

Abstract
A distinguishing feature of rod arrestin is its ability to form oligomers at physiological concentrations. Using visible light scattering, we show that rod arrestin forms tetramers in a cooperative manner in solution. To investigate the structure of the tetramer, a nitroxide side chain (R1) was introduced at 18 different positions. The effects of R1 on oligomer formation, EPR spectra, and inter-spin distance measurements all show that the structures of the solution and crystal tetramers are different. Inter-subunit distance measurements revealed that only arrestin monomer binds to light-activated phosphorhodopsin, whereas both monomer and tetramer bind microtubules, which may serve as a default arrestin partner in dark-adapted photoreceptors. Thus, the tetramer likely serves as a ‘storage' form of arrestin, increasing the arrestin-binding capacity of microtubules while readily dissociating to supply active monomer when it is needed to quench rhodopsin signaling.
Keywords: arrestin, EPR, oligomer, photoreceptor, signaling
Introduction
The phototransduction cascade has long served as a model of G protein-coupled receptor (GPCR) signaling. Essentially the same mechanisms of activation, signal amplification, and termination are used by the majority of the ∼1000 GPCRs, but in many ways the rod photoreceptor cell has no equal: it has incredibly low dark noise despite containing up to three billion molecules of the light receptor rhodopsin, it can produce reliable signals after capturing single photons, and upon adaptation is capable of extending the dynamic range of light sensitivity up to five orders of magnitude (Burns and Baylor, 2001). This level of perfection is achieved through several unique features. Rods have a specialized signaling compartment, the outer segment, where rhodopsin molecules are tightly packed in membranous discs, which is separated from the inner segment, where soluble signaling proteins are stored until needed. Rhodopsin has a covalently attached inverse agonist, 11-cis-retinal, that is converted by light into a potent, covalently attached agonist, all-trans-retinal. There is also the massive light-dependent translocation of major signaling molecules (reviewed in Calvert et al, 2006). The visual G protein transducin moves to the outer segment in the dark to amplify the signal, and moves away from the receptor to the inner segment in bright light (Sokolov et al, 2002). The key quenching protein, visual arrestin, moves in the opposite direction (Nair et al, 2005).
Here we explore another specific feature of the visual system: the self-association of rod arrestin. The arrestin molecule consists of two domains each containing a seven-stranded beta sandwich. Visual arrestin crystallizes as a tetramer (dimer of dimers) (Granzin et al, 1998; Hirsch et al, 1999). Previous studies using sedimentation equilibrium (Schubert et al, 1999) and small-angle X-ray scattering (SAXS) (Imamoto et al, 2003; Shilton et al, 2002) to investigate arrestin self-association in solution yielded contradictory results. More importantly, the structure of the arrestin oligomer in solution and its function in rod photoreceptors were not determined. Here, we employed multiangle visible light scattering to show that visual arrestin cooperatively forms tetramers in solution. Site-directed spin labeling (SDSL) and double electron–electron resonance (DEER) spectroscopy were used to show that the crystal and solution tetramers are different. The functional capabilities of the monomer and tetramer were also compared for the first time. We demonstrate that only monomeric arrestin binds rhodopsin, whereas microtubules bind both arrestin monomer and tetramer.
Results
Visual arrestin cooperatively forms tetramers at physiological concentrations
In a previous SAXS study, Imamoto et al (2003) concluded that visual arrestin forms tetramers according to 2M⇆D (K1), 2D⇆T (K2), where M, D, and T are monomer, dimer, and tetramer, respectively (MDT model). The oligomerization was found to be cooperative in the sense that the association constant K2>K1. These results are at odds with earlier equilibrium sedimentation (Schubert et al, 1999) and SAXS (Shilton et al, 2002) studies, which concluded that arrestin only forms dimers at physiological concentrations. We re-examined the oligomerization of visual arrestin in solution using multiangle laser light scattering to determine the weight-average molecular weight (MWav) as a function of total monomer concentration. This method has been previously employed to study protein–protein association (Woodbury et al, 2002; Mogridge, 2004; Attri and Minton, 2005). Its advantages include high resolution to within a few hundred daltons, wide molecular mass range, relatively small sample size, and high sample throughput. Importantly, because the wavelength of light is large compared with the dimensions of proteins, the scattering is independent of molecular shape (Mogridge, 2004).
Figure 1A shows the experimental MWav of wild-type (WT) visual arrestin as a function of total monomer concentration between 1 and 80 μM. The data are well fit by the MDT model, with K1 and K2 as the only adjustable parameters (Figure 1A, solid curve, K1=2.7±0.1 × 104, K2=1.3±0.1 × 105). Simple dimerization cannot account for the data, as molecular weights in excess of the size of the dimer (90 kDa) are observed at concentrations above 20 μM. Even at low concentrations, the data do not fit a monomer–dimer model (Figure 1A, dotted trace). The data also cannot be accounted for by the model of direct tetramer formation from monomers (Figure 1A, dashed trace). Based on the experimental values of K1 and K2, the tetramer is the dominant species at the physiological concentration of arrestin in rods (>1 mM) (Broekhuyse et al, 1985; Hamm and Bownds, 1986; Strissel et al, 2006; Hanson et al, 2007) (Figure 1B).
Figure 1.
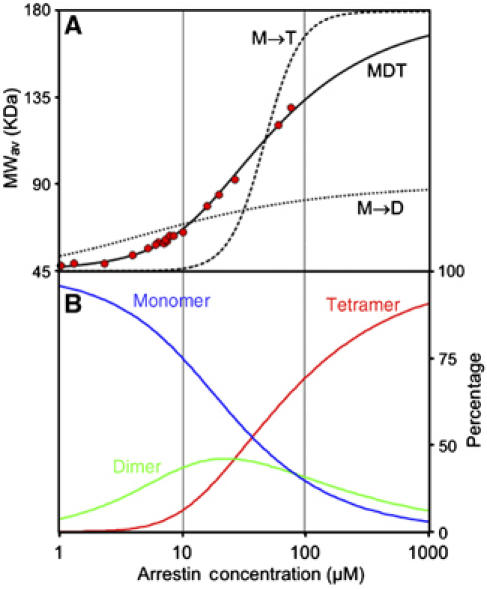
Arrestin cooperatively forms tetramers at physiological concentrations. (A) The average molecular weight of WT visual arrestin as a function of total arrestin concentration (red circles) was determined from the light scattering data as described in Materials and methods. The solid curve is a least-squares fit of the data to the MDT model with K1=2.7 × 104 and K2=1.3 × 105. For comparison, best-fit curves for dimer formation (M → D, K=1.3 × 105, dotted black line) and direct formation of tetramers (M → T, K=2.5 × 1012, dashed line) are shown. (B) The percentage of arrestin molecules in monomer (blue), dimer (green), and tetramer (red) as a function of total arrestin concentration computed for the MDT model, where K1=2.7 × 104 and K2=1.3 × 105.
Selection of sites for the introduction of a spin-labeled side chain
Visual arrestin invariably crystallizes as a tetramer (Figure 2A) (Granzin et al, 1998; Hirsch et al, 1999). Three interfaces are apparent in the crystal lattice. Within the tetramer, the C domain of one molecule contacts the N domain of another (CN interface) and two C domains make contact (CC interface; Figure 2B). The third is between tetramers, where the N domains make contact (NN interface; Figure 2C) (Schubert et al, 1999). SDSL was employed to determine whether the solution interfaces correspond to those in the crystal. To this end, unique cysteines were introduced on the background of a fully functional cysteine-less arrestin (Hanson et al, 2006a, 2006b) at selected locations at or near each of the putative interfaces and modified with a sulfhydryl-specific spin label to generate the R1 nitroxide side chain (Figure 2D). The spin-labeled proteins were used to detect direct contact interactions of the R1 side chain through perturbations in self-association and through changes in nitroxide mobility reflected in the EPR spectra.
Figure 2.
