

Abstract
Processing of the amyloid precursor protein (APP) by β- and γ-secretases leads to the generation of amyloid-β (Aβ) peptides with varying lengths. Particularly Aβ42 contributes to cytotoxicity and amyloid accumulation in Alzheimer's disease (AD). However, the precise molecular mechanism of Aβ42 generation has remained unclear. Here, we show that an amino-acid motif GxxxG within the APP transmembrane sequence (TMS) has regulatory impact on the Aβ species produced. In a neuronal cell system, mutations of glycine residues G29 and G33 of the GxxxG motif gradually attenuate the TMS dimerization strength, specifically reduce the formation of Aβ42, leave the level of Aβ40 unaffected, but increase Aβ38 and shorter Aβ species. We show that glycine residues G29 and G33 are part of a dimerization site within the TMS, but do not impair oligomerization of the APP ectodomain. We conclude that γ-secretase cleavages of APP are intimately linked to the dimerization strength of the substrate TMS. The results demonstrate that dimerization of APP TMS is a risk factor for AD due to facilitating Aβ42 production.
Keywords: amyloid Aβ, amyloid precursor protein (APP), dimerization, GxxxG, secretase
Introduction
Amyloid precursor protein (APP) is a type I transmembrane (TM) protein, which undergoes proteolytic processing by several secretases (Annaert and De Strooper, 2002). First, the bulk of the ectodomain needs to be removed by membrane-bound α- or β-secretases, leading to secreted forms of APP and membrane-bound C-terminal fragments α-CTF or β-CTF, respectively (Vassar et al, 1999; Annaert and De Strooper, 2002). Regulated intramembrane proteolysis (RIP) of the β-CTF by γ-secretase occurs only after ectodomain shedding and releases the amyloid-β (Aβ) peptide from the membrane. The active γ-secretase complex consists of the four subunits presenilin-1 (PS-1) (Rogaev et al, 1995), APH-1, PEN-2 and nicastrin (Edbauer et al, 2003). PS-1 contains two catalytically active aspartates in TM sequence (TMS)-6 and TMS-7 (Wolfe et al, 1999). PS-1 is endoproteolytically cleaved (Thinakaran et al, 1996) and the N- and C-terminal fragments were shown to exist as dimers in the catalytic core of γ-secretase (Cervantes et al, 2001, 2004; Schroeter et al, 2003). The amino termini of CTF stubs are recognized by nicastrin, which functions as a γ-secretase substrate receptor (Shah et al, 2005). The γ-secretase has been reported to cleave at variable sites, thus generating Aβ peptides of varying lengths (Qi-Takahara et al, 2005; Zhao et al, 2005). Aβ42 peptides are the pathologically most relevant form, as they form the core of amyloid plaques in the brains of Alzheimer's disease (AD) patients (Iwatsubo et al, 1994). AD patients with inherited mutations in PS-1 and APP show enhanced Aβ42 generation (Borchelt et al, 1996; Scheuner et al, 1996). Aβ42 is more prone to aggregation than Aβ40 (Jarrett and Lansbury, 1993; Schmechel et al, 2003), and in animal models low levels of Aβ42 result in amyloid plaque pathology even when total Aβ levels are reduced but the Aβ42/Aβ40 ratio is increased (McGowan et al, 2005; Wang et al, 2006). Although distinct oligomeric structures are discussed to be neurotoxic (Bitan et al, 2003; Chen and Glabe, 2006), factors that regulate the cleavage mechanism of the γ-secretase and determine the level of Aβ42 have largely remained unknown.
Recently, an explanation for the multiple cleavages of γ-secretase was provided, indicating a sequential proteolytic cleavage mechanism to release Aβ (Qi-Takahara et al, 2005; Zhao et al, 2005). Accordingly, the first cut occurs at the cytoplasmic edge of the TMS at the ɛ-site, that is, residue 49 or 48 of β-CTF (Sastre et al, 2001; Weidemann et al, 2002). The products Aβ49/Aβ48 remain membrane bound and are further processed in a sequential mode into Aβ46/Aβ45, that is, the ζ-site (Zhao et al, 2004). Aβ46 is further processed into Aβ43 and finally Aβ40, whereas Aβ45 is the direct precursor of Aβ42 (Qi-Takahara et al, 2005; Zhao et al, 2005). Notch was recently reported to be processed by γ-secretase in a very similar action mode (Chandu et al, 2006). Certain non-steroidal anti-inflammatory drugs (NSAIDS) were found to modulate the γ-secretase cleavage, either to longer or shorter forms of Aβ, when added to a cell culture system or in mice models. For example, fenofibrate was found to raise Aβ42 and to lower Aβ38 levels (Kukar et al, 2005), whereas ibuprofen had the inverse effect (Weggen et al, 2001) and Aβ40 remained unaffected. This indicates an inverse regulation of Aβ production between longer and shorter Aβ forms with Aβ42 and Aβ38 being the most prominent forms. The underlying mechanism for this regulation is uncertain, but alterations in Aβ production may be reflected by the dynamic nature of the substrate and/or altered enzyme/substrate interactions (Eriksen et al, 2003; Weggen et al, 2003).
We and others have previously shown that APP, amyloid precursor like proteins (APLPs) and other substrates of the γ-secretase, for example, ErbB-4 and E-cadherin can form homodimers (Beher et al, 1996; Rossjohn et al, 1999; Ferguson et al, 2000; Scheuermann et al, 2001; Marambaud et al, 2002; Wang and Ha, 2004; Soba et al, 2006). APP homodimerization is likely to be mediated by two different sites of the ectodomain, the loop region encompassing residues 91–111 and a second site overlapping with the collagen binding site spanning residues 448–465 (Beher et al, 1996; Rossjohn et al, 1999; Scheuermann et al, 2001; Wang and Ha, 2004).
By further studying homodimerization of APP, we found that the TMS of APP contains three consecutive GxxxG motifs encompassing residues 621–633 of APP695 or Aβ residues 25–37. GxxxG motifs are known to mediate TM helix–helix interactions within the membrane (Brosig and Langosch, 1998; Russ and Engelman, 2000; Senes et al, 2004). In the γ-secretase substrate β-CTF, effects of GxxxG mutations could be independent of the dimerization sites within the APP ectodomain.
In this study, we investigated the function of the APP GxxxG motifs by analyzing the effect on (1) APP–APP interactions of yellow fluorescent protein (YFP)- and cyan fluorescent protein (CFP)-tagged full-length proteins, (2) the dimerization strength of the TMS and (3) APP processing. We show that the consecutive GxxxG motifs provide a third dimerization site of APP and that this site within the membrane is secondary to the two sites of the ectodomain. Most importantly, GxxxG mutations destabilizing the dimerization strength determine the γ-secretase cleavage sites. We demonstrate an inverse generation of Aβ42 and Aβ38 levels that can be regulated by concerted mutations within the APP TMS itself and the generation of the shorter species Aβ37, Aβ35 and Aβ34. Finally, we present a model mechanism for γ-secretase cleavages on APP TMS based on our findings. We hypothesize that dimerization of APP TMS facilitates Aβ42 generation and is thus central to the onset of AD.
