaken eIF1A binding and increase mobility of the CTT in AUG and UUG complexes in the presence of WT or SUI5/G31R forms of eIF5. These findings fit with the Ssu− and leaky scanning phenotypes of these eIF1A NTT mutations in vivo.
Figure 6.
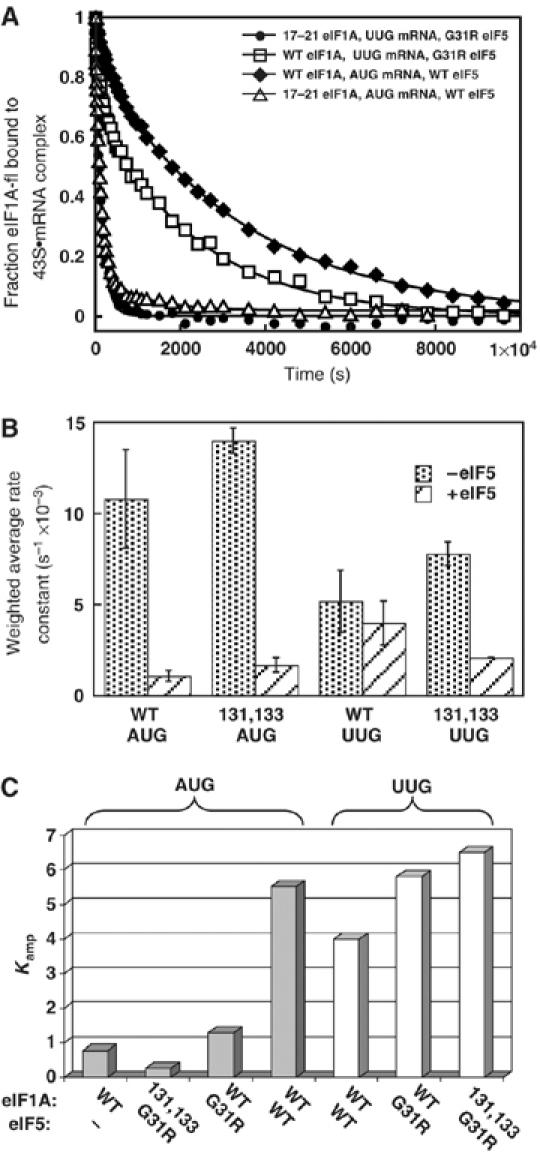
Sui− and Ssu− mutations in eIF1A affect the rate of eIF1A dissociation from reconstituted 48S PICs. (A) The FL-17–21 mutation accelerates eIF1A dissociation from 43S·mRNA(AUG) complexes in the presence of WT eIF5 and from 43S·mRNA(UUG) complexes in the presence of eIF5-G31R. Dissociation of fluorescein-conjugated FL-tagged mutant or untagged WT eIF1A was monitored by anisotropy changes when chased from the 43S·mRNA·eIF5 complex with unlabeled eIF1A. (B) Weighted-average rate constants calculated from data in Table I by multiplying the rate constant of each phase by the amplitude of that phase and summing the resulting numbers. Weighted rate constants are misleading when there is a very fast phase with a small amplitude, but this is not the case in comparisons made here. (C) Plot of selected Kamp values from Table I for the indicated combinations of untagged eIF1A (WT or 131,133 mutant) and eIF5 (WT or G31R mutant) proteins.
We also examined the untagged 131,133 CTT mutant to determine whether its Sui− phenotype involves tighter binding of eIF1A at UUG complexes. The 131,133 mutant clearly differs from WT in showing monophasic dissociation from the UUG complex, with a rate constant intermediate between the WT values of k1 and k2 for the UUG complex (Table I, row 25 versus rows 1–2). Comparing the monophasic rate constant for this mutant to a weighted-average rate constant for WT eIF1A suggests that the 131,133 mutation strengthens eIF1A binding at UUG in the presence (but not absence) of eIF5 (Figure 6B; last four bars), but has little effect on eIF1A dissociation from AUG complexes in the presence of WT eIF5 (Figure 6B, first four bars; Table I, rows 25–28 versus 1–4, AUG). The 131,133 mutation also decreases mobility of the CTT in complexes with UUG to the point where the anisotropy exceeds that seen for AUG with 131,133 or WT eIF1A (Table I, r values).