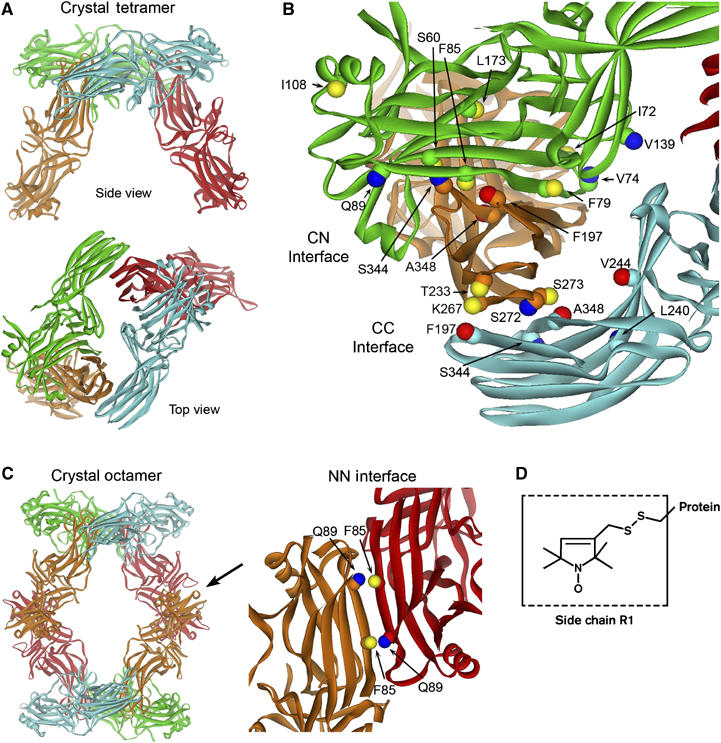
The interfaces of the arrestin crystal tetramer. (A) The 2.8 Å crystal structure of the visual arrestin tetramer (Hirsch et al, 1999), where monomers are colored differently for clarity. (B) The CN interface between the C domain of one monomer (orange) and the N domain of another (green) and the CC interface between two C domains (orange and cyan) are indicated. (C) The crystal octamer is shown highlighting the NN interface (arrow) between the N domain of one monomer (red) and the N domain of another (orange) in the adjacent tetramer. The sites examined in this study are labeled. Both the Cα and Cβ carbons are shown to indicate the direction in which the side chain projects. The Cβ atoms are color-coded based on the effect of R1 on oligomer formation: blue, no perturbation or small increases in Ko; yellow, moderate decreases in Ko; red, large decreases in Ko (see text). (D) The structure of the R1 spin label side chain.
Sites where the spin label was introduced are shown in Figure 2. Residues S60, F79, F85, F197, and S344 are in the CN interface (Figure 2B); F197, K267, S272, S273, and A348 are in the CC interface (Figure 2B); F85 and Q89 are in the NN interface (Figure 2C). Other residues substituted by R1 that do not make direct van der Waals contacts at any crystallographic interface include I72, V74, I108, V139, L173, T233, L240, and V244 (Figure 2B). Modeling of the R1 side chain at these sites confirms that it does not make direct contacts with a neighboring subunit.
The inter-subunit contact surfaces in solution are different from the crystal
The self-association of spin-labeled arrestins was compared with that of WT in Figure 3. The data were fit to the MDT model and individual K1 and K2 values are plotted in Figure 4 as K2 versus K12 so that all mutants with the same overall tetramerization constant (Ko=K12K2) lie on a line with slope of −1 and y-intercept of Ko (three such lines are shown). Numerical values are provided in Supplementary Table 1. R1 at the majority of sites produces some level of perturbation (Figures 3 and 4). For convenience, the mutants were divided into three groups according to the effect on Ko.
Figure 3.
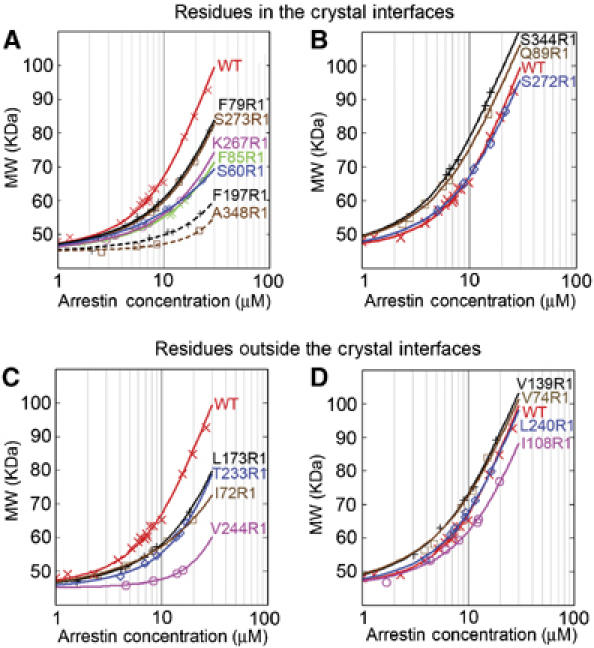
The effects of spin label at different positions on arrestin self-association. The average molecular weight of the indicated spin-labeled (R1) arrestin mutants as a function of total arrestin concentration (symbols) was fit to the MDT model (solid lines). The wild-type (WT) arrestin data (X) are shown for comparison. The data for arrestins with R1 in one or more of the crystal interfaces are shown in (A) and (B). The data for arrestins with R1 outside the crystal interfaces are shown in (C) and (D). Self-association constants for each mutant derived from the fit are summarized in Supplementary Table 1.
Figure 4.
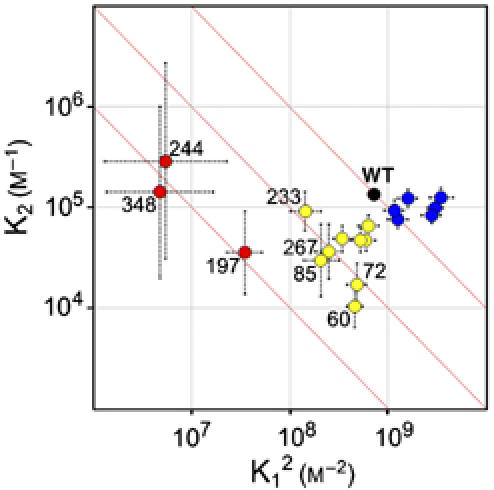
Comparison of the self-association constants of WT and spin-labeled arrestins. K12 versus K2 values for WT and spin-labeled arrestins are plotted on a logarithmic scale with estimated errors. The abscissa is plotted as K12 so that all mutants with the same Ko (K12K2; the overall tetramerization constant) lie on a line with slope of −1 and intercept of Ko. Three such lines (red) are shown for reference, one of which passes through the point for the WT protein (black dot). For convenience, the mutants are categorized into three groups, colored according to the effect on Ko; group 1 (red), large reduction in Ko; group 2 (yellow), moderate reduction in Ko; and group 3 (blue), no change or moderate increase in Ko. The large errors in K1 and K2 for group 1 are due to the limited concentration range explored and the large reduction in K1 produced by these mutants.
In the first group (197, 244, 348; Figure 4, red), the spin label produced the largest perturbations, roughly two orders of magnitude reduction in Ko. F197 and A348 are located in flexible sequences at both the CN and CC interfaces (Figure 2B). Interestingly, the perturbation resulting from the V244R1 mutation strongly suggests that this site is at a direct contact surface, but V244R1 does not make any contacts in the crystal (Figure 2B). In the second group (60, 72, 79, 85, 108, 173, 233, 267, and 273; Figure 4, yellow), R1 produced smaller reductions in Ko by a factor of 2–20. Notably, 72, 79, 173, and 233 are not at any of the crystal contacts, yet the measurable perturbation due to R1 at these sites suggests that they are at or near an interface in the solution tetramer. In the third group (74, 89, 139, 240, 272, and 344; Figure 4, blue), the spin label produced either no change or small increases in Ko by a factor of 2–5. Whereas 74, 139, and 240 are not at contact sites in the crystal tetramer, 89, 272, and 344 are deeply buried at contact interfaces and might be expected to produce large perturbations.