Results
APP TMS dimerizes via the GxxxG motifs
To selectively analyze the ability of the GxxxG motifs as part of the TMS to promote helix self-interaction, we applied the ToxR system (Langosch et al, 1996; Russ and Engelman, 1999). This assay allows to measure the mutual affinity of short TMSs as monitored by transcription activation of a reporter gene. To this end, the APP TMS and several point mutants were expressed in the context of membrane-anchored chimeric proteins composed of the extracellularly located maltose binding protein (MBP), the TMS under study and the intracellular ToxR transcription activator protein. In this assay, the dimerization of the TMS under study is required for transcription activation and its strength is reported by β-galactosidase activity. TMS residues Aβ 29–42 gave a strong signal similar to that obtained with residues 75–87 of glycophorin A (GpA) (Figure 1A and B), a well-investigated self-interacting TMS (Langosch et al, 1996). Dimerization strength was gradually reduced from Aβ TMS mutant constructs G29A and G33A to mutation G29/33A and was abolished by the mutation G33I (Figure 1B). The mutants G37A and G38A distal to the central GxxxG motif did not cause any effects. The influence of the juxtamembrane residue G25 could not be measured by this system as only TMS sequences can be analyzed. To separate the effects of APP TMS dimerization from other structural changes, we analyzed the impact of the mutations I31V, I32V and L34A centered around glycine G33. None of these mutants was able to reduce the dimerization strength of the TMS (Figure 1B). Thus, we conclude that the GxxxG motif determines the association of Aβ TMS, with G29 and G33 being the key residues. The differences between the double mutation G29/33A and mutation G33I might directly reflect the varying affinities of the two helices. While the mutation G29/33A might still allow the two TMS to approximate each other, stabilizing van der Waals bonding could occur. In contrast, the branched amino acid isoleucine then prevents this approximation and thus abolishes dimerization. To exclude the possibility that the observed differences in transcription activation are due to differences in efficiencies of membrane integration, we determined the ability of the ToxR proteins to complement the MBP deficiency of the Escherichia coli deletion strain PD28 (Supplementary Figure 1A). Furthermore, we analyzed protein expression by Western blotting of total cell lysates to ensure equal expression levels (Supplementary Figure 1B). Thus, we generated a set of GxxxG mutations and appropriate controls with which we were able to gradually reduce the dimerization strength of the APP TMS.
Figure 1.
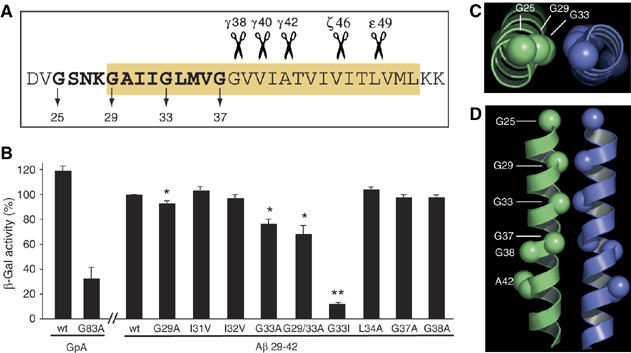
The APP TMS dimerizes via the G29xxxG33 motif. (A) Sequence of the APP TMS. Yellow box marks membrane-embedded amino-acid residues. The three consecutive GxxxG motifs extending into the TMS are highlighted in bold, and Aβ numbering is indicated for the glycine residues. Scissors represent the main cleavage sites of γ-secretase, for example, the ɛ-, ζ- and γ-cleavages at positions 49, 46, 42, 40 and 38. (B) ToxR assay. APP TMS residues 29−42 were analyzed. Measured β-galactosidase activity of the wt sequence was set as 100% (mean±s.e.m., n=4–6). Mutations to alanine at position G29 and especially G33 reduce dimerization strength. The double mutant G29/33A enhances the disrupting effects of the single mutants and G33I severely impairs dimerization. Non-glycine mutants and G37A and G38A do not impair dimerization strength. Asterisks indicate significant differences to Aβ29–42wt (*P<0.01, **P<0.0001, Student's t-test). (C, D) Structural model of dimeric APP TMS encompassing residues 25−46 with a low-energy conformation. (C) View down the dimer axis showing the close proximity of G29/G33. (D) View along the dimer interface highlighting G25, G29, G33, G37, G38 and A42. G29 and G33 are closely packed and form the core of the helix–helix interface.
A helix–helix interface with G29 and G33 is conformationally favored
In order to test whether a helix–helix interaction through the GxxxG motif is energetically and conformationally favored, we performed a computational search of conformations of Aβ residues 25–46. We observed a low-energy conformation with a right-handed crossing angle and all four glycine residues of the three consecutive GxxxG motifs at the helix–helix interface (Figure 1C and D). According to this model, residues G29 and G33 constitute the core of the interface, with Cα–Cα distances of less than 5 Å for each pair compared with the distances of G25 (6.7 Å) and G37 (5.7 Å). This computational model supports the data obtained from the ToxR assay, indicating that the TMS of APP can adopt a dimeric state with G29 and G33 being the key mediators of dimerization.
Impact of the GxxxG motifs on APP homodimerization
To examine whether APP full-length dimerization also depends on the GxxxG motif, we used a fluorescence resonance energy transfer (FRET) approach (Figure 2). C-terminally YFP-tagged full-length APP695 wild-type (wt) or mutant constructs G33A, G33I or G29/33A showed identical patterns of subcellular distribution (Figure 2A). In living cells, a FRET efficiency of 11% was obtained for APPwt-YFP and APPwt-CFP (Figure 2B–E). FRET signals originated from specific interactions between APP molecules, because FRET efficiencies were concentration-dependently reduced upon coexpression of non-fluorescent APPwt-Flag and negligible for coexpression of APP-YFP with a colocalized membrane protein (EGFR-CFP; Figure 2B). Thus, these data demonstrate homodimerization of APP in living higher eukaryotic cells for the first time. FRET efficiencies for APP G33A, APP G29/33A and APP G33I were all similar to the wt (Figure 2C). We conclude that a disturbance of the GxxxG motif does not necessarily influence full-length APP homodimerization and that GxxxG-mediated interactions may be crucial only for β- or α-secretase cleavage products of APP (Kaden et al, manuscript in preparation).
Figure 2.
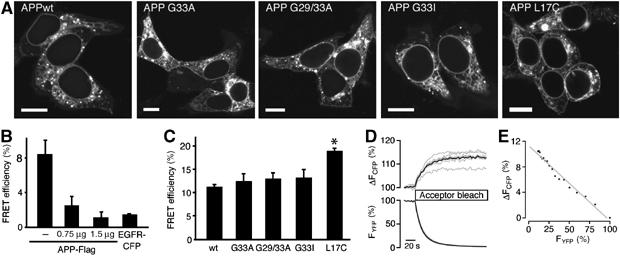
APP full-length dimerization. (A) Confocal microscopy images of APPwt-YFP and APP-YFP mutants show equal subcellular localization in HEK 293 cells. Bars, 10 μm. (B) FRET approach: competition of fluorescent APPwt dimer formation with increasing amounts of APPwt-Flag (means±s.e.m., n=4 with 3–6 cells each). As a negative control, APPwt-YFP does not interact with EGFR-CFP. (C) FRET efficiencies of wt and mutant APP (means±s.e.m., n=5 evaluated in quadruplicate). GxxxG mutants do not affect FRET efficiencies, whereas the mutant L17C forms covalent disulfide-bridged dimers and increases FRET efficiency significantly, *P<0.0001 Student's t-test. Note that APPwt FRET efficiency in (B) is lower compared with (C) owing to lower amounts of transfected plasmids. (D) Time course of donor fluorescence recovery (ΔFCFP) during selective photobleaching of the acceptor (FYFP). Data represent single cell (gray lines) and mean fluorescence traces (black lines) of a representative cell group. (E) Linear regression analysis of donor recovery (ΔFCFP) versus fractional acceptor (YFP) photobleach. Depicted is the mean of single cells from a representative experiment shown in (C).