When combined with SUI5/G31R, the eIF1A-131,133 mutant slightly decreases the rate of dissociation at UUG, enhancing the tighter binding of eIF1A to UUG complexes produced by SUI5, and also exacerbates the effect of SUI5/G31R in weakening eIF1A interaction in AUG complexes. (Table I, rows 5–8 versus 29–32). A graph of Kamp for selected combinations of eIF1A and eIF5 proteins (Figure 6C) reveals that the combination of eIF1A-131,133 and SUI5/G31R reverses the normal effects of AUG and UUG on the rate of eIF1A dissociation. These biochemical data are consistent with the synthetic lethality of combining the SUI5 and 131,133 mutations in vivo.
Discussion
In this paper, we provide new insights into the distinct roles of the unstructured tails of eIF1A in PIC assembly. The 131,133 CTT mutation lowers the level of native 43S PICs in extracts and derepresses GCN4 translation in a manner partially suppressed by overexpressing TC. This mutation also decreases the rate of TC loading onto reconstituted PICs in vitro, consistent with its Gcd− phenotype. Ala substitutions of NTT residues 7–11, 12–16, or 17–21 in FL-tagged eIF1A conferred lethality or slow growth (for 17–21), and these phenotypes were suppressed by eIF1 overexpression. Overexpressing eIF1 also restored 40S association of MFC in FL-17–21 cells. By contrast, eIF1 overproduction does not suppress the phenotypes of the CTT mutation 131,133 (Figure 3B) or of other (non-NTT) mutations we tested that impair native PIC assembly (data not shown).
In the presence of the N-terminal FL tag (which exacerbates NTT mutations), the 17–21 and 7–11 mutations decrease the rate of TC loading on reconstituted 48S PICs in vitro. The untagged 7–11 mutant is also defective for this function in the absence of eIF1, but is not detectably different from WT in the presence of eIF1. Thus, the 7–11 substitutions reduce the rate of TC loading in a manner overcome by eIF1. By contrast, the CTT mutation 131,133 impairs TC loading only in the presence of eIF1. Clearly, mutations in the unstructured NTT and CTT impair PIC assembly by different mechanisms.
One way to explain suppression of the eIF1A NTT mutations by eIF1 overexpression is to propose that they disrupt a function of eIF1A in recruiting TC and other MFC components that is redundant with eIF1 activity. Consistent with this, eIF1A can bind to eIF2 and eIF3, and their interactions in the PIC require the eIF1A NTT (Olsen et al, 2003). eIF1 interacts with eIF2β, eIF3c, and eIF5 and is critical for MFC association with 40S subunits in vivo (Asano et al, 2001a; Singh et al, 2004), and eIF1 binds directly to the 40S (Lomakin et al, 2000). Hence, overexpressing eIF1 could drive 40S–MFC interactions and compensate for a reduced ability of eIF1A NTT mutants to promote MFC recruitment. In addition, binding of eIF1A and eIF1 to the 40S subunit is thermodynamically coupled (Maag and Lorsch, 2003; Majumdar et al, 2003), and overexpression of eIF1 increases 40S binding of the FL-17–21 protein in vivo (Figure 3E). This effect may contribute to suppressing the growth defect of this mutant, and also the lethality of the FL-7–11 mutation, considering that 7–11 confers a marked 40S binding defect in vitro (Figure 2E).
To explain why the CTT mutation 131,133 impairs TC loading in vitro only in the presence of eIF1, it could be proposed that 131,133 eliminates a functional interaction between eIF1A and eIF1 required for the stimulatory effect of eIF1 on TC loading. This would be another instance of their interdependence in PIC assembly revealed previously by the thermodynamic coupling of their 40S binding (Maag and Lorsch, 2003; Majumdar et al, 2003), which is not disrupted by the 131,133 mutation. The fact that 131,133 more seriously impedes TC loading in the presence versus absence of mRNA could indicate that it also impairs the ability of mRNA to stimulate TC loading in the reconstituted system (Maag et al, 2005).
The ΔC and 131,133 CTT mutations confer Sui− phenotypes in vivo, and the C-terminal HA3 tag on WT eIF1A exacerbates the Sui− phenotype of eIF2β mutation SUI3–2. Thus, altering the eIF1A CTT enhances initiation at UUG. Remarkably, the 7–11 and 17–21 substitutions in the NTT, and also the N-terminal FL tag, have the opposite effect and suppress the Sui− phenotypes of mutations in eIF2β or eIF5. The CTT and NTT eIF1A mutations also have opposing effects on AUG selection, with CTT mutations decreasing and NTT mutations increasing leaky scanning of the GCN4 uORF1 AUG codon. Remarkably, the CTT mutation ΔC, and other Sui− mutations (SUI3-2 and SUI5) suppress the increased leaky scanning phenotype of FL-17–21. Thus, the CTT mutations enhance UUG and AUG selection, while the NTT mutations suppress both events.