A significant reduction of Ko suggests that the spin label is at or near a contact interface in the oligomer. However, the lack of an effect is inconclusive because R1 may be accommodated in the structure without perturbation. Indeed, the docking of arrestin to rhodopsin (Hanson et al, 2006b) and SecB to SecA (Crane et al, 2005) appears to be little perturbed by R1 located directly at contact sites. If this is the case in the arrestin oligomers, interaction will be evident by an immobilization of the R1 residue reflected in the EPR spectra (Crane et al, 2005; Kusnetzow et al, 2006). To assess this possibility, the EPR spectra of 10 μM spin-labeled arrestin with and without 180 μM WT arrestin were compared (Figure 5). At 10 μM, the tetramer population for the WT protein is 8%, with dimer at 25% and monomer at 67% (Figure 1B). For destabilizing mutations, the monomer population would be even higher, and the corresponding EPR spectra primarily reflect the monomer structure. Even for mutations that increase Ko, the monomer is still the dominant species at 10 μM (⩾55%). At 190 μM total arrestin, the amount of tetramer in the WT is 70% (Figure 1B). WT protein was used to increase the total arrestin concentration to minimize perturbations due to R1. At a ratio of spin-labeled to WT arrestin of 1:18, the probability of perturbing more than one interface out of four is small, whereas the fraction of spin labels in the tetramer is maximized. The difference between the spectra in the presence and absence of WT arrestin reveals changes in the R1 environment due to oligomer formation. Note that arrestin is of sufficiently high molecular weight so that changes in the overall rotational diffusion due to oligomerization do not significantly affect the EPR spectra. Although this procedure is not quantitative, a spectral change positively identifies residues at a contact surface or in a region where the conformation is modified by the contact. A complete absence of change upon oligomerization indicates that a site is not at a contact surface.
Figure 5.
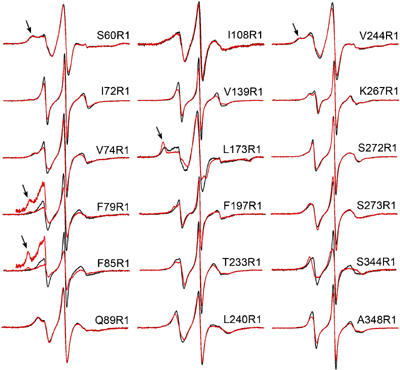
Concentration-dependent changes in the EPR spectra of R1-labeled arrestins. For each site, the spectrum was recorded for 10 μM spin-labeled arrestin (black trace) and in the presence of 180 μM WT arrestin (red trace). For 60, 79, 85, 173, and 244, arrows indicate components of the spectra corresponding to immobilized states of R1. The spectra are normalized to represent the same number of spins for each overlaid pair. The insets for F79R1 and F85R1 show a magnified view of the low-field region to more clearly reveal the immobilized components.
All 18 positions selected for introduction of the spin label are at solvent-exposed sites in the monomer. At 10 μM arrestin, the EPR spectra (Figure 5) are compatible with the crystal structure of the arrestin monomer (Hirsch et al, 1999) and known correlations between β-sheet structures and spin label mobility (Lietzow and Hubbell, 2004). The spectra of R1 at 72, 79, 85, 139, 197, 233, 267, 272, 273, 344, and 348, which are located in the edge strands of β-sheets or in loops, reflect rapid isotropic or weakly ordered (anisotropic) motions. The unusually high mobility of R1 at these sites suggests a flexible backbone structure. Indeed, residues 72, 79, 139, 344, and 348 reside in sequences with a high B factor, and 344 and 348 are in the ‘plastic' sequences that adopt distinct conformations in different monomers (Hirsch et al, 1999). The spectra of R1 at 60, 173, and 244 have broad components, reflecting low mobility (Figure 5, arrows). Modeling of the R1 side chain at these sites suggests that the constraints are due to interactions with neighboring side chains on the sterically crowded surfaces of the concave β-sheets (Lietzow and Hubbell, 2004).
In the presence of 180 μM WT (190 μM total protein), R1 residues located at or near contact sites are expected to show immobilization of some fraction of the population, the extent of which depends on the structure and populations of the oligomeric states present. The EPR spectra of 79R1 and 85R1 in the presence of excess WT reveal the appearance of a new immobilized population (Figure 5, arrows), whereas an existing relatively immobile population becomes strongly immobilized in 173R1, suggesting that these sites make direct contact even though these mutations induce relatively small perturbations in self-association (Figures 3 and 4).
Surprisingly, R1 at the most strongly perturbing sites, 197, 244, and 348, experiences only small changes in mobility. This is apparently not due to a large reduction in Ko and the resulting small populations of dimers and tetramers; the similar Ko for these mutants in the pure spin-labeled protein (∼1 × 1012) predicts a lower limit of ∼50% for arrestin in the form of tetramers at 190 μM. The presence of R1 may cause a local reorganization of the flexible interfaces, removing R1 from steric interference at the interface with a concomitant reduction in Ko. Alternatively, because only one out of 18 arrestin molecules carries an R1, it is also possible that this monomer will preferentially occupy a position in the tetramer where the substitution is the least detrimental for self-association, that is, away from direct contacts.
Small changes in R1 mobility are observed at 72, 74, 139, 233, 240, 267, and 344 (groups 2 and 3), with no evidence of strongly immobilized populations (Figure 5). These small spectral changes may be due to the presence of R1 in the vicinity of interacting surfaces. R1 residues at sites 60, 89, 108, 272, and 273 show no significant changes in mobility (Figure 5). Based on the values of K1 and K2 for the pure spin-labeled proteins (Supplementary Table S1), there should be at least 40–70% tetramer present at a total concentration of 190 μM for all of the above spin-labeled derivatives. Thus, none of the sites are at direct contact surfaces of the solution oligomer. The observed changes in K1 and K2 (Figures 3 and 4) may be due to the absence of the native residue near the interface rather than the interactions of the R1 side chain.
Collectively, the light scattering and EPR data indicate that residues 79, 85, 173, 197, 244, and 348 are involved in inter-subunit interactions in the solution tetramer. Of these, 79, 85, 197, and 348 are anticipated from the crystal structure (Figure 2). In contrast, the strong immobilization of 173R1 (Figure 5) and the strong perturbation owing to 244R1 (Figures 3 and 4) are not predicted by the crystal structure. Neither the native Leu173 and Val244 nor the R1 side chain modeled at these positions in the crystal structure make contacts with neighboring subunits. The strong immobilization of 173R1 upon tetramer formation could not occur without significant rearrangement at the CN interface.
Other inconsistencies with the crystal tetramer include relatively small perturbations and lack of immobilization of R1 at sites 60, 272, and 344, which are deeply buried at the CN, CC, and CN interfaces, respectively (Figure 2). Residue 344 is buried to the extent that the R1 side chain cannot be modeled without major rearrangement of the structure. Remarkably, the S344R1 mutation does not perturb the formation of oligomers (Figures 3 and 4). The EPR spectrum of 344R1 in the presence of excess WT arrestin reveals the appearance of a new dynamic state, but one that reflects changes in the order of R1 motion without immobilization. Residues 267R1 and 273R1 are located at the CC interface, but show only small changes in mobility upon tetramer formation. Modeling of R1 at these sites shows that it points away from the interface and the nitroxide may not be immobilized by the interaction. Nevertheless, the involvement of the backbone in this interaction would be expected to significantly damp the motion of these flexible sequences. The weak perturbation and lack of spectral change of 89R1, located directly at the NN interface, does not support its existence. These results clearly indicate that the tetramer in solution is not the same as that observed in the crystal.