To estimate the extent of APP dimerization, we determined the FRET efficiency for the mutation APP L17C that constitutively formed disulfide-linked dimers (Figure 3C) and showed a subcellular distribution comparable to the wt (Figure 2A). The FRET efficiency was increased by about 7% compared with APPwt (Figure 2C). Densitometric analysis of Western blots (see Figure 3C for a representative blot) showed that covalent dimers account for about 30% of total APP L17C. Assuming that the relative orientation of CFP and YFP is unchanged in covalent dimers, we estimate that the increase in FRET efficiency by 7% correlates to an increased dimerization of about 30%. Accordingly, it may be inferred from the APPwt FRET efficiency that approximately half of the APPwt molecules are homodimeric complexes. Thus, a significant portion of APPwt may be present in a dimeric conformation in living cells.
Figure 3.

Expression and processing of APP and SPA4CT in transfected SH-SY5Y cells. (A) Schematic representation of the processing of APP and β-CTF derived from the construct SPA4CT. SP: signal peptide; loop: loop region residues 91–111; CBP: collagen binding site residues 448–465; Aβ: Aβ domain, part of the ectodomain and the TMS; α- or β-secretase cleaves within the juxtamembrane region to generate soluble APP (sAPPα or sAPPβ, respectively) and C-terminal fragments (α-CTF or β-CTF, respectively). γ-Secretase cleaves β-CTF within the membrane and generates the C-terminal end of Aβ peptides. (B) Western blot analysis of APP-transfected cells. Several lanes were mirrored to adjust figure labels indicated by bars. Antibody 22C11 was used to detect full-length APP (APP). Antibody W0-2 (epitope Aβ residues 5–8) was used to label immunoprecipitated Aβ (Aβ). Note that antibody W0-2 captures Aβ species with varying C termini, showing that all APP mutants generate equal levels of total Aβ. (C) Western blot analysis of APP L17C-transfected cells. APP L17C forms covalent S–S-bridged APP dimers (antibody 22C11), which can be reduced by the addition of β-mercaptoethanol (MCE). Densitometric analysis of the dimer revealed that approximately 30% of APP L17C is dimeric. This mutant is processed to dimeric, S–S-linked Aβ peptides (antibody W0-2). (D) Quantification of sAPPα by ELISA. sAPPα from conditioned media of APP-transfected cells was captured using an anti-Myc antibody and detected by biotinylated W0-2. Single alanine mutations do not alter sAPPα levels, whereas G33I shows a nonsignificant increase of sAPPα. L17C yields a 53% reduction in sAPPα. The wt was set as 100% (means±s.e.m., n=5–7). (E) Western blot analysis of SPA4CT-transfected cells. Expression control of immunoprecipitated β-CTF and immunoprecipitated Aβ. Western blots were probed with antibody W0-2. Lanes showing Aβ of G29/33A and G33I were mirrored for better illustration.
To investigate the dimerization properties of APP-CTF, we expressed the mutant constructs L17C and L17C/G33I in transiently transfected SH-SY5Y cells. The analysis by Western blot revealed that both proteins constitutively formed disulfide-linked dimers, but L17C/G33I showed a decreased dimer formation of about 25% (Supplementary Figure 2D).
GxxxG mutations do not impair cleavage efficiency
We next examined whether the GxxxG mutations alter APP processing and generation of Aβ. SH-SY5Y cells stably expressing APP695wt or mutants G25A, G29A, G33A, G29/33A or G33I, or transiently expressing mutants I31V, I32V, L34A and L17C were generated. To avoid any influence of the APP ectodomain dimerization sites, we also stably transfected SH-SY5Y cells with signal-peptide-A4CT fragment (SPA4CT) (Dyrks et al, 1992). This construct releases β-CTF (A4CT) after cleavage of the signal peptide (SP) and is thus independent from α- or β-secretase cleavages (Figure 3A). All mutant constructs showed similar subcellular distribution patterns determined by immunofluorescence analysis (Supplementary Figure 2A and B). For all cell lines, similar levels of corresponding analytes were observed by Western blots, that is, full-length APP and Aβ from APP- and SPA4CT-transfected cells (Figure 3B, C and E and Supplementary Figure 2C). Note that total secreted Aβ levels are unchanged.
In addition, we quantified the sAPPα (secreted APP ectodomain α) levels in a sandwich enzyme-linked immunosorbent assay (ELISA) to detect possible effects of GxxxG mutants on α- or β-secretase cleavage activities (Figure 3D). There was no change in sAPPα levels for all single alanine mutants and I31V and I32V compared with the wt sequence. Solely, the mutants G29/33A and G33I showed a slight increase of sAPPα of about 5 and 14%, which is negligible compared with the effects on Aβ levels (see below and results shown in Figure 4). Thus, single alanine GxxxG mutations neither affected shedding by α- or β-secretases nor interfered with maturation and surface expression of APP as analyzed by Western blots. The mutant L17C produced 47% less sAPPα compared with the wt. Western blot analysis of this mutant revealed the presence of APP and Aβ dimers under non-reducing conditions and the amount of total Aβ generated remained comparable with the wt (Figure 3C).
Figure 4.
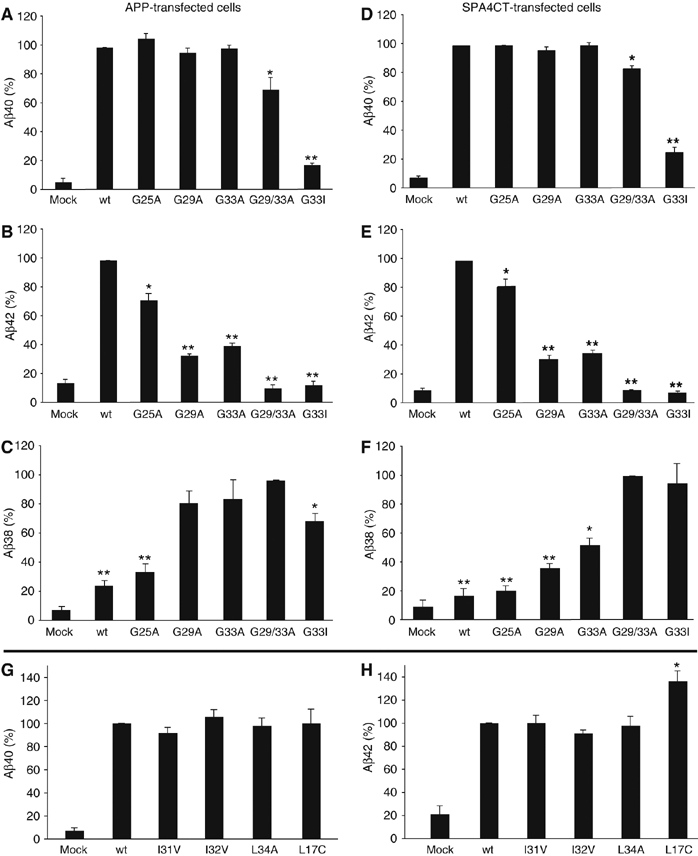
Disturbed GxxxG motifs reduce Aβ42 and increase Aβ38 levels. (A–C) Aβ40-, Aβ42- and Aβ38-specific ELISAs, respectively, with Aβ precipitated from media of stably APP-transfected SH-SY5Y cells. (D–F) Aβ40-, Aβ42- and Aβ38-specific ELISAs, respectively, with Aβ precipitated from media of stably SPA4CT-transfected SH-SY5Y cells. (A, B, D and E) The wt was set as 100% (means±s.e.m., n=3–5). Asterisks indicate significant differences to wt (*P<0.01 and **P<0.0001, Student's t-test). Single alanine mutants do not affect Aβ40 levels, but reduce the Aβ42 level significantly. The mutations G29/33A and G33I reduce Aβ40 as well as Aβ42 levels. The mutations G37A and G38A could not be analyzed by ELISA as the mutations altered epitope recognition by the monoclonal antibodies specific for Aβ40 or Aβ42. (C, F) The mutant G29/33A was set as 100% in Aβ38 ELISAs (means±s.e.m., n=3–5). Asterisks indicate significant differences to G29/33A (*P<0.01 and **P<0.0001, Student's t-test). GxxxG motif-disturbing mutations gradually increase Aβ38 levels depending on the individual glycine substitution. (G, H) Aβ40- and Aβ42-specific ELISAs from transiently or stably transfected SH-SY5Y cells. The wt was set as 100% (means±s.e.m., n=3–9). Non-glycine mutants do not affect Aβ40 or Aβ42 levels. Mutant L17C specifically increased Aβ42 levels by ∼36%.