The increased leaky scanning of uORF1 in the NTT mutants likely contributes to their impaired ability to derepress translation of GCN4 (Gcn− phenotype) when all four uORFs are present, as uORF1 translation is required for the subsequent bypass of uORFs 2–4 by reinitiating 40S subunits when TC levels are reduced. This defect can help explain why the FL-17–21 NTT mutation does not produce a Gcd− phenotype (derepression of GCN4 in non-starvation conditions) despite a reduced rate of TC loading on 40S subunits and decreased PIC levels in vivo, as observed previously for eIF5 mutations (Singh et al, 2005).
Our recent in vitro analysis of eIF1A interactions with reconstituted 48S complexes revealed that AUG in the P-site and eIF5 collaborate to produce tighter binding of eIF1A. Moreover, the ΔC and SUI5 Sui− mutations tighten eIF1A binding to complexes with UUG start codons, but weaken binding to AUG complexes. This led us to propose that eIF1A binds less tightly to the open, scanning-conducive conformation of the PIC than to the closed, scanning-incompetent conformation of the IC at AUG (see model in Figure 7). The fast and slow phases of eIF1A dissociation would reflect the different affinities of eIF1A for open and closed conformations, which exist in equilibrium (Maag et al, 2006). (It could also be proposed that eIF1A does not dissociate from the closed complex and the slow phase reflects the transition from closed to open conformation. The probability of shifting from closed to open conformation, favored at UUG and disfavored at AUG, would then dictate the fast and slow rates of eIF1A dissociation, respectively, from these two complexes.)
Figure 7.
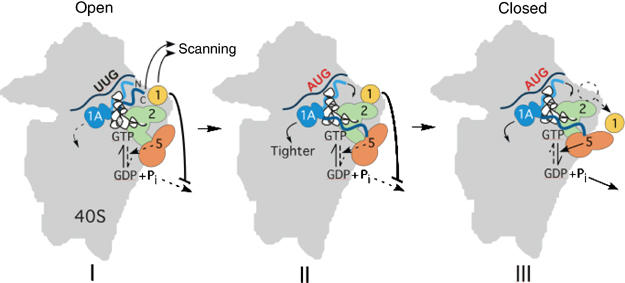
Hypothetical model for functions of eIF1A and eIF1 in scanning and AUG selection. (I) 48S PIC in the open, scanning-conducive conformation with a non-AUG in the P-site and the β-barrel fold of eIF1A occupying the A-site. GTP in the TC is partially hydrolyzed in a manner stimulated by eIF5, but Pi release from eIF2-GDP-Pi is blocked by eIF1 bound near the P-site. The CTT of eIF1A (dark-blue wavy line) is in proximity to eIF1 and both factors promote scanning. (II) Pairing of the anticodon of Met-tRNAiMet with AUG in the P-site elicits a rapid conformational change that increases the distance between eIF1A-CTT and eIF1 and results in tighter binding of eIF1A to the IC. eIF1A–IC interaction is weakened by eIF1A-CTT, which in turn is antagonized by eIF5 (perhaps through direct CTT–eIF5 interaction as suggested here) to strengthen eIF1A-IC association. The eIF1A NTT (light blue wavy line) also strengthens eIF1A–IC interaction. Hence, eIF1A–CTT mutations favor the closed complex, allowing increased initiation at UUG (Sui− phenotype), whereas NTT mutations favor the open complex, suppressing initiation at UUG (Ssu− phenotype). (III) Dissociation of eIF1 from its location near the P-site allows release of Pi from eIF2-GDP-Pi, an irreversible step that drives GTP hydrolysis to completion and finalizes start codon selection.