Long-range distance measurements of the solution tetramer
The pulse EPR technique, DEER (Pannier et al, 2000; Jeschke, 2002), is a powerful method for measuring distances between paramagnetic centers in the range of ∼19–80 Å, complementing continuous wave (CW) methods that determine distances between 10 and 20 Å (Rabenstein and Shin, 1995; Altenbach et al, 2001). To probe the structure of the solution tetramer, we measured inter-molecular distances between spin labels at eight non-perturbing or mildly perturbing sites in the tetramer (74, 108, 139, 173, 240, 272, 273, and 344) using DEER. To ensure that the proportion of tetramer exceeds 70% (Figure 1B), we performed the experiments at 200 μM spin-labeled arrestin. In all cases, there is a clearly measurable interaction between the nitroxides in the tetramer as revealed by the dipolar evolution function (Figure 6A). The Fourier transform of the dipolar evolution function gives a sum of ‘Pake patterns' (Jeschke, 2002) (Figure 6B); the splitting between resolved extrema is proportional to r−3, where r is an inter-spin distance. Fitting of the dipolar evolution function (see Materials and methods) gives the experimental inter-spin distance distribution (Figure 6C). The inter-nitroxide distances predicted by modeling of the R1 side chain in the crystal tetramer are shown as shaded bars for comparison.
Figure 6.
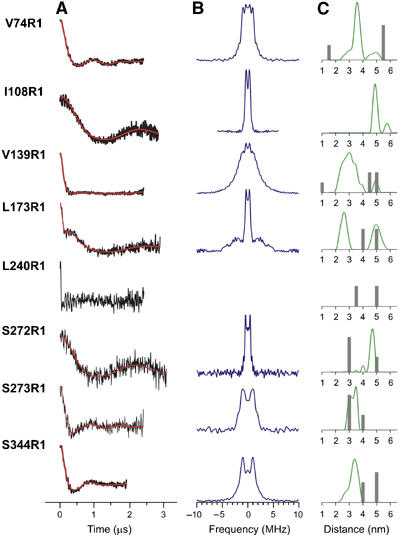
Inter-subunit distance measurements of the solution tetramer are inconsistent with the crystal tetramer. The inter-nitroxide distances between spin labels at eight distinct positions in arrestin were measured by DEER spectroscopy as described in Materials and methods. The DEER experiment measures the magnetic dipolar interaction between nitroxides as a modulation of the electron spin echo decay; the frequency of modulation is directly proportional to r−3, where r is the inter-spin distance. The primary data are the echo amplitude as a function of time. The dipolar evolution function (A, black) is obtained after subtraction of an exponentially decaying background owing to spins with randomly distributed inter-spin distances (e.g. interactions between adjacent tetramers). Fourier transformation of the dipolar evolution function gives the Pake pattern (B, purple); splitting between the extrema of the Pake pattern is proportional to r−3. Fitting of the dipolar evolution function (A, red line) yields the experimental inter-spin distance distribution (C, green). Estimates of the inter-nitroxide distances, predicted from modeling the R1 side chain in the crystal tetramer (see Materials and methods), are shown as shaded bars for comparison. For each mutant, there are six inter-nitroxide distances in a tetramer, some degenerate owing to symmetry. Distances beyond 70 Å do not contribute significantly to the derived distance distribution for the collection times of ⩽3 μs used in these experiments; therefore, only the predicted distances within the range of the DEER data are shown. The relative height of the bars reflects the fraction of the predicted total interacting spin population in the crystal tetramer within the measurable range. For 240R1, the decay of the echo is too rapid for further analysis, but shows a strong interaction between spins separated by ⩽18 Å.
Only in one case (273R1) are the experimentally determined inter-spin distances close to the predictions based on the crystal tetramer, whereas the data for the other sites are clearly incompatible with this structure. The broad distance distribution from 20 to 40 Å for 139R1 may reflect flexibility of the sequence in which 139 is located (Figure 5). Of particular note is the short inter-spin distance of 240R1 (⩽18 Å), where the echo decay was too rapid for further analysis, while the shortest predicted distance is ∼30–40 Å.
Collectively, the position-dependent effects of R1 on arrestin self-association (Figures 3 and 4), the concentration-dependent EPR spectral changes (Figure 5), and the distance measurements (Figure 6) clearly demonstrate that the structure of visual arrestin tetramers in solution and in the crystal is dramatically different.
The visual arrestin tetramer binds microtubules but not rhodopsin
Visual arrestin terminates the phototransduction cascade by binding phosphorylated light-activated rhodopsin (P-Rh*) with high affinity, thus preventing further transducin activation. Microtubules (MTs), which are extremely abundant in the rod inner segment, bind arrestin and apparently sequester it away from rhodopsin in dark-adapted photoreceptors (Nair et al, 2004; Nair et al, 2005). Direct binding assays performed at 1–2 nM arrestin (where no self-association can occur) demonstrate that the monomer effectively binds both P-Rh* and MTs (Gurevich and Benovic, 1993; Hanson et al, 2006a).
To explore the possibility that the tetramer also binds these partners, inter-subunit distance measurements were made under conditions where arrestin was bound to either P-Rh* or MTs (Figures 7 and 8). No change in distances is expected if the intact tetramer binds either partner. However, if only a monomer binds a given partner or if the binding causes significant changes in the tetramer structure, the characteristic distances would be absent or modified. The concentrations of rhodopsin and microtubules used in these experiments were sufficient to bind virtually all (in the case of P-Rh*) or most (⩾65–70% in the case of MTs) of the spin-labeled arrestin (Figures 7A and 8A). Arrestin binding to MTs did not change the dipolar evolution, and hence the distance distribution, for four out of the five spin-labeled arrestins (74R1, 139R1, 173R1, and 272R1) and produced only a minor change for 344R1 (Figure 7B). Thus, the arrestin tetramer binds to microtubules.
Figure 7.
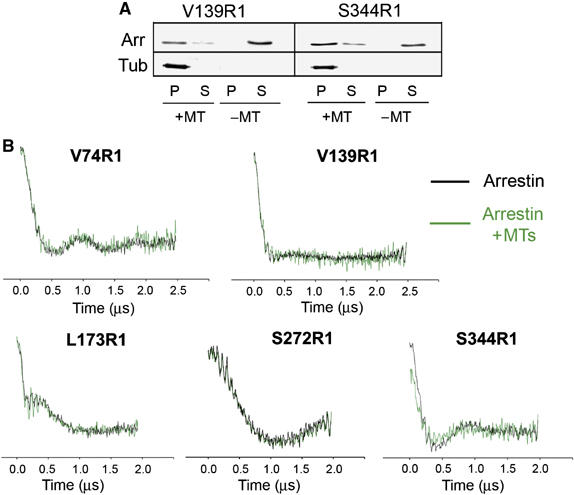
The arrestin tetramer binds microtubules. (A) The amount of spin-labeled arrestin bound to microtubules (MTs) under the conditions used in the DEER experiments was measured for V139R1 and S344R1 as described in Materials and methods. Arrestin (Arr) and tubulin (Tub) in the MT pellet (P) and soluble fraction (S) were quantified by Western blot. The amount of MTs present was sufficient to bind 65–70% of arrestin. (B) Dipolar evolution functions of arrestin spin labeled at five different positions were obtained in the absence (black) or presence (green) of MTs as described in Materials and methods. Dipolar evolution does not appreciably change for four out of the five mutants tested (V74R1, V139R1, L173R1, and S272R1) and showed only small changes for 344R1.
Figure 8.