GxxxG mutations reduce Aβ42 levels and increase Aβ38 levels
We quantified the levels of different Aβ species by the ELISA using monoclonal antibodies specific for Aβ42, Aβ40 and Aβ38. Strikingly, we observed significant differences in Aβ levels between the GxxxG mutants compared with the wt sequence. Whereas the single alanine mutants G25A, G29A and G33A did not show altered Aβ40 levels, Aβ42 was significantly reduced by 28, 67 and 60%, respectively, compared with cell lines expressing APPwt (wt set as 100%; Figure 4A and B). As the G25A mutant significantly reduced Aβ42 levels, this extracellular juxtamembrane position appears to be part of an extended GxxxG dimerization motif. For the double mutant G29/33A, Aβ40 generation was slightly reduced and the Aβ42 level dropped to the background of mock-transfected cells. The generation of Aβ40 or Aβ42 was abolished for the mutant G33I (Figure 4A and B).
Although the amount of total Aβ levels remained constant for all mutations as indicated by Western blots (Figure 3B and E), we suspected shorter Aβ species to be produced at the expense of longer forms of Aβ, which comigrated with Aβ42 and Aβ40. First, we determined the level of Aβ38 by ELISA and found an increase of Aβ38, whereas Aβ42 levels were decreased (Figure 4C). The mutations G29/33A and G33I showed the highest amount of Aβ38 produced (mutation G29/33A was set as 100%) corresponding to their abolished Aβ42 generation and reduced Aβ40 levels. The single mutants G29A and G33A reduced Aβ42 levels and increased Aβ38 secretion to a similar extent, whereas in the ToxR assay, G33A attenuated dimerization of the TMS more efficiently than G29A (Figure 1B). This slight variation from the consistency between effects on dimerization and Aβ production of these GxxxG mutants is most likely due to the position of bacterial membrane insertion of Aβ residues 29–42 with G29 located at the boundary of the constructs used in the ToxR assay (Figure 1B). Note that the mutant G33I led to decreased Aβ38 levels compared with G29/33A (Figure 4C), which is in favor of detection of increasing amounts of even shorter Aβ species (see MALDI-MS analysis; Figure 5).
Figure 5.

MALDI-MS spectra of secreted Aβ from APPwt- or mutant-transfected cells. Note that each alanine substitution leads to a unique mass shift of 14 Da, the G33I mutant increases by 56 Da and L34A mutant decreases by 42 Da. External calibration yielded standard deviations of less than 250 p.p.m. (A) Mass spectra showing Aβ peptides corresponding to Aβ34 (calculated monoisotopic mass 3785.9) and Aβ35 (calculated monoisotopic mass 3916.9). Note that for the wt sequence and L34A, Aβ34 and Aβ35, signals were barely above the background noise. In contrast, the relative amount of Aβ34 is significantly increased for all GxxxG mutants compared with the wt. The generation of Aβ35 is also increased except for the mutant G33I. (B) Mass spectra showing Aβ peptides corresponding to Aβ37 and Aβ40. Calculated monoisotopic mass for Aβ37: 4073.0, Aβ38: 4130.0, Aβ39: 4229.0 and Aβ40: 4328.2. The GxxxG mutants show increased Aβ37 and Aβ38 peak intensities compared with Aβ40 peaks, whereas Aβ39 remains stable. The mutant G33I generates only little Aβ40 and high amounts of Aβ38 and Aβ37. Non-glycine mutant L34A shows an Aβ pattern similar to the wt. Note that mass shifts are due to the amino-acid substitutions of the mutants, thus allowing to differentiate between Aβ40 from GxxxG mutants and minor amounts of endogenous sources as indicated by arrows. (C) Mass spectra showing Aβ42. The calculated monoisotopic mass for Aβ42 is 4512.3. Aβ42* represents the sodium adduct of Aβ42. The mutants G29/33A and G33I generated only negligible amounts of Aβ42, whereas Aβ42 of L34A was easily detected. The mass shifts are due to the amino-acid substitutions.
To investigate whether the altered quantity of Aβ species depends on altered dimerization strength or a possible structural effect, we have performed ELISAs to quantify Aβ40 and Aβ42 produced from I31V, I32V and L34A. As shown in Figure 4G and H, none of the mutants significantly affected Aβ40 or Aβ42 production in either direction.
To reverse the effects of the GxxxG mutants, we have also analyzed the engineered APP dimer L17C. This mutant produced 136% of Aβ42 and 100% of Aβ40 (Figure 4G and H). The specific increase of Aβ42 production without affecting Aβ40 levels confirms the effects of G29A and G33A mutants, which attenuated dimerization and decreased Aβ42 levels, but also left Aβ40 levels unimpaired.
Effects of GxxxG mutants on Aβ production are independent of the APP ectodomain
To exclude any influence of the β-secretase or of the APP ectodomain on Aβ generation caused by the APP GxxxG mutants, we analyzed Aβ of SH-SY5Y cells stably transfected with SPA4CT. We quantified levels of Aβ38, Aβ40 and Aβ42 by ELISA and obtained very similar results as those for the corresponding Aβ38, Aβ40 and Aβ42 levels of APP-transfected cells (Figure 4D–F). This indicates that the effect of the GxxxG motifs on Aβ production is independent of the APP ectodomain and β-secretase cleavage. Rather the GxxxG motif mutants specifically affect the γ-secretase cleavage mechanism, which is particularly evident from the single alanine mutants (Figures 3 and 4 and Supplementary Figure 2C).
Thus, we conclude that there is an inter-relationship between TMS dimerization and the production of shorter Aβ species including the inverse production of Aβ42 and Aβ38. The gradually decreasing or increasing levels of Aβ42 and Aβ38 clearly depend on the individual glycine substitutions and can be attributed to conservative or non-conservative mutations. Also, conservative or non-conservative mutations decide if Aβ40 production is additionally affected or not. The data indicate that Aβ38 production is intimately linked to the dimerization strength of the TMS–TMS interaction as indicated by the ToxR assay.
GxxxG mutants increase Aβ37 and Aβ35/Aβ34 levels
To gain further information about the cleavage sites of γ-secretase following the production of Aβ38, we used MALDI-MS analysis. Aβ from conditioned media of APP-transfected cells was immunoprecipitated with the monoclonal antibody W0-2, which recognizes amino-acid residues Aβ5–8 (Figure 5). Note that the mass shift of Aβ peaks of GxxxG mutants is due to the individual amino-acid substitutions.