The new Sui− mutation 131,133 strengthens eIF1A binding and restricts motion of the CTT in UUG complexes, consistent with the idea that it promotes a closed conformation at non-AUGs. Strikingly, the Ssu− eIF1A NTT mutations FL-17–21 and 7–11 have the opposite effects in UUG complexes with either WT or G31R/SUI5 forms of eIF5. These results help explain why NTT mutations suppress initiation at UUG in Sui− mutants and they support our model that tighter binding of eIF1A to the IC is an important step in start site selection (Figure 7). The FL-17–21 and 7–11 mutations also weaken eIF1A binding to AUG complexes, which fits with the increased leaky scanning of the uORF1 AUG codon in FL-17–21 cells. The fact that the Sui− mutations SUI3-2, SUI5, and ΔC (in eIF1A itself) diminish this leaky scanning phenotype suggest that they stimulate the transition from open to closed conformations with either AUG or UUG in the P-site.
Two aspects of the eIF1A dissociation data are at odds with the in vivo results. The Sui− mutations SUI5 and ΔC weaken eIF1A binding at AUG complexes while strengthening eIF1A binding to non-AUG complexes (Maag et al, 2006) (Table I), and the CTT mutation 131,133 exacerbates both trends. This suggests that these Sui− mutations disfavor the closed conformation at AUG while enhancing the closed conformation at UUG. Indeed, on the basis of eIF1A dissociation rates (and anisotropy values), one would predict a higher rate of initiation at UUG versus AUG in the SUI5 and ΔC mutants, which is not observed. One would also predict that these mutations increase leaky scanning of the uORF1 AUG, whereas 131,133 and ΔC decrease leaky scanning in otherwise WT cells, and SUI5 and ΔC suppress this phenotype in FL-17–21 cells. These discrepancies suggest that the altered eIF1A affinity for the initiation complex, and the underlying conformational change, involves only one step in the transition to the closed, scanning-arrested IC from the scanning PIC (Figure 7).
We suggest that this transition includes several conformational changes in the 40S subunit that restrict the mRNA binding channel or strengthen interactions of TC with mRNA to impede scanning, make the P-site more accommodating to Met-tRNAiMet, or enhance the ability of eIF5 to stimulate GTP hydrolysis or Pi release from eIF2-GDP-Pi. eIF1 has been implicated in opposing many of these steps as a negative effector of non-AUG selection (Pestova and Kolupaeva, 2002; Unbehaun et al, 2004; Valasek et al, 2004; Algire et al, 2005), and eIF1 dissociation is thought to be required for AUG selection (Maag et al, 2006). These different steps may combine additively or synergistically to account for the ∼30-fold difference between initiation rates at AUG versus UUG observed in vivo. By overcoming the inhibitory effect of eIF1 on these other steps, AUG in the P-site may override the effects of SUI5 and ΔC in weakening eIF1A binding to AUG complexes. In fact, as these mutations produce Sui− phenotypes, they likely stimulate the additional steps that promote scanning arrest, GTP hydrolysis, and Pi release, neutralizing the inhibitory effects on the eIF1A-related conformational change at AUG. This could explain why UUG initiation remains lower than AUG initiation in SUI5, ΔC, and 131,133 mutants, and also why SUI5, ΔC, and 131,133 decrease, rather than increase, leaky scanning at uORF1.
It is also noteworthy that the eIF1A NTT mutations (especially 7–11) evoke a stronger reduction in eIF1A affinity for AUG complexes than the SUI5, ΔC, and 131,133 mutations. In addition, their suppression of Sui− phenotypes suggests that NTT mutations may inhibit one of the other steps leading to IC formation that the Sui− mutations likely stimulate in order to enhance UUG initiation. In fact, we found recently that the NTT mutation FL-17–21 slows down both the conformational change that increases eIF1A–eIF1 separation and the dissociation of eIF1 from its 40S binding site upon AUG recognition (unpublished observations). We propose that the cumulative effect of multiple impediments to IC formation at a start codon caused by FL-17–21 accounts for its increased leaky scanning of AUGs.
It could be argued that if our reconstituted system displayed the proper selectivity, then 43S·mRNA(UUG) complexes would be too unstable for measurements of eIF1A dissociation at the rates we observe (∼5–50 h−1). It is possible that we are missing in our system a selectivity factor that destabilizes non-AUG complexes (e.g. eIF4G). However, it is also possible that the UUG complexes are dynamic and slide along the mRNA, but that UUG is highly favored over the other triplets for P-site occupancy, so that the dissociation reaction involves an ensemble of different complexes that is heavily dominated by the UUG complex. Indeed, we have evidence from TC-binding data that the UUG complex is more stable than nearly all other non-AUG complexes, whereas less stable than the AUG complex (unpublished observations). Moreover, the fact that Sui− and Ssu− mutations in eIF1A have opposite effects on the kinetics of eIF1A dissociation from UUG complexes clearly indicates that the reconstituted system recapitulates at least some critical elements governing AUG selectivity in vivo.