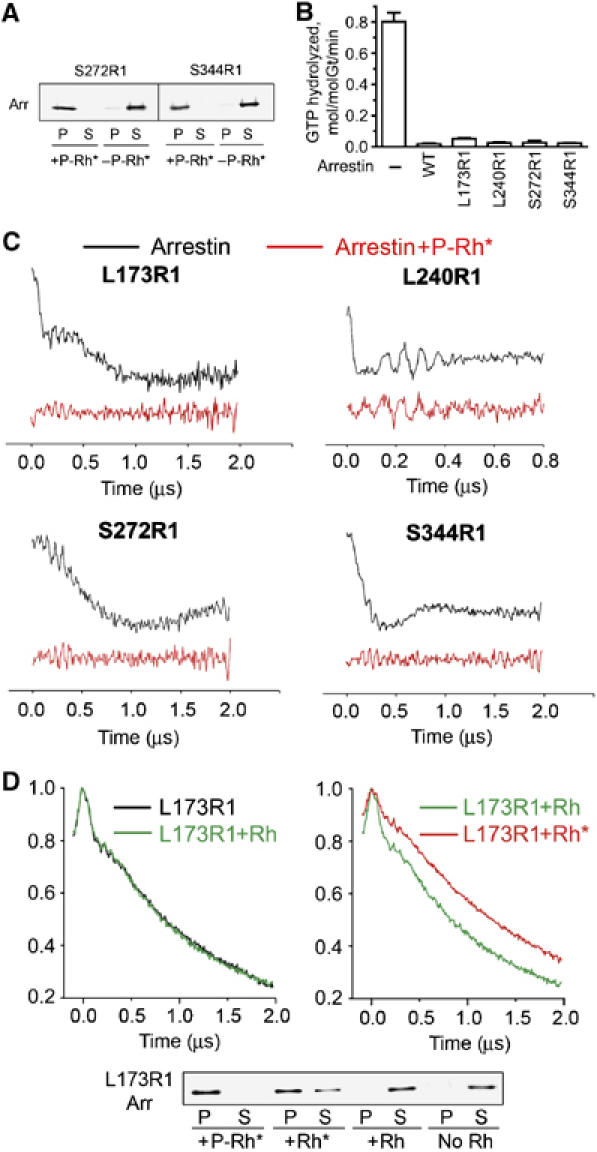
Arrestin binding to light-activated rhodopsin induces tetramer dissociation. (A) The amount of spin-labeled arrestin bound to light-activated phosphorylated rhodopsin (P-Rh*) under the conditions used in the DEER experiments was measured for S272R1 and S344R1 as described in Materials and methods. Arrestin (Arr) in the pellet (P) and soluble fraction (S) was quantified by Western blot. The amount of P-Rh* present was sufficient to bind >98% of arrestin. (B) The effect of WT and spin-labeled arrestin on transducin activation in photoreceptor membranes containing phosphorylated rhodopsin. Light-dependent GTPase activity was measured after a dim flash of light as described in Materials and methods. Average results (+s.d.) from two experiments, each performed in duplicate, are shown after subtracting transducin-independent GTPase activity measured in the dark (0.12+0.01 min−1). (C) Dipolar evolution functions for arrestin spin labeled at four positions obtained in the absence (black) or presence (red) of P-Rh*. (D) Top panels: echo decays (uncorrected for background) for L173R1 arrestin in the absence of rhodopsin (black), in the presence of dark rhodopsin (Rh) (green), or light-activated unphosphorylated rhodopsin (Rh*) (red). The uncorrected echo decays are shown rather than the dipolar evolution functions because of an uncertainty in the shape of the background correction, which is different for arrestin bound to a membrane surface (two dimensional) and in solution (three dimensional). For the case of arrestin in equilibrium with membrane binding sites, a mixed background would be required. Bottom panel: the amount of L173R1 bound to the different forms of rhodopsin at the concentrations used in the DEER experiments was determined by Western blot. Virtually no binding of arrestin to dark Rh was observed, whereas 56% bound to Rh* and >95% bound to P-Rh*.
In contrast, we found that arrestin binding to P-Rh* resulted in the complete disappearance of inter-subunit spin–spin interactions for each of the arrestins tested (Figure 8C), demonstrating that only the monomer binds P-Rh*. This important conclusion is based on the assumption that spin-labeled arrestins interact with P-Rh* normally. We verified this in two control experiments. First, we demonstrated that spin-labeled arrestins bind rhodopsin in a light- and phosphorylation-dependent manner in membrane sedimentation assays (Figure 8A and D). Second, we compared the ability of WT arrestin and the four spin-labeled mutants (173R1, 240R1, 272R1, and 344R1) used in these experiments to inactivate P-Rh* in a transducin activation assay (Figure 8B). Using photoreceptor membranes in which >95% of rhodopsin was phosphorylated, we measured transducin activation under conditions in which its rate was limited by the amount of available Rh* (Fung et al, 1981; Kuhn et al, 1981), so that the effects of Rh* phosphorylation and arrestin binding on transducin activation could be easily detected (Arshavsky et al, 1985; Wilden et al, 1986). Because transducin activation is followed by the hydrolysis of bound GTP, the light-dependent GTPase activity is directly proportional to the amount of transducin activated by Rh* and can be used as a measure of transducin activation. A robust transducin activation was observed in the absence of arrestin (Figure 8B), which is consistent with Rh* phosphorylation inhibiting but not abolishing transducin activation (Arshavsky et al, 1986; Wilden et al, 1986). However, the addition of arrestin (either WT or mutant) caused virtually the same drastic reduction in transducin activation rate, approximately equal to the fraction of phosphorylated rhodopsin in these membranes. This indicates that the mutants were fully functional with regard to P-Rh* inactivation.
Although arrestin preferentially binds P-Rh*, it has a measurable affinity for unphosphorylated Rh*, but little affinity for dark rhodopsin (Rh) (Gurevich and Benovic, 1993). A weak binding of the arrestin monomer to Rh* would increase the fraction of arrestin present as monomer at the expense of tetramer. This would be revealed as a decrease in the depth of modulation of the echo decay, because the number of interacting spin pairs is decreased (Hilger et al, 2005). The echo decays of L173R1 in the presence or absence of dark Rh are identical (Figure 8D, left panel), showing that membranes containing rhodopsin have no effect on the arrestin oligomeric equilibria. However, in the presence of Rh*, the depth of modulation is decreased relative to that in the presence of Rh (Figure 8D, right panel), as evident by the larger asymptotic value of the echo amplitude. This is consistent with limited binding of monomeric arrestin (about 55% bound; Figure 8D, lower panel). Collectively, these results demonstrate that any form of light-activated rhodopsin binds the arrestin monomer preferentially.
Discussion
Self-association of arrestin was discovered in 1977 (Wacker et al, 1977), but significant interest in this phenomenon was stirred only recently when visual arrestin was shown to form dimers and tetramers in the physiological concentration range (Schubert et al, 1999). The authors of two follow-up studies using SAXS came to contradictory conclusions interpreting the predominant arrestin oligomers either as dimers (Shilton et al, 2002) or as tetramers (Imamoto et al, 2003). Imamoto et al (2003) provided a likely explanation for these conflicting conclusions: at the non-physiologically high salt used by Shilton et al (2002), tetramer formation is inhibited, so that the dimer predominates.
We used laser light scattering (Mogridge, 2004; Attri and Minton, 2005) to determine the composition of the arrestin oligomer at the physiological salt concentration (150 mM) and pH 7.2 and found that the tetramer predominates (Figure 1), in agreement with Imamoto et al (2003). Our data corroborate the conclusion that tetramer formation is cooperative, that is, the KD for the dimer–tetramer equilibrium (1/K2=7.5 μM) is lower than that for the monomer–dimer equilibrium (1/K1=37 μM). However, the absolute values of the constants determined in our study are ∼10 times higher for K1 and ∼2 times lower for K2 compared with the K2/K1=100 model by Imamoto et al (2003). The difference may be attributable to assumptions involved in the analysis of SAXS data in terms of MW owing to the dependence of X-ray scattering on molecular shape. In contrast, the wavelength of visible light is higher than the dimensions of proteins, making light scattering independent of molecular shape (Mogridge, 2004).
Because visual arrestin crystallizes as a tetramer (Granzin et al, 1998; Hirsch et al, 1999), we asked whether the solution and crystal tetramers have the same inter-subunit contact interfaces. To answer this question, the nitroxide side chain R1 was introduced at selected sites in the molecule and its effects on self-association together with the concentration dependence of the EPR spectra were employed to identify protein–protein interaction sites. The dramatic reduction of arrestin self-association by R1 at positions 197, 244, and 348 (Figures 3 and 4) and the concentration-dependent immobilization of R1 at 79, 85, and 173 (Figure 5) imply that these residues make direct contacts with another subunit in the tetramer. We found that neither the native residues at 173 and 244, nor R1 modeled at these sites, make contacts in the crystal tetramer. Residues Gln89, Ser272, and Ser344 are buried in crystal interfaces, but R1 at these positions did not significantly perturb arrestin self-association (Figures 3 and 4) or become immobilized upon tetramer formation (Figure 5). In addition, direct distance measurements for seven of the eight mutants examined yielded results inconsistent with the crystal tetramer (Figure 6). These key observations indicate that the crystal and solution tetramer structures are different.
The difference between the solution and crystal tetramer is not surprising. First, light scattering (Figure 1) and previous SAXS data (Imamoto et al, 2003) indicate that tetramer formation is cooperative (K1<K2). This cooperativity means that the free energy for tetramer formation is more favorable than that for dimer formation, implying a greater area of contact between dimers. This is not the case in the crystal tetramer: the dimer–dimer interface (CC) is smaller than the monomer–monomer (CN) interface by a factor of ∼2 (Figure 2). Second, the crystal structure has an ‘open' configuration with two exposed N domains that are unrestricted for association with other arrestin molecules, but higher-order oligomers are not observed even at high concentrations of arrestin (Imamoto et al, 2003). Thus, it is likely that the solution tetramer has a closed ‘circular' configuration, although extensive modeling is necessary to determine the exact orientation of the subunits and the contact surfaces involved.
Among arrestin subtypes, only rod arrestin crystallizes as a tetramer (Hirsch et al, 1999; Han et al, 2001; Sutton et al, 2005). Its spontaneous self-association at physiological concentrations in solution suggests that the tetramer has a specific biological role in rod photoreceptors. The major function of arrestin is to terminate the photoresponse by binding to P-Rh*. A unique feature of rod arrestin is its light-dependent movement between two specialized compartments of the cell. Light causes massive translocation of arrestin to the outer segment, whereas in the dark it localizes to the inner segment (Broekhuyse et al, 1985; Nair et al, 2005). This movement was proposed to play a role in light and dark adaptation (Arshavsky, 2003; Calvert et al, 2006). Recent studies suggest that arrestin exclusion from the rod outer segment in the dark may be mediated by its dynamic interactions with microtubules (Nair et al, 2005). Arrestin binding to both P-Rh* (Gurevich and Benovic, 1993) and microtubules (Hanson et al, 2006a) is observed at nanomolar concentrations (five orders of magnitude lower than dimerization constant), demonstrating that the monomer interacts with both partners. Here we tested the ability of the arrestin tetramer to bind rhodopsin and microtubules.
Our results indicate that arrestin binds rhodopsin only as a monomer, because the binding to P-Rh* caused the dissociation of the tetramer, as evidenced by the complete loss of the inter-subunit spin–spin interactions (Figure 8C). In contrast, arrestin binding to microtubules did not appreciably change the distance distribution between R1 residues in adjacent subunits (Figure 7), suggesting that both monomer and tetramer bind microtubules. The biological significance of the tetramer binding to microtubules may be suggested as follows: at concentrations present in rods, most arrestin would exist as a tetramer (Figure 1B), so the binding of this predominant form to microtubules would make arrestin sequestration away from rhodopsin in the dark more efficient owing to the requirement of smaller number of binding sites.
Taken together, these data suggest that the tetramer likely serves as a ‘storage' form of arrestin, preventing premature signal termination at low light by buffering the amount of free monomeric arrestin available to quench rhodopsin signaling. The tetramer is likely a part of this regulatory mechanism because it significantly increases the arrestin-binding capacity of microtubules (‘binding four for the price of one'). This would keep arrestin away from the outer segment when maximum signaling efficiency is necessary. Arrestin affinity for P-Rh* is very high (KD=0.7 nM) (Osawa et al, 2000), whereas its affinity for self-association (Supplementary Table S1) and for microtubules (Hanson et al, 2006a) is several orders of magnitude lower. Therefore, at increased light intensities, ‘stored' tetrameric microtubule-bound arrestin becomes readily available to quench light-activated rhodopsin via its rapid release from MTs and the dissociation of the tetramer.
Materials and methods
Site-directed mutagenesis and arrestin purification
Site-directed mutagenesis and arrestin expression and purification were performed as described (Gurevich and Benovic, 1995; Gurevich and Benovic, 2000). All mutations were generated on the background of fully functional cysteine-less arrestin: ASA-CL (C63A, C128S, and C143A) or VSV-CL (C63V, C128S, and C143V) (Hanson et al, 2006a, 2006b).
Light scattering
Light scattering measurements were made with a DAWN EOS detector coupled to an Optilab refractometer (Wyatt Technologies) following gel filtration on a 7.8 mm (ID) × 15.0 cm (L) QC-PAK GFC 300 column (Tosoh Bioscience). The arrestin samples (100 μl) at different concentrations were injected onto the column at 25°C at a flow rate of 0.8 ml/min in 50 mM MOPS and 100 mM NaCl, pH 7.2. The column used did not resolve oligomeric species, but simply acted as a filter to remove highly scattering particulates. Light scattering at 18 angles (15–160°), absorbance at 280 nm, and refractive index (at 690 nm) for each sample were taken for a narrow slice at the peak of the elution profile (Woodbury et al, 2002).
The relationships describing the arrestin equilibria according to the MDT model (Imamoto et al, 2003) are as follows:
where M, D, and T are the monomer, dimmer, and tetramer concentrations, respectively, and C is the total protein concentration. From these relationships and particular values for K1, K2, and C, the concentrations of monomer, dimer, and tetramer may be obtained as solution to equations (1), (2) and (3):
Using the values of M, D, and T thus determined, MWav is computed as
where Mm is the monomer molecular weight (45 000).
The value for C in the light scattering cell at the point where data were collected was determined from the refractive index increment (0.184 g/ml) and A280, both of which were in agreement. The experimental Mav values are then obtained from the concentration and light scattering data using Astra for Windows 4.90 software (Wyatt Technologies). Experimental data for MWav as a function of C were least-squares fit to equations (1), (2), (3) and (4), with only K1 and K2 as adjustable parameters. The probable error associated with any given MWav point was taken as the standard deviation of the best fit from the extensive data for the WT protein (±1000). Errors in K1 and K2 for the mutants were determined using this value and the least-squares fit.
Sample preparation, EPR spectroscopy, and modeling of the R1 side chain
Arrestin cysteine mutants in 50 mM MOPS and 100 mM NaCl, pH 7.2, buffer were labeled with a 10-fold molar excess of 2,2,5,5-tetramethylpyrroline-3-yl-methanethiosulfonate spin label (MTSL, Toronto Research Chemicals) overnight at 4°C followed by removal of excess label, as described (Hanson et al, 2006a, 2006b). CW EPR spectroscopy was carried out at X-band on a Bruker EleXsys 500 fitted with a super-high Q cavity. Samples (10 μl) were collected in a glass capillary and spectra were recorded at room temperature over 100 G at a microwave power of 10 mW and modulation amplitude of 1 G, and typically were signal averaged 25–36 times.
For estimation of the interspin distances in the crystal tetramer, the R1 side chain was modeled using Discovery Studio Pro (Accelrys, San Diego). For solvent-exposed sites in loops where steric clashes could be avoided, the favored rotamer observed in crystal structures was built (X1, X2=−60) (Langen et al, 2000) (M Fleissner, D Cascio, and WL Hubbell, unpublished data). The disulfide dihedral was selected as ±90 to avoid atomic overlap. For sites in β-sheets, R1 was modeled as described by Lietzow and Hubbell (2004). The width of the distance distribution is estimated to be ∼10 Å based on variation of the X4, X5 dihedral angles and the width of experimentally measured distributions in model systems (Borbat et al, 2002). For sites 272 and 344, directly at contact surfaces between subunits in the tetramer, R1 could not be built without atomic overlap, and the distances were estimated using the rotamer with the least interference.
Four-pulse DEER measurements and data analysis
DEER measurements were performed using a Bruker EleXsys 580 spectrometer equipped with a 2 mm split-ring resonator at a temperature of 50 K. Before insertion into the resonator, samples (200 μM) containing 20% glycerol as a cryoprotectant were flash-frozen in sealed quartz capillaries (1.5 mm ID × 1.8 mm OD) using a liquid–solid slurry of chlorodifluoromethane (Freon 22).
Rhodopsin membranes (200 μg) (prepared as described by Gurevich and Benovic, 1993) were pelleted at 16 000 g for 30 min. The pellet was resuspended with spin-labeled arrestins in the dark to final concentrations of 450 μM rhodopsin and 200 μM arrestin. P-Rh* and Rh* samples were then exposed to room light for 5 min at room temperature before freezing. Microtubules were prepared from purified tubulin (Cytoskeleton Inc.) according to the manufacturer's instructions. Microtubules (1 mg) were pelleted at 100 000 g for 20 min at 30°C. The microtubule pellet was resuspended with spin-labeled arrestins to final concentrations of 450 μM tubulin and 200 μM arrestin. The samples were incubated for 20 min at 37°C before freezing. The amount of arrestin bound to rhodopsin and microtubules in each case was quantified under the same conditions as in the DEER experiments. These samples were centrifuged at 100 000 g for 10 min to separate bound and free arrestin. The pellet containing bound arrestin was resuspended in Laemmli's sample buffer (Sigma) and quantified by Western blotting.
The four-pulse DEER pulse sequence (π/2)ν1–τ1–(π)ν1–t–(π)ν2–τ1+τ2–t–(π)ν1–τ2–echo was used in each of the arrestin tetramer experiments (Pannier et al, 2000; Jeschke, 2002). The ELDOR pulse (ν2, 16 ns) was positioned at the centerfield maxima of the echo-detected nitroxide spectrum, whereas the π/2 and π observe pulses (ν1, 8 and 16 ns) were positioned at the low field line of the spectrum (ν1−ν2=70 MHz). A phase cycle +x/−x was applied to the first π/2 observe pulse to remove baseline offsets. Echo decay data were analyzed using the DeerAnalysis 2004 package (available at http://www.mpip-mainz.mpg.de/~jeschke/distance.html). For arrestin in solution and bound to microtubules, background echo decay was corrected using a homogeneous three-dimensional spin distribution. For arrestin bound to rhodopsin in membranes, the background was corrected using a two-dimensional spin distribution. The distance distribution was calculated by fitting the corrected dipolar evolution data using Tichonov regularization as implemented in DeerAnalysis 2004.
Rhodopsin phosphorylation and transducin GTPase activation assays
Rod outer segments were purified from frozen bovine retinas (WL Lawson Co., Lincoln, NE) as described (McDowell, 1993). The membrane pellet was resuspended in 100 mM K-phosphate buffer, pH 7.4, to a final rhodopsin concentration of 0.5 mg/ml and rhodopsin was phosphorylated by the endogenous rhodopsin kinase in the presence of 3 mM ATP and 2 mM MgCl2 in bright light for 180 min according to Wilden and Kuhn (1982). To maximize the yield of phosphorylated rhodopsin, 1 mM GTP was added to the reaction mixture to prevent transducin from ‘shielding' light-activated rhodopsin from rhodopsin kinase. Rhodopsin was regenerated by 11-cis-retinal, as described (McDowell, 1993). The membranes were urea-stripped, as described (Li et al, 1995). Rhodopsin concentration in the resulting preparation was determined by difference spectroscopy (Bownds et al, 1971). The degree of rhodopsin phosphorylation was assessed by mass spectrometry (Lee et al, 2002) and showed that >95% of rhodopsin contained two or more phosphates.
Transducin activation was measured under infrared illumination after reconstituting phosphorylated membranes with purified bovine transducin (Ting et al, 1993). Transducin activation was assessed by the level of the light-stimulated GTPase activity, which is a linear function of the amount of transducin activated by Rh*. In order to assure that transducin activation by Rh* rather than other steps of the transducin activation/inactivation cycle was rate limiting in these assays, we initiated the reaction by bleaching a small rhodopsin fraction by a flash from a calibrated light source (Kuhn et al, 1981; Arshavsky et al, 1985; Wilden et al, 1986). The reaction mixture contained 100 mM NaCl, 8 mM MgCl2, 10 mM Tris–HCl, pH 7.8, 12 μM total rhodopsin, 1 μM transducin, 10 μM [γ-32P]GTP (∼0.1 μCi/sample), and 3 μM of WT or mutant arrestin, where indicated. The reaction was started by illuminating the mixture with a 20 ms flash bleaching 0.5% rhodopsin and continued for 1 min at 22°C. Control experiments showed that this bleaching level causes ∼40% of the maximal transducin activation and is within the linear range of the dependency of transducin activation on Rh* concentration. 32P formation was measured as described by Cowan et al (2000).
Supplementary Material
Supplementary Table S1
Acknowledgments
We are grateful to Drs MJ Kennedy and JB Hurley for mass spectrometric characterization of phosphorhodopsin and Mrs Cherie Hubbell and Mr Nicholas J Bessman for technical assistance. This work was supported by NIH grants EY11500 (VVG), EY05216 and the Jules Stein Professorship Endowment (WLH), AI58024 and GM70642 (CSK), and EY10336 (VYA). SMH was supported by Training Grant GM07628.
References
- Altenbach C, Oh KJ, Trabanino RJ, Hideg K, Hubbell WL (2001) Estimation of inter-residue distances in spin labeled proteins at physiological temperatures: experimental strategies and practical limitations. Biochemistry 40: 15471–15482 [DOI] [PubMed] [Google Scholar]
- Arshavsky VY (2003) Protein translocation in photoreceptor light adaptation: a common theme in vertebrate and invertebrate vision. Sci STKE PE43 [DOI] [PubMed] [Google Scholar]
- Arshavsky VY, Antoch MP, Philippov PP (1986) Transducin activation by individual phosphorylated forms of rhodopsin in a detergent solution. Biol Membr 3: 1197–1203 [Google Scholar]
- Arshavsky VY, Dizhoor AM, Shestakova IK, Philippov P (1985) The effect of rhodopsin phosphorylation on the light-dependent activation of phosphodiesterase from bovine rod outer segments. FEBS Lett 181: 264–266 [DOI] [PubMed] [Google Scholar]
- Attri AK, Minton AP (2005) New methods for measuring macromolecular interactions in solution via static light scattering: basic methodology and application to nonassociating and self-associating proteins. Anal Biochem 337: 103–110 [DOI] [PubMed] [Google Scholar]
- Borbat PP, Mchaourab HS, Freed JH (2002) Protein structure determination using long-distance constraints from double-quantum coherence ESR: study of T4 lysozyme. J Am Chem Soc 124: 5304–5314 [DOI] [PubMed] [Google Scholar]
- Bownds D, Gordon-Walker A, Gaide-Huguenin AC, Robinson W (1971) Characterization and analysis of frog photoreceptor membranes. J Gen Physiol 58: 225–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekhuyse RM, Tolhuizen EF, Janssen AP, Winkens HJ (1985) Light induced shift and binding of S-antigen in retinal rods. Curr Eye Res 4: 613–618 [DOI] [PubMed] [Google Scholar]
- Burns ME, Baylor DA (2001) Activation, deactivation, and adaptation in vertebrate photoreceptor cells. Annu Rev Neurosci 24: 779–805 [DOI] [PubMed] [Google Scholar]
- Calvert PD, Strissel KJ, Schiesser WE, Pugh EN, Arshavsky VY (2006) Light-driven translocation of signaling proteins in vertebrate photoreceptors. Trends Cell Biol 16: 560–568 [DOI] [PubMed] [Google Scholar]
- Cowan CW, Wensel TG, Arshavsky VY (2000) Enzymology of GTPase acceleration in phototransduction. Methods Enzymol 315: 524–538 [DOI] [PubMed] [Google Scholar]
- Crane JM, Mao C, Lilly AA, Smith VF, Suo Y, Hubbell WL, Randall LL (2005) Mapping of the docking of SecA onto the chaperone SecB by site-directed spin labeling: insight into the mechanism of ligand transfer during protein export. J Mol Biol 353: 295–307 [DOI] [PubMed] [Google Scholar]
- Fung BK, Hurley JB, Stryer L (1981) Flow of information in the light-triggered cyclic nucleotide cascade of vision. Proc Natl Acad Sci USA 78: 152–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzin J, Wilden U, Choe HW, Labahn J, Krafft B, Buldt G (1998) X-ray crystal structure of arrestin from bovine rod outer segments. Nature 391: 918–921 [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL (1993) Visual arrestin interaction with rhodopsin: sequential multisite binding ensures strict selectivity towards light-activated phosphorylated rhodopsin. J Biol Chem 268: 11628–11638 [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL (1995) Visual arrestin binding to rhodopsin: diverse functional roles of positively charged residues within the phosphorylation-recognition region of arrestin. J Biol Chem 270: 6010–6016 [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL (2000) Arrestin: mutagenesis, expression, purification, and functional characterization. Methods Enzymol 315: 422–437 [DOI] [PubMed] [Google Scholar]
- Hamm HE, Bownds MD (1986) Protein complement of rod outer segments of frog retina. Biochemistry 25: 4512–4523 [DOI] [PubMed] [Google Scholar]
- Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C (2001) Crystal structure of beta-arrestin at 1.9 Å: possible mechanism of receptor binding and membrane translocation. Structure 9: 869–880 [DOI] [PubMed] [Google Scholar]
- Hanson SM, Francis DJ, Vishnivetskiy SA, Klug CS, Gurevich VV (2006a) Visual arrestin binding to microtubules involves a distinct conformational change. J Biol Chem 281: 9765–9772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Francis DJ, Vishnivetskiy SA, Kolobova EA, Hubbell WL, Klug CS, Gurevich VV (2006b) Differential interaction of spin labeled arrestin with inactive and active phosphorhodopsin. Proc Natl Acad Sci USA 103: 4900–4905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Gurevich EV, Vishnivetskiy SA, Ahmed MR, Song X, Gurevich VV (2007) Each rhodopsin molecule binds its own arrestin. Proc Natl Acad Sci USA, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilger D, Jung H, Padan E, Wegener C, Vogel KP, Steinhoff HJ, Jeschke G (2005) Assessing oligomerization of membrane proteins by four-pulse DEER: pH-dependent dimerization of NhaA Na+/H+ antiporter of E. coli. Biophys J 89: 1328–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JA, Schubert C, Gurevich VV, Sigler PB (1999) The 2.8 Å crystal structure of visual arrestin: a model for arrestin's regulation. Cell 97: 257–269 [DOI] [PubMed] [Google Scholar]
- Imamoto Y, Tamura C, Kamikubo H, Kataoka M (2003) Concentration-dependent tetramerization of bovine visual arrestin. Biophys J 85: 1186–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschke G (2002) Distance measurements in the nanometer range by pulse EPR. Chemphyschem 3: 927–932 [DOI] [PubMed] [Google Scholar]
- Kuhn H, Bennett N, Michel-Villaz M, Chabre M (1981) Interactions between photoexcited rhodopsin and GTP-binding protein: kinetic and stoichiometric analyses from light-scattering changes. Proc Natl Acad Sci USA 78: 6873–6877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusnetzow AK, Altenbach C, Hubbell WL (2006) Conformational states and dynamics of rhodopsin in micelles and bilayers. Biochemistry 45: 5538–5550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langen R, Oh KJ, Cascio D, Hubbell WL (2000) Crystal structures of spin labeled T4 lysozyme mutants: implications for the interpretation of EPR spectra in terms of structure. Biochemistry 39: 8396–8405 [DOI] [PubMed] [Google Scholar]
- Lee KA, Craven KB, Niemi GA, Hurley JB (2002) Mass spectrometric analysis of the kinetics of in vivo rhodopsin phosphorylation. Protein Sci 11: 862–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Franson WK, Gordon JW, Berson EL, Dryja TP (1995) Constitutive activation of phototransduction by K296E opsin is not a cause of photoreceptor degeneration. Proc Natl Acad Sci USA 92: 3551–3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lietzow MA, Hubbell WL (2004) Motion of spin label side chains in cellular retinol-binding protein: correlation with structure and nearest-neighbor interactions in an antiparallel beta-sheet. Biochemistry 43: 3137–3151 [DOI] [PubMed] [Google Scholar]
- McDowell JH (1993) Preparing rod outer segment membranes, regenerating rhodopsin, and determining rhodopsin concentration. Methods Neurosci 15: 123–130 [Google Scholar]
- Mogridge J (2004) Using light scattering to determine the stoichiometry of protein complexes. Methods Mol Biol 261: 113–118 [DOI] [PubMed] [Google Scholar]
- Nair KS, Hanson SM, Kennedy MJ, Hurley JB, Gurevich VV, Slepak VZ (2004) Direct binding of visual arrestin to microtubules determines the differential subcellular localization of its splice variants in rod photoreceptors. J Biol Chem 279: 41240–41248 [DOI] [PubMed] [Google Scholar]
- Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, Vishnivetskiy SA, Chen J, Hurley JB, Gurevich VV, Slepak VZ (2005) Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein–protein interactions. Neuron 46: 555–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa S, Raman D, Weiss ER (2000) Heterologous expression and reconstitution of rhodopsin with rhodopsin kinase and arrestin. Methods Enzymol 315: 411–422 [DOI] [PubMed] [Google Scholar]
- Pannier M, Veit S, Godt A, Jeschke G, Spiess HW (2000) Dead-time free measurement of dipole–dipole interactions between electron spins. J Magn Reson 142: 331–340 [DOI] [PubMed] [Google Scholar]
- Rabenstein MD, Shin YK (1995) Determination of the distance between two spin labels attached to a macromolecule. Proc Natl Acad Sci USA 92: 8239–8243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert C, Hirsch JA, Gurevich VV, Engelman DM, Sigler PB, Fleming KG (1999) Visual arrestin activity may be regulated by self-association. J Biol Chem 274: 21186–21190 [DOI] [PubMed] [Google Scholar]
- Shilton BH, McDowell JH, Smith WC, Hargrave PA (2002) The solution structure and activation of visual arrestin studied by small-angle X-ray scattering. Eur J Biochem 269: 3801–3809 [DOI] [PubMed] [Google Scholar]
- Sokolov M, Lyubarsky AL, Strissel KJ, Savchenko AB, Govardovskii VI, Pugh EN Jr, Arshavsky VY (2002) Massive light-driven translocation of transducin between the two major compartments of rod cells: a novel mechanism of light adaptation. Neuron 34: 95–106 [DOI] [PubMed] [Google Scholar]
- Strissel KJ, Sokolov M, Trieu LH, Arshavsky VY (2006) Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J Neurosci 26: 1146–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Vishnivetskiy SA, Robert J, Hanson SM, Raman D, Knox BE, Kono M, Navarro J, Gurevich VV (2005) Crystal structure of cone arrestin at 2.3 Å: evolution of receptor specificity. J Mol Biol 354: 1069–1080 [DOI] [PubMed] [Google Scholar]
- Ting TD, Goldin SB, Ho Y-K (1993) Purification and characterization of bovine transducin and its subunits. Methods Neurocsi 15: 180–195 [Google Scholar]
- Wacker WB, Donoso LA, Kalsow CM, Yankeelov JA Jr, Organisciak DT (1977) Experimental allergic uveitis. Isolation, characterization, and localization of a soluble uveitopathogenic antigen from bovine retina. J Immunol 119: 1949–1958 [PubMed] [Google Scholar]
- Wilden U, Kuhn H (1982) Light-dependent phosphorylation of rhodopsin: number of phosphorylation sites. Biochemistry 21: 3014–3022 [DOI] [PubMed] [Google Scholar]
- Wilden U, Hall SW, Kuhn H (1986) Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein in rod outer segments. Proc Natl Acad Sci USA 83: 1174–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbury RL, Hardy SJ, Randall LL (2002) Complex behavior in solution of homodimeric SecA. Protein Sci 11: 875–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1