The increase of Aβ38 and the concomitant decrease of Aβ42, as shown by ELISA, were confirmed by the relative intensities of the corresponding mass peaks (Figure 5B and C). Aβ42 was hardly detected by MS from G29/33A and G33I mutants, in agreement with the ELISA results (Figure 5C). In addition to increased Aβ38 generation, MS analysis revealed a gradual increase of Aβ37 peak intensities for those GxxxG mutants that strongly disrupted helix–helix interactions (Figure 5B). The mutant G33I primarily generated Aβ38 and Aβ37 in considerable amounts and little amounts of Aβ40. Aβ42 generated was below the detection limit (Figure 5B and C). This indicates that for G33I the γ-secretase products Aβ40 and Aβ42 might act as intermediate species and may easily be further processed into shorter forms. We further detected the shorter Aβ species Aβ35 and Aβ34, which are hardly generated from the wt (Figure 5A). Interestingly, Aβ34 was produced from all GxxxG mutants analyzed. Aβ35 was derived from all GxxxG mutants except the G33I mutant. The cleavage patterns of the non-glycine Aβ mutants were very similar to the wt as exemplarily shown by the spectrum of L34A in Figure 5.
Taken together, the GxxxG mutants not only modulate Aβ42, Aβ40 and Aβ38 levels, but also promote further processing of Aβ into several shorter species, for example, Aβ37, Aβ35 and Aβ34.
Discussion
To gain more insight into the importance and the role of the TMS of APP in amyloid Aβ production, we used site-directed mutagenesis to study the role of the GxxxG motifs in the TM segment. GxxxG motifs can have essential functions in the homo- and heterodimerization of a number of TM proteins. This motif forms the basis for helix–helix interactions of which GpA is the best-characterized model protein (Brosig and Langosch, 1998; Russ and Engelman, 2000; Senes et al, 2004). Mutations usually lead to impaired protein functions, such as for the Helicobacter pylori vacuolating toxin, the yeast ATP synthase, the ATP-binding cassette transporters (ABCG2), the copper-uptake transporter CTR, the yeast α-factor receptor STE2 and the peroxin Pex14p (Arselin et al, 2003; McClain et al, 2003; Overton et al, 2003; Aller et al, 2004; Polgar et al, 2004; Itoh and Fujiki, 2006).
Our analyses demonstrate that the G29xxxG33 motif in APP plays an important role in the dimerization of APP TMS and the processing of β-CTF into Aβ by the γ-secretase complex. Interestingly, a GxxxG motif has a critical role in the assembly of the γ-secretase complex. In Caenorhabditis elegans, a point mutation of the first GxxxG motif in TMS-4 of APH-1 (G123D) leads to a loss-of-function anterior pharynx-defective (Aph) phenotype (Goutte et al, 2002). The corresponding mutation in mammalian APH-1 disrupts γ-secretase complex assembly and reduces proteolytic activity (Lee et al, 2004). Thus, we propose that the GxxxG motif might also be required to support the interaction of APP with other proteins within the γ-secretase complex.
Based on a FRET approach, our results provide evidence that APP forms dimers in living cells. Similar to APP, other γ-secretase substrates, which have a single GxxxG motif in their TMS, were described to form full-length homodimers such as ErbB-4, E-cadherin, Nectin-1 and APLP-1 (Ferguson et al, 2000; Miyahara et al, 2000; Troyanovsky et al, 2003; Soba et al, 2006).
Furthermore, our work shows that the APP TMS can form homodimers and that the interaction is primarily mediated by the crucial glycine residues G29 and G33 of the three consecutive GxxxG motifs similar to the TMS of E-cadherin, ErbB-receptors and Notch (Huber et al, 1999; Mendrola et al, 2002; Vooijs et al, 2004). We demonstrate that dimeric (wt) as well as dimer-impaired substrates, that is, the mutant G33I, are utilized equally well as substrates by the γ-secretase, as we observed equal levels of total Aβ. It has previously been published that the γ-secretase complex contains two PS-1 molecules and therefore possesses two catalytically active sites (Cervantes et al, 2001, 2004; Schroeter et al, 2003). Thus, it is conceivable that the γ-secretase complex can cleave dimeric substrates, which might yield two different product lines, that is, encompassing Aβ42, 38, 35 and Aβ40, 37, 34. Thus, our data contradict a previous study, that suggested that dimeric substrates cannot be cleaved by γ-secretase (Struhl and Adachi, 2000). The authors used chimeric constructs to claim that dimeric γ-secretase substrates cannot be processed. However, as they do not provide biochemical evidence that the chimeras dimerize and do not affect recognition or binding sites of γ-secretase, their conclusion was rather of indirect evidence and possibly invalid (Struhl and Adachi, 2000). Meanwhile, Sardi et al (2006) reported that ErbB4 is cleaved by γ-secretase upon neuregulin stimulation. Neuregulin has been known to induce dimerization of ErbB4, which was required for γ-secretase cleavage according to the authors either as a homodimer or an ErbB4/2 heterodimer.
Our results establish a crucial functional role of the GxxxG motifs in APP processing through the γ-secretase complex. For GxxxG mutants, we observed an inverse relationship between Aβ42 and Aβ38 generation. Aβ40 levels remained unaffected except for the mutants G29/33A and G33I, which may adversely affect the helix-packing capacity of APP TMS to a greater extent. Of the two glycine residues G29 and G33, our data support a more critical role of residue G33, which relative to G29 is located more toward the center of the TM helix of APP. Substitution of G33 by a bulkier residue, that is, isoleucine, was deleterious for the stability of the APP TMS. A more conservative mutation of G33 into alanine did not have such a pronounced adverse effect. Although the steady-state levels of Aβ40 appeared not to be as significantly reduced by single Ala mutations of G29 or G33, we observed that the processing into Aβ42 was clearly compromised in G29A and G33A mutants. Specifically, we demonstrated that the APP TMS bearing the mutation G33I was less able to support the dimerization of APP TMS and that cells harboring this mutant in APP or β-CTF displayed a reduced capacity to produce Aβ42 and Aβ40 compared with the wt proteins. To exclude the possibility that structural changes of residues within the TMS could have an influence on recognition and cleavage by the γ-secretase, we have analyzed single point mutations neighboring the key glycine residue 33, that is, I31V, I32V and L34A. These results indicate that TMS dimerization directly alters γ-secretase processing rather than that processing is influenced by structural changes of GxxxG mutants.
Furthermore, GxxxG mutants led to an increased level of shorter Aβ species, that is, Aβ37, Aβ35 and Aβ34. The exchange of the glycine residues for hydrophobic ones with bulkier side chains not only interfered with the packing interface of neighboring APP helices and prevented the dimerization, but also affected substrate/enzyme interaction of APP and thus γ-secretase activity. Similarly, the introduction of an extra residue between glycine residues of the GxxxG motif might have prevented helix–helix associations as shown in a previous study, in which Lichtenthaler et al (2002) varied the APP TMS length. Two of the mutants described obviously affected the G25xxxG29 and G29xxxG33 motifs and generated elevated levels of Aβ38, Aβ37, Aβ34 and Aβ33.
We propose that GxxxG mutants allow the γ-secretase to cleave sequentially from the ɛ-cut to positions Aβ38/Aβ37 and further to Aβ35/Aβ34 when the stability of the TMS dimer is severely affected (Figure 6B). Given that there are ∼3.6 residues per α-helical turn, the glycine residues of the motif would be arranged on the same face of the helix, and the number of three-spacer residues separating them would preserve this arrangement. These results support a recently presented model proposing that γ-secretase processing is a continuous process starting with the ɛ-cleavage from the C-terminal end of the TMS at position 49/48 followed by cleavages at position 46/45 (ζ-cleavages) and 42/40 (γ-cleavages) thus removing helix turn by helix turn from β-CTF (Qi-Takahara et al, 2005; Zhao et al, 2005) (Figure 6A). According to our data, we expand the sequential cleavage model by proposing that under normal conditions processing of β-CTF is largely arrested at the γ-cut 42/40, allowing one to detect high levels of the intermediate products Aβ42 and Aβ40. We assume that this arrest at Aβ42/Aβ40 is due to the dimeric state of the substrate β-CTF, which has a cross-point at residues G29 and G33, affecting both the stability and the molecular organization of TMS helices (Figure 6A). APP TMS dimerization is unique insofar as it is the only known γ-secretase substrate with a GxxxG motif in triplicate. Similar to the GxxxG mutants, NSAIDs like sulindac sulfide and flurbiprofen were reported to affect the generation of different Aβ species (Weggen et al, 2001; Eriksen et al, 2003). Depending on the specific NSAID tested, Aβ42 levels were reduced and Aβ38 levels selectively increased or vice versa (Kukar et al, 2005). As shown for conservative GxxxG mutations, most NSAIDs left the Aβ40 levels unaffected. Hence, we conclude from our data that some NSAIDs might directly affect the dimerization strength of the substrate β-CTF. However, any effects of NSAIDs on GxxxG motifs of TMS-7 of PS-1 or TMS-4 or TMS-6 of APH-1 altering the active site conformation might also be possible (Lleo et al, 2004).
Figure 6.

Model of the γ-secretase cleavage mechanism on dimeric APP TMSs. (A) Sequential γ-secretase cleavages proceed from the C terminal end to the N terminus. The ɛ- and ζ-cleavage are suggested to occur independently of the dimeric or monomeric form of the substrate (green scissors). G29 and G33 form the cross point of two APP–TMS helices and represent a sterical hindrance for the proceeding γ-secretase such that the main final cleavages occur after residue 42/40 and produce mainly Aβ42 and Aβ40 (orange scissors). Thus, the following cleavages only generate Aβ38 and Aβ37 if not inhibited by the helix–helix interactions of the dimer (red scissors). (B) Perturbed dimers (indicated by G29/G33 to alanine substitutions) resolve the sterical hindrance and allow the γ-secretase to proceed to further N-terminal sites, yielding Aβ38 and Aβ37, and Aβ34 and Aβ35 (green scissors). Thus, the site of the final cleavage is determined by the strength of interaction of the two helices of two APP molecules mediated by the TMS residues G29 and G33.
In the case of APP, dimerization of TMS through the residues G29 and G33 of the GxxxG motifs determines where the final γ-secretase cleavage site might occur. This advanced model predicts that γ-secretase is not only involved in AICD production (Sastre et al, 2001) but has also a function in Aβ clearance by cleaving off turn by turn until more hydrophilic forms of Aβ are released from the membrane, such as Aβ38/Aβ37 and Aβ35/Aβ34. Taken together, we conclude that a strong dimerization of the substrate β-CTF is central to AD as any events stabilizing dimerization would increase the production of Aβ42. It could be considered that AD risk factors like high cholesterol levels or disturbed lipid homeostasis, oxidative stress, disturbed metal homeostasis or familial AD mutations in PS1 and APP affect the dimer stability of β-CTF and thus influence Aβ production. Compounds targeting the dimerization of the APP TMS and interfering with the G29xxxG33 interaction motif of two γ-secretase substrate molecules may be useful as therapeutics to prevent generation of Aβ42.
Materials and methods
Plasmids and transfections
Generation of ToxR system plasmids has been described (Langosch et al, 1996). Briefly, the TMS under study was fused between the MBP and the ToxR transcription activator domain using a cassette cloning approach.
APP695 with N-terminal Myc tag and C-terminal Flag tag or SPA4CT with C-terminal Flag tag were used as a template to introduce L17C, G25A, G29A, I31V, I32V, G33A, L34A, G29/33A or G33I mutations by site-directed mutagenesis. The cDNAs were inserted into pCEP4 (Invitrogen), which contains a hygromycine resistance gene. For transient or stable expression, plasmids (2 μg) were transfected into SH-SY5Y cell line (ATCC number: CRL-2266) using Transfectine (Bio-Rad) following the manufacturer's instructions. All sequences were confirmed by dideoxy sequencing.
ToxR assay
The TMS under study is fused between the MBP and the ToxR transcription activator domain and inserted into the inner membrane of FHK12 cells. The TMS-driven dimerization leads to close proximity of the ToxR domains, which are able to initiate transcription of the lacZ gene encoding β-galactosidase only in the dimeric state. Aliquots of E. coli FHK12 cells expressing chimeric constructs were transferred to 96-well dishes, lysed and β-galactosidase activity was measured using the substrate ortho-nitrophenyl-β-D-galactopyranoside (Langosch et al, 1996).
Molecular modeling
For both helices, five different, non-symmetrical starting conformations with identical backbone but varying random side-chain conformations were generated. From each starting conformation, 65 000 structures were generated by randomly rotating the helix 0–360° around the long axis; translating it −2 to +6 Å toward or away from geometric center, respectively; translating each helix ±10 Å along its long axis; and tilting the helices relative to the diad axis by ±20°. A dielectric constant of 20 was used for the electrostatics. An energy map was created for the rotation, tilt, longitudinal and translational shifts independently. The rotation was sampled in 36° bins, the tilt in 2.5° bins, the longitudinal shift in 2 Å bins and the translational shift was sampled in 0.8 Å bins. The average energy of each bin was evaluated independently by using Boltzmann averaging of all conformations within this bin. The energy map was graphically evaluated and a symmetric structure was assumed so that energy minima corresponding to asymmetric structures were disregarded. A related procedure has been shown to reliably model helix bundles (Gottschalk, 2004).
FRET
FRET efficiencies were determined using an acceptor photobleach protocol essentially as described previously (Voigt et al, 2005). HEK293 cells were transiently transfected with plasmids encoding CFP and YFP fusions of APP using Fugene 6 transfection reagent (Roche Molecular Biochemicals). For competition FRET experiments, the amount of plasmid encoding fluorescent proteins was reduced to allow addition of up to 1.5 μg of empty vector or plasmid encoding the competitor. Expression plasmids were generated by in-frame ligation of APP695 cDNA in custom-made vectors pcDNA3-CFP and pcDNA3-YFP.
Sandwich ELISA, immunoprecipitation and Western blots
Stably or transiently transfected cells were plated at a density of 2 × 106 cells per 60-mm dish or 3.6 × 106 cells per 12-well dish. The day after splitting, 3 ml or 350 μl of fresh media was added and incubated for 24 h. For Aβ40- and Aβ42-specific ELISAs, 50 μl of media was analyzed according to the manufacturer's instructions (The Genetics company). The same protocol was applied to determine Aβ38 and sAPPα levels, except that the antibody BAP-29 (provided by M Brockhaus, Roche) or the anti-Myc antibody (Cell Signaling Technology) was used. For Western blot analysis, Aβ was precipitated from equal volumes of conditioned media with polyclonal rabbit anti-serum (generated against residues 1–40). Aliquots of conditioned media were directly analyzed for sAPP. For full-length APP, cells were lysed in a buffer containing 50 mM Tris, pH 7.4, 150 mM NaCl, 2 mM EDTA, 2% NP-40 and 2% Triton X-100. For detection of C-terminal fragments (β-CTF) of APP- and SPA4CT-transfected cells, cells were treated with γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl-L-alanyl)]-S-phenylglycine t-butyl ester (DAPT) for 24 h, lysed and equal amounts of protein were immunoprecipitated with a polyclonal rabbit anti-serum (directed against APP cytosolic domain). Samples were separated by SDS–PAGE, transferred to nitrocellulose and immunolabelled with either antibody W0-2 to Aβ residues 5–8 (Ida et al, 1996), 879 (sAPPβ, provided by P Paganetti, Novartis) or antibody 22C11 (Hilbich et al, 1993).
MALDI-MS
Aβ was immunoprecipitated from conditioned media with W0-2. Sepharose was washed first in PBS, then in the presence of high salt (500 mM NaCl) followed by PBS and finally 100 mM ammonium chloride (pH 7.4). Aβ was eluted twice with 500 μl of 50% acetic acid and vacuum-dried. The sample was resuspended in 10 μl of 20% acetonitrile containing 0.1% tri-fluoric acetic acid and ultrasonicated. MALDI-MS analysis was carried out on sinapinic acid matrix with an UltraflexII TOF/TOF (Bruker Daltonics).
Supplementary Material
Supplementary Information
Acknowledgments
We thank P Paganetti (Novartis) for kindly providing the antibody 879, M Brockhaus (Roche) for kindly providing the Aβ38-specific antibody BAP-29, SO Deininger (Bruker Daltonics) for MALDI-MS measurements and the Deutsche Forschungs Gemeinschaft (DFG) and the Hans und Ilse Breuer Stiftung for financial support.
References
- Aller SG, Eng ET, De Feo CJ, Unger VM (2004) Eukaryotic CTR copper uptake transporters require two faces of the third transmembrane domain for helix packing, oligomerization, and function. J Biol Chem 279: 53435–53441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annaert W, De Strooper B (2002) A cell biological perspective on Alzheimer's disease. Annu Rev Cell Dev Biol 18: 25–51 [DOI] [PubMed] [Google Scholar]
- Arselin G, Giraud MF, Dautant A, Vaillier J, Brethes D, Coulary-Salin B, Schaeffer J, Velours J (2003) The GxxxG motif of the transmembrane domain of subunit e is involved in the dimerization/oligomerization of the yeast ATP synthase complex in the mitochondrial membrane. Eur J Biochem 270: 1875–1884 [DOI] [PubMed] [Google Scholar]
- Beher D, Hesse L, Masters CL, Multhaup G (1996) Regulation of amyloid protein precursor (APP) binding to collagen and mapping of the binding sites on APP and collagen type I. J Biol Chem 271: 1613–1620 [DOI] [PubMed] [Google Scholar]
- Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB (2003) Amyloid beta-protein (Abeta) assembly: Abeta 40 and Abeta 42 oligomerize through distinct pathways. Proc Natl Acad Sci USA 100: 330–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS (1996) Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1–42/1–40 ratio in vitro and in vivo. Neuron 17: 1005–1013 [DOI] [PubMed] [Google Scholar]
- Brosig B, Langosch D (1998) The dimerization motif of the glycophorin A transmembrane segment in membranes: importance of glycine residues. Protein Sci 7: 1052–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes S, Gonzalez-Duarte R, Marfany G (2001) Homodimerization of presenilin N-terminal fragments is affected by mutations linked to Alzheimer's disease. FEBS Lett 505: 81–86 [DOI] [PubMed] [Google Scholar]
- Cervantes S, Saura CA, Pomares E, Gonzalez-Duarte R, Marfany G (2004) Functional implications of the presenilin dimerization: reconstitution of gamma-secretase activity by assembly of a catalytic site at the dimer interface of two catalytically inactive presenilins. J Biol Chem 279: 36519–36529 [DOI] [PubMed] [Google Scholar]
- Chandu D, Huppert SS, Kopan R (2006) Analysis of transmembrane domain mutants is consistent with sequential cleavage of Notch by gamma-secretase. J Neurochem 96: 228–235 [DOI] [PubMed] [Google Scholar]
- Chen YR, Glabe CG (2006) Distinct early folding and aggregation properties of alzheimer amyloid-beta peptide Abeta 40 and Abeta 42: Stable trimer or tetramer formation by Abeta 42. J Biol Chem 281: 24414–24422 [DOI] [PubMed] [Google Scholar]
- Dyrks T, Dyrks E, Masters C, Beyreuther K (1992) Membrane inserted APP fragments containing the beta A4 sequence of Alzheimer's disease do not aggregate. FEBS Lett 309: 20–24 [DOI] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C (2003) Reconstitution of gamma-secretase activity. Nat Cell Biol 5: 486–488 [DOI] [PubMed] [Google Scholar]
- Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, Golde TE (2003) NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Invest 112: 440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson KM, Darling PJ, Mohan MJ, Macatee TL, Lemmon MA (2000) Extracellular domains drive homo- but not hetero-dimerization of erbB receptors. EMBO J 19: 4632–4643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk KE (2004) Structure prediction of small transmembrane helix bundles. J Mol Graph Model 23: 99–110 [DOI] [PubMed] [Google Scholar]
- Goutte C, Tsunozaki M, Hale VA, Priess JR (2002) APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc Natl Acad Sci USA 99: 775–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilbich C, Monning U, Grund C, Masters CL, Beyreuther K (1993) Amyloid-like properties of peptides flanking the epitope of amyloid precursor protein-specific monoclonal antibody 22C11. J Biol Chem 268: 26571–26577 [PubMed] [Google Scholar]
- Huber O, Kemler R, Langosch D (1999) Mutations affecting transmembrane segment interactions impair adhesiveness of E-cadherin. J Cell Sci 112 (Part 23): 4415–4423 [DOI] [PubMed] [Google Scholar]
- Ida N, Hartmann T, Pantel J, Schroder J, Zerfass R, Forstl H, Sandbrink R, Masters CL, Beyreuther K (1996) Analysis of heterogeneous A4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J Biol Chem 271: 22908–22914 [DOI] [PubMed] [Google Scholar]
- Itoh R, Fujiki Y (2006) Functional domains and dynamic assembly of the peroxin pex14p, the entry site of matrix proteins. J Biol Chem 281: 10196–10205 [DOI] [PubMed] [Google Scholar]
- Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y (1994) Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron 13: 45–53 [DOI] [PubMed] [Google Scholar]
- Jarrett JT, Lansbury PT Jr (1993) Seeding ‘one-dimensional crystallization' of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell 73: 1055–1058 [DOI] [PubMed] [Google Scholar]
- Kukar T, Murphy MP, Eriksen JL, Sagi SA, Weggen S, Smith TE, Ladd T, Khan MA, Kache R, Beard J, Dodson M, Merit S, Ozols VV, Anastasiadis PZ, Das P, Fauq A, Koo EH, Golde TE (2005) Diverse compounds mimic Alzheimer disease-causing mutations by augmenting Abeta42 production. Nat Med 11: 545–550 [DOI] [PubMed] [Google Scholar]
- Langosch D, Brosig B, Kolmar H, Fritz HJ (1996) Dimerisation of the glycophorin A transmembrane segment in membranes probed with the ToxR transcription activator. J Mol Biol 263: 525–530 [DOI] [PubMed] [Google Scholar]
- Lee SF, Shah S, Yu C, Wigley WC, Li H, Lim M, Pedersen K, Han W, Thomas P, Lundkvist J, Hao YH, Yu G (2004) A conserved GXXXG motif in APH-1 is critical for assembly and activity of the gamma-secretase complex. J Biol Chem 279: 4144–4152 [DOI] [PubMed] [Google Scholar]
- Lichtenthaler SF, Beher D, Grimm HS, Wang R, Shearman MS, Masters CL, Beyreuther K (2002) The intramembrane cleavage site of the amyloid precursor protein depends on the length of its transmembrane domain. Proc Natl Acad Sci USA 99: 1365–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lleo A, Berezovska O, Herl L, Raju S, Deng A, Bacskai BJ, Frosch MP, Irizarry M, Hyman BT (2004) Nonsteroidal anti-inflammatory drugs lower Abeta42 and change presenilin 1 conformation. Nat Med 10: 1065–1066 [DOI] [PubMed] [Google Scholar]
- Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK (2002) A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J 21: 1948–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain MS, Iwamoto H, Cao P, Vinion-Dubiel AD, Li Y, Szabo G, Shao Z, Cover TL (2003) Essential role of a GXXXG motif for membrane channel formation by Helicobacter pylori vacuolating toxin. J Biol Chem 278: 12101–12108 [DOI] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T (2005) Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 47: 191–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendrola JM, Berger MB, King MC, Lemmon MA (2002) The single transmembrane domains of ErbB receptors self-associate in cell membranes. J Biol Chem 277: 4704–4712 [DOI] [PubMed] [Google Scholar]
- Miyahara M, Nakanishi H, Takahashi K, Satoh-Horikawa K, Tachibana K, Takai Y (2000) Interaction of nectin with afadin is necessary for its clustering at cell–cell contact sites but not for its cis dimerization or trans interaction. J Biol Chem 275: 613–618 [DOI] [PubMed] [Google Scholar]
- Overton MC, Chinault SL, Blumer KJ (2003) Oligomerization, biogenesis, and signaling is promoted by a glycophorin A-like dimerization motif in transmembrane domain 1 of a yeast G protein-coupled receptor. J Biol Chem 278: 49369–49377 [DOI] [PubMed] [Google Scholar]
- Polgar O, Robey RW, Morisaki K, Dean M, Michejda C, Sauna ZE, Ambudkar SV, Tarasova N, Bates SE (2004) Mutational analysis of ABCG2: role of the GXXXG motif. Biochemistry 43: 9448–9456 [DOI] [PubMed] [Google Scholar]
- Qi-Takahara Y, Morishima-Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, Kametani F, Maeda M, Saido TC, Wang R, Ihara Y (2005) Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. J Neurosci 25: 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, Mar L, Sorbi S, Nacmias B, Piacentini S, Amaducci L, Chumakov I, Cohen D, Lannfelt L, Fraser PE, Rommens JM, St-George-Hyslop PH (1995) Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature 376: 775–778 [DOI] [PubMed] [Google Scholar]
- Rossjohn J, Cappai R, Feil SC, Henry A, McKinstry WJ, Galatis D, Hesse L, Multhaup G, Beyreuther K, Masters CL, Parker MW (1999) Crystal structure of the N-terminal, growth factor-like domain of Alzheimer amyloid precursor protein. Nat Struct Biol 6: 327–331 [DOI] [PubMed] [Google Scholar]
- Russ WP, Engelman DM (1999) TOXCAT: a measure of transmembrane helix association in a biological membrane. Proc Natl Acad Sci USA 96: 863–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ WP, Engelman DM (2000) The GxxxG motif: a framework for transmembrane helix–helix association. J Mol Biol 296: 911–919 [DOI] [PubMed] [Google Scholar]
- Sardi SP, Murtie J, Koirala S, Patten BA, Corfas G (2006) Presenilin-dependent ErbB4 nuclear signaling regulates the timing of astrogenesis in the developing brain. Cell 127: 185–197 [DOI] [PubMed] [Google Scholar]
- Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, Teplow DB, Haass C (2001) Presenilin-dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep 2: 835–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuermann S, Hambsch B, Hesse L, Stumm J, Schmidt C, Beher D, Bayer TA, Beyreuther K, Multhaup G (2001) Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer's disease. J Biol Chem 276: 33923–33929 [DOI] [PubMed] [Google Scholar]
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S (1996) Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med 2: 864–870 [DOI] [PubMed] [Google Scholar]
- Schmechel A, Zentgraf H, Scheuermann S, Fritz G, Pipkorn R, Reed J, Beyreuther K, Bayer TA, Multhaup G (2003) Alzheimer beta-amyloid homodimers facilitate A beta fibrillization and the generation of conformational antibodies. J Biol Chem 278: 35317–35324 [DOI] [PubMed] [Google Scholar]
- Schroeter EH, Ilagan MX, Brunkan AL, Hecimovic S, Li YM, Xu M, Lewis HD, Saxena MT, De Strooper B, Coonrod A, Tomita T, Iwatsubo T, Moore CL, Goate A, Wolfe MS, Shearman M, Kopan R (2003) A presenilin dimer at the core of the gamma-secretase enzyme: insights from parallel analysis of Notch 1 and APP proteolysis. Proc Natl Acad Sci USA 100: 13075–13080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senes A, Engel DE, DeGrado WF (2004) Folding of helical membrane proteins: the role of polar, GxxxG-like and proline motifs. Curr Opin Struct Biol 14: 465–479 [DOI] [PubMed] [Google Scholar]
- Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CE III, Sudhof T, Yu G (2005) Nicastrin functions as a gamma-secretase-substrate receptor. Cell 122: 435–447 [DOI] [PubMed] [Google Scholar]
- Soba P, Eggert S, Wagner K, Zentgraf H, Siehl K, Kreger S, Lower A, Langer A, Merdes G, Paro R, Masters CL, Muller U, Kins S, Beyreuther K (2006) Homo- and hetero-dimerization of APP family members promotes intercellular adhesion. EMBO J 25: 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G, Adachi A (2000) Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol Cell 6: 625–636 [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS (1996) Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17: 181–190 [DOI] [PubMed] [Google Scholar]
- Troyanovsky RB, Sokolov E, Troyanovsky SM (2003) Adhesive and lateral E-cadherin dimers are mediated by the same interface. Mol Cell Biol 23: 7965–7972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M (1999) Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286: 735–741 [DOI] [PubMed] [Google Scholar]
- Voigt P, Brock C, Nurnberg B, Schaefer M (2005) Assigning functional domains within the p101 regulatory subunit of phosphoinositide 3-kinase gamma. J Biol Chem 280: 5121–5127 [DOI] [PubMed] [Google Scholar]
- Vooijs M, Schroeter EH, Pan Y, Blandford M, Kopan R (2004) Ectodomain shedding and intramembrane cleavage of mammalian Notch proteins is not regulated through oligomerization. J Biol Chem 279: 50864–50873 [DOI] [PubMed] [Google Scholar]
- Wang R, Wang B, He W, Zheng H (2006) Wild-type presenilin 1 protects against Alzheimer disease mutation-induced amyloid pathology. J Biol Chem 281: 15330–15336 [DOI] [PubMed] [Google Scholar]
- Wang Y, Ha Y (2004) The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol Cell 15: 343–353 [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH (2001) A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414: 212–216 [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Ozols V, Fauq A, Golde TE, Koo EH (2003) Evidence that nonsteroidal anti-inflammatory drugs decrease amyloid beta 42 production by direct modulation of gamma-secretase activity. J Biol Chem 278: 31831–31837 [DOI] [PubMed] [Google Scholar]
- Weidemann A, Eggert S, Reinhard FB, Vogel M, Paliga K, Baier G, Masters CL, Beyreuther K, Evin G (2002) A novel epsilon-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry 41: 2825–2835 [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ (1999) Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 398: 513–517 [DOI] [PubMed] [Google Scholar]
- Zhao G, Cui MZ, Mao G, Dong Y, Tan J, Sun L, Xu X (2005) Gamma-Cleavage is dependent on zeta-cleavage during the proteolytic processing of amyloid precursor protein within its transmembrane domain. J Biol Chem 280: 37689–37697 [DOI] [PubMed] [Google Scholar]
- Zhao G, Mao G, Tan J, Dong Y, Cui MZ, Kim SH, Xu X (2004) Identification of a new presenilin-dependent zeta-cleavage site within the transmembrane domain of amyloid precursor protein. J Biol Chem 279: 50647–50650 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information