Finally, it is notable that the entire set of phenotypes exhibited by the eIF1A NTT mutations described above are displayed by the DGNK53−56AANA mutation (Fekete et al, 2005) in conserved surface residues of the β-barrel. The 53–56 mutation impairs initiation and reduces native PIC levels in a manner diminished by hc SUI1, suppresses the Sui− phenotype of SUI5 (Ssu−), increases leaky scanning of uORF1 and impairs derepression of GCN4 (Gcn−), and its Gcn− and Slg− phenotypes are partially suppressed by SUI3-2 (Supplementary Figures S2 S3). Thus, this region of the β-barrel surface (between β strands 2–3) (Battiste et al, 2000) may cooperate with the NTT in PIC assembly and regulating AUG selection.
Materials and methods
Plasmids and yeast strains used are listed in Tables II and III of Supplementary data along with descriptions of their constructions and all relevant biochemical methods.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary data
Acknowledgments
We thank Tom Dever and our laboratories for helpful suggestions, Stephen Blakely and Azadeh Esmaeli for technical assistance, Ernie Hannig, Tom Donahue, Jon Warner, and Fan Zhang for antibodies or strains. This work was supported in part by the Intramural Research Program of the NICHD, NIH.
References
- Algire MA, Maag D, Lorsch JR (2005) Pi release from eIF2, not GTP hydrolysis, is the step controlled by start-site selection during eukaryotic translation initiation. Mol Cell 20: 251–262 [DOI] [PubMed] [Google Scholar]
- Algire MA, Maag D, Savio P, Acker MG, Tarun SZ Jr, Sachs AB, Asano K, Nielsen KH, Olsen DS, Phan L, Hinnebusch AG, Lorsch JR (2002) Development and characterization of a reconstituted yeast translation initiation system. RNA 8: 382–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano K, Phan L, Valasek L, Schoenfeld LW, Shalev A, Clayton J, Nielsen K, Donahue TF, Hinnebusch AG (2001a) A multifactor complex of eIF1, eIF2, eIF3, eIF5, and tRNA(i)Met promotes initiation complex assembly and couples GTP hydrolysis to AUG recognition. Cold Spring Harb Symp Quant Biol 66: 403–415 [DOI] [PubMed] [Google Scholar]
- Asano K, Shalev A, Phan L, Nielsen K, Clayton J, Valášek L, Donahue TF, Hinnebusch AG (2001b) Multiple roles for the carboxyl terminal domain of eIF5 in translation initiation complex assembly and GTPase activation. EMBO J 20: 2326–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battiste JB, Pestova TV, Hellen CUT, Wagner G (2000) The eIF1A solution structure reveals a large RNA-binding surface important for scanning function. Mol Cell 5: 109–119 [DOI] [PubMed] [Google Scholar]
- Donahue T (2000) Genetic approaches to translation initiation in Saccharomyces cerevisiae. In Translational Control of Gene Expression, Sonenberg N, Hershey JWB, Mathews MB (eds), pp 487–502. Cold Spring Harbor: Cold Spring Harbor Laboratory Press [Google Scholar]
- Fekete CA, Applefield DJ, Blakely SA, Shirokikh N, Pestova T, Lorsch JR, Hinnebusch AG (2005) The eIF1A C-terminal domain promotes initiation complex assembly, scanning and AUG selection in vivo. EMBO J 24: 3588–3601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershey JWB, Merrick WC (2000) Pathway and mechanism of initiation of protein synthesis. In Translational Control of Gene Expression, Sonenberg N, Hershey JWB, Mathews MB (eds), pp 33–88. Cold Spring Harbor: Cold Spring Harbor Laboratory Press [Google Scholar]
- Hinnebusch AG (2000) Mechanism and regulation of initiator methionyl-tRNA binding to ribosomes. In Translational Control of Gene Expression, Sonenberg N, Hershey JWB, Mathews MB (eds), pp 185–243. Cold Spring Harbor: Cold Spring Harbor Laboratory Press [Google Scholar]
- Hinnebusch AG (2005) Translational regulation of gcn4 and the general amino acid control of yeast. Annu Rev Microbiol 59: 407–450 [DOI] [PubMed] [Google Scholar]
- Huang H, Yoon H, Hannig EM, Donahue TF (1997) GTP hydrolysis controls stringent selection of the AUG start codon during translation initiation in Saccharomyces cerevisiae. Genes Dev 11: 2396–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jivotovskaya AV, Valasek L, Hinnebusch AG, Nielsen KH (2006) Eukaryotic translation initiation factor 3 (eIF3) and eIF2 can promote mRNA binding to 40S subunits independently of eIF4G in yeast. Mol Cell Biol 26: 1355–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolupaeva VG, Unbehaun A, Lomakin IB, Hellen CU, Pestova TV (2005) Binding of eukaryotic initiation factor 3 to ribosomal 40S subunits and its role in ribosomal dissociation and anti-association. RNA 11: 470–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin IB, Hellen CU, Pestova TV (2000) Physical association of eukaryotic initiation factor 4G (eIF4G) with eIF4A strongly enhances binding of eIF4G to the internal ribosomal entry site of encephalomyocarditis virus and is required for internal initiation of translation. Mol Cell Biol 20: 6019–6029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin IB, Kolupaeva VG, Marintchev A, Wagner G, Pestova TV (2003) Position of eukaryotic initiation factor eIF1 on the 40S ribosomal subunit determined by directed hydroxyl radical probing. Genes Dev 17: 2786–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maag D, Algire MA, Lorsch JR (2006) Communication between eukaryotic translation initiation factors 5 and 1A within the ribosomal pre-initiation complex plays a role in start site selection. J Mol Biol 356: 724–737 [DOI] [PubMed] [Google Scholar]
- Maag D, Fekete CA, Gryczynski Z, Lorsch JR (2005) A conformational change in the eukaryotic translation preinitiation complex and release of eIF1 signal recognition of the start codon. Mol Cell 17: 265–275 [DOI] [PubMed] [Google Scholar]
- Maag D, Lorsch JR (2003) Communication between eukaryotic translation initiation factors 1 and 1A on the yeast small ribosomal subunit. J Mol Biol 330: 917–924 [DOI] [PubMed] [Google Scholar]
- Majumdar R, Bandyopadhyay A, Maitra U (2003) Mammalian translation initiation factor eIF1 functions with eIF1A and eIF3 in the formation of a stable 40 S preinitiation complex. J Biol Chem 278: 6580–6587 [DOI] [PubMed] [Google Scholar]
- Olsen DS, Savner EM, Mathew A, Zhang F, Krishnamoorthy T, Phan L, Hinnebusch AG (2003) Domains of eIF1A that mediate binding to eIF2, eIF3 and eIF5B and promote ternary complex recruitment in vivo. EMBO J 22: 193–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova TV, Borukhov SI, Hellen CUT (1998) Eukaryotic ribosomes require initiation factors 1 and 1A to locate initiation codons. Nature 394: 854–859 [DOI] [PubMed] [Google Scholar]
- Pestova TV, Kolupaeva VG (2002) The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev 16: 2906–2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh CR, Curtis C, Yamamoto Y, Hall NS, Kruse DS, He H, Hannig EM, Asano K (2005) Eukaryotic translation initiation factor 5 is critical for integrity of the scanning preinitiation complex and accurate control of GCN4 translation. Mol Cell Biol 25: 5480–5491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh CR, He H, Ii M, Yamamoto Y, Asano K (2004) Efficient incorporation of eukaryotic initiation factor 1 into the multifactor complex is critical for formation of functional ribosomal preinitiation complexes in vivo. J Biol Chem 279: 31910–31920 [DOI] [PubMed] [Google Scholar]
- Unbehaun A, Borukhov SI, Hellen CU, Pestova TV (2004) Release of initiation factors from 48S complexes during ribosomal subunit joining and the link between establishment of codon-anticodon base-pairing and hydrolysis of eIF2-bound GTP. Genes Dev 18: 3078–3093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valášek L, Nielsen KH, Hinnebusch AG (2002) Direct eIF2-eIF3 contact in the multifactor complex is important for translation initiation in vivo. EMBO J 21: 5886–5898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valasek L, Nielsen KH, Zhang F, Fekete CA, Hinnebusch AG (2004) Interactions of eukaryotic translation initiation factor 3 (eIF3) subunit NIP1/c with eIF1 and eIF5 promote preinitiation complex assembly and regulate start codon selection. Mol Cell Biol 24: 9437–9455 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary data