

Abstract
We examined interactions in a soluble tapasin (TPN)/HLA-B*0801 complex to gain mechanistic insights into the functions of TPN. Results show that TPN acts as a chaperone by increasing the ratio of active-to-inactive peptide-deficient HLA-B*0801 molecules in solution. TPN causes peptides to associate and dissociate faster owing to its effect on widening the binding groove of HLA-B*0801 molecules. Our data indicate that a TPN-assisted mechanism of peptide selection relies on disruption of conserved hydrogen bonds at the C-terminal end of the groove. Peptide sequence-dependent interactions along the entire length of the groove also play a role in this mechanism. We suggest that TPN influences presentation of antigenic peptides according to a mechanistically complicated process in which bound candidate peptides that are unable to conformationally disengage TPN from class I molecules are excluded from the repertoire. Overall, these studies unify our understanding of the functions of TPN.
Keywords: class I assembly complex, class I assembly pathway, peptide kinetics, peptide repertoire, tapasin
Introduction
The cell-surface presentation of antigenic peptides by class I major histocompatibility (MHC) molecules is the culmination of a complex series of events that take place in the endoplasmic reticulum (ER) and involve specialized proteins: calnexin, calreticulin, tapasin (TPN), ERp57, and the transporter associated with antigen processing (TAP). The recognition of class I/peptide molecules by CD8+ TCRs is important for controlling pathological states such as viral infection and tumor progression.
Evidence have been provided that TPN enhances the ability of TAP to transport peptides into the ER (Lehner et al, 1998). It was also suggested that TPN stabilizes the peptide-deficient, open form of class I molecules (Ortmann et al, 1997; Schoenhals et al, 1999; Barnden et al, 2000). Other experiments suggested that TPN edits the peptide repertoire in favor of high-affinity binders (Barnden et al, 2000; Garbi et al, 2000; Barber et al, 2001; Williams et al, 2002). This function was attributed to TPN based on findings that class I/peptide molecules expressed on the surface of TPN-deficient cells were less stable than those expressed on the surface of normal cells. However, an analysis of peptides eluted from class I molecules on normal and TPN-deficient cells indicated that no correlation could be established between the decreased stability of class I/peptide molecules in TPN-deficient cells and the binding of low-affinity peptides (Zarling et al, 2003). Instead, the authors suggested that the apparent ability of TPN to stabilize conformationally immature class I molecules in the ER broadens the repertoire of bound peptides in terms of both its complexity and binding affinities (Zarling et al, 2003). More recently, a key role for TPN in shaping the cell-surface presentation of peptides according to their intrinsic half-lives has also been demonstrated (Howarth et al, 2004). At present, although it is uncontested that TPN plays a vital role in the class I antigen presentation pathway, a unifying understanding of the function of TPN is lacking.
Previous kinetics studies of class I/peptide molecules carried out in the absence of TPN have contributed greatly in elucidating mechanistic pathways for the assembly/disassembly of these molecules (Springer et al, 1998; Gakamsky et al, 2000; Binz et al, 2003). Here, we carried out a rigorous molecular analysis of interactions in a recombinant, soluble TPN/class I complex to develop a mechanistic understanding of the functions of TPN. For this, HLA-B*0801 was selected, as this allele was shown to be dependent on TPN for normal cell-surface expression (Peh et al, 1998). Moreover, a large number of antigenic peptides restricted to HLA-B*8 have been reported (Burrows et al, 1992; DiBrino et al, 1994), allowing to establish an interesting pool of peptides for our studies. We used a strategy in which the ER-lumenal domains of TPN and HLA-B*0801 heavy chain were fused at their C-termini to Jun and Fos leucine peptides, respectively. Based on our results, we propose that in the class I assembly complex, TPN stabilizes the open forms of peptide-deficient and peptide-filled class I molecules, thereby providing an energy barrier that bound candidate peptides must overcome in order to induce maturing conformational changes in the class I binding groove.
Results
Weak interaction between the ER-lumenal domains of TPN and HLA-B*0801 molecules
Interaction between recombinant, soluble forms of TPN and HLA-B*0801 molecules was monitored by native gel (Figure 1A). Results show that incubation of TPN with peptide-filled (loaded with the FLRGRAYGL peptide from Epstein–Barr virus; Burrows et al, 1992) and peptide-deficient HLA-B*0801 molecules (1:1 molar ratio) failed to produce new bands (lanes 4 and 5, respectively) that migrated distinctively from the uncomplexed proteins (lanes 1–3). Analytical ultracentrifugation and gel filtration chromatography similarly failed to detect interaction between soluble TPN and peptide-filled and peptide-deficient HLA-B*0801 molecules (data not shown). We also examined by fluorescence anisotropy the effect of soluble TPN on dissociation of the short-lived EIYK*RWIIL peptide, derived from HIV-1 (DiBrino et al, 1994), from HLA-B*0801 (Figure 1B). The decay curve for the mixture of soluble TPN and HLA-B*0801/EIYK*RWIIL is essentially identical to that of HLA-B*0801/EIYK*RWIIL alone. Together, these results suggest weak interaction between the ER-lumenal domains of TPN and class I molecules, a conclusion consistent with the transient nature of the class I assembly complex in the ER. To circumvent the problem of weak protein–protein interaction, we used a strategy in which Jun and Fos leucine peptides are fused at the C-terminus of TPN and HLA-B*0801 heavy chain. A similar tethering of soluble HLA-DM and class II molecules led to a functional complex (Busch et al, 2002).
Figure 1.
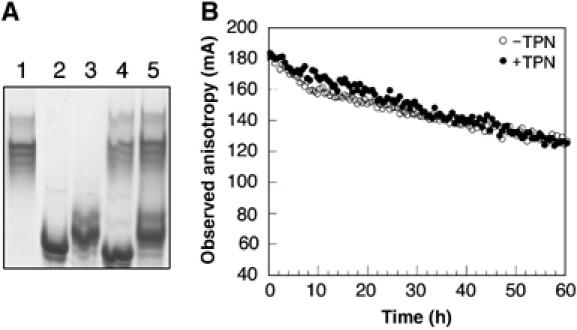
Interaction between recombinant, soluble TPN and HLA-B*0801. (A) TPN (10 μg) and HLA-B*0801 (10 μg) (1:1 molar ratio) were incubated on ice in 20 mM Tris, 150 mM NaCl, and 10% glycerol (pH 7.5) for 30 min. Samples were analyzed on a native gel (8%): lane 1, TPN; lane 2, peptide-filled HLA-B*0801; lane 3, peptide-deficient HLA-B*0801; lane 4, mixture of TPN and peptide-filled HLA-B*0801; and lane 5, mixture of TPN and peptide-deficient HLA-B*0801. The gel was run at 4°C in 25 mM Tris and 200 mM glycine (pH 8.3). (B) At time=0, HLA-B*0801 (40 nM) loaded with EIYK*RWIIL (○) was added under stirring to a 1-cm cuvette containing 1000-fold molar excess of the nontagged peptide and 100-fold molar excess of β2m in 20 mM Hepes and 150 mM NaCl (pH 7.5). Peptide dissociation was monitored at 20°C (excitation and emission wavelengths were 495 and 524 nm, respectively). The experiment was repeated in the presence of TPN (400 nM) (•), which was pre-mixed in the buffer before adding HLA-B*0801. Data points were collected for 60 h.
Characterization of TPNjun and HLA-B*0801fos molecules
The ER-lumenal domain (residues 1–392) of TPN (Figure 2A), extended at its C-terminus with a short linker, thrombin site, Jun peptide, and (His)6 sequence, was engineered as described in Materials and methods (hereafter referred to as TPNjun). The GGSGG linker was inserted to increase rotational freedom, the thrombin site to permit release of the protein, and the (His)6 sequence to facilitate purification. TPNjun was purified (Figure 2B, lanes 1–7) to moderate yields (∼0.5 mg/l culture) (Chen et al, 2002). Purified TPNjun eluted from the gel filtration column (Figure 2C) as a single peak with no sign of aggregates. Note that TPNjun migrated slightly faster than expected based on its molecular weight (50 kDa); this may be explained by the weak tendency of Jun to homodimerize in solution (O'Shea et al, 1989). This behavior may also account for the smear produced by TPNjun on native gel (Figure 2D, lane 1). Importantly, note that soluble TPN migrates as a series of closely spaced bands on native gel (lane C; see also Figure 1, lane 1).
Figure 2.
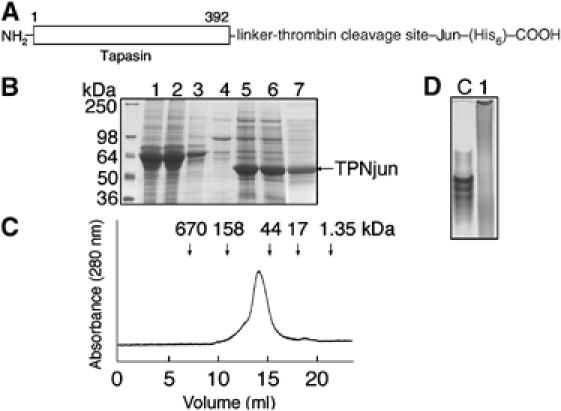
Characterization of TPNjun. (A) The ER-lumenal domain of TPN was extended at its C-terminus with a linker (GGSGG), a thrombin site, a Jun leucine peptide, and a (His)6 sequence. (B) SDS–PAGE (12%) analysis of TPNjun. Lane 1, supernatant from High Five insect cells at 60 h after infection; lane 2, flow-through after washing column with 10 mM imidazole; lane 4, flow-through after washing column with 25 mM imidazole; lane 5, Ni-NTA matrix beads showing bound TPNjun; lane 6, flow-through after washing column with 250 mM imidazole to elute TPNjun; and lane 7, TPNjun after purification on a Superdex 200 HR 10/300 column at 10°C in 20 mM Tris and 150 mM NaCl (pH 7.5). (C) Gel filtration chromatogram of purified TPNjun (see (B) for conditions). The column was calibrated using protein standards that elute at positions corresponding to arrows at the top of the chromatogram. (D) Native PAGE (8%) analysis of purified TPNjun (lane 1) showing a protein smear while soluble TPN (lane C) migrates as a series of closely spaced bands.
The ER-lumenal domain (residues 1–275) of HLA-B*0801 heavy chain (Figure 3A), extended at its C-terminus with a linker, thrombin site, and Fos peptide, was engineered as described in Materials and methods. The HLA-B*0801fos heavy chain was expressed as inclusion bodies (Figure 3B), which were purified as described previously (Bouvier and Wiley, 1998). HLA-B*0801fos heavy chain was refolded in the presence of β2m and a synthetic peptide (see Materials and methods). Analysis of an aliquot of purified peptide-filled HLA-B*0801fos molecules by gel filtration chromatography (Figure 3C) revealed a major peak with no sign of aggregates. Note that peptide-filled HLA-B*0801fos molecules eluted from the gel filtration column with a slightly larger size than expected based on its molecular weight (50 kDa). Here again, the very weak tendency of the Fos peptide to homodimerize (O'Shea et al, 1989) may account for this behavior. Peptide-deficient HLA-B*0801fos molecules were generated from the chemical denaturation of peptide-filled molecules as described previously (Bouvier and Wiley, 1998). The activity of peptide-deficient molecules varied between 10 and 60% (see Materials and methods).
Figure 3.
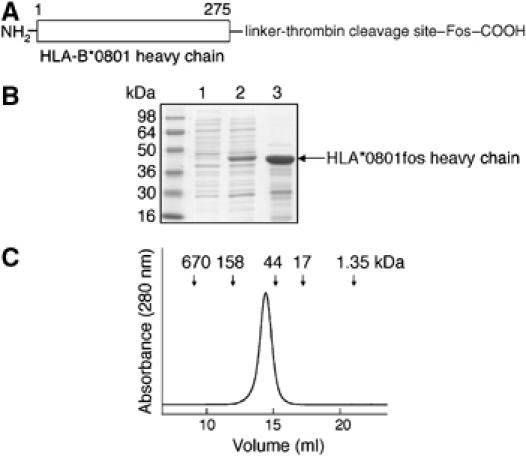
Characterization of the ER-lumenal domain of HLA-B*0801fos heavy chain. (A) HLA-B*0801 heavy chain was extended at its C-terminus with a linker (GGSGG), a thrombin site, and a Fos leucine peptide. (B) SDS–PAGE (12%) analysis of HLA-B*0801fos heavy chain from bacterial cells. Lane 1, before induction; lane 2, after induction with IPTG; and lane 3, purified inclusion bodies. (C) Gel filtration chromatogram of purified peptide-filled HLA-B*0801fos (see Figure 2B for conditions).
Formation of the TPNjun/HLA-B*0801fos complex
The zippering of TPNjun and HLA-B*0801fos molecules was monitored by native gel electrophoresis (Figure 4). TPNjun (lane 1) was incubated with peptide-filled (loaded with FLRGRAYGL) (lane 2) or peptide-deficient (lane 3) HLA-B*0801fos molecules (1:1 molar ratio) yielding new bands (lane 4 and 5, respectively). These bands were analyzed by N-terminal amino-acid sequencing and results confirmed the presence of four components in lane 4—TPNjun (G1PAVIE( )WFV), HLA-B*0801fos heavy chain (G1SHSM(R)Y), β2m (M1IQRTPK), and peptide (F1L(R)GR(A)Y))—and of two proteins in lane 5—TPNjun (G1PAVIE( )WFV) and HLA-B*0801fos heavy chain (G1SHSM(R)Y). It was expected that β2m would be absent in the new band of lane 5 owing to the intrinsic instability of peptide-deficient class I molecules, causing β2m to dissociate during electrophoresis. Thus, results from native gel unambiguously show that TPNjun spontaneously paired with both peptide-filled and peptide-deficient HLA-B*0801fos molecules.
Figure 4.
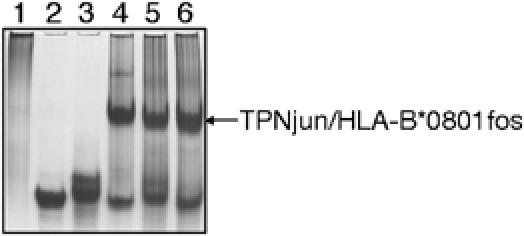
Formation of the TPNjun/HLA-B*0801fos complex. TPNjun (10 μg) and HLA-B*0801fos (10 μg) (1:1 molar ratio) were incubated as described in Figure 1A. Samples were analyzed on native gel (8%): lane 1, TPNjun; lane 2, peptide-filled HLA-B*0801fos; lane 3, peptide-deficient HLA-B*0801fos; lane 4, mixture of TPNjun and peptide-filled HLA-B*0801fos; lane 5, mixture of TPNjun and peptide-deficient HLA-B*0801fos; and lane 6, TPNjun/peptide-deficient HLA-B*0801fos complex incubated with an excess of FLRGRAYGL. The gel was run as described in Figure 1A.
For the purpose of our studies, we confirmed that peptide-deficient HLA-B*0801fos molecules associated with TPNjun can bind peptides (Figure 4). The incubation of an excess of FLRGRAYGL with the TPNjun/peptide-deficient HLA-B*0801fos complex yielded a new band (lane 6), which, after analysis by N-terminal amino-acid sequencing, confirmed the presence of TPNjun (G1PAVIEYWFV), HLA-B*0801fos heavy chain (G1SHSMRYFDT), β2m (M1IQRTP), and the peptide (F1L(R)G(R)AY). These results ascertain that the TPNjun/peptide-deficient HLA-B*0801fos complex can effectively bind peptides.
Influence of TPNjun on peptide dissociation kinetics
The effect of TPNjun on the kinetics of peptide dissociation from HLA-B*0801fos molecules was monitored by fluorescence anisotropy (Figure 5). The decay curve of the short-lived EIYK*RWIIL peptide obtained in the absence of TPNjun (Figure 5A) is best described by two phases (Table I): a fast phase (7% of the reaction) with a dissociation half-life, τ1, of 2.4 h, and a slower phase with τ2=109 h (93% of the reaction). In the presence of TPNjun (Figure 5A), the anisotropy profile is considerably steeper and best described by a monophasic behavior with τ=1.4 h (Table I). These results indicate that (1) leucine peptides play an important role in mediating a functional interaction between the ER-lumenal domains of TPN and HLA-B*0801, (2) TPNjun converted the biphasic nature of the dissociation reaction to a monophasic dissociation pattern. This implies that TPNjun altered the dissociation pathway of EIYK*RWIIL (see Discussion), and (3) TPNjun accelerated the release of EIYK*RWIIL by 78-fold (Table I).
Figure 5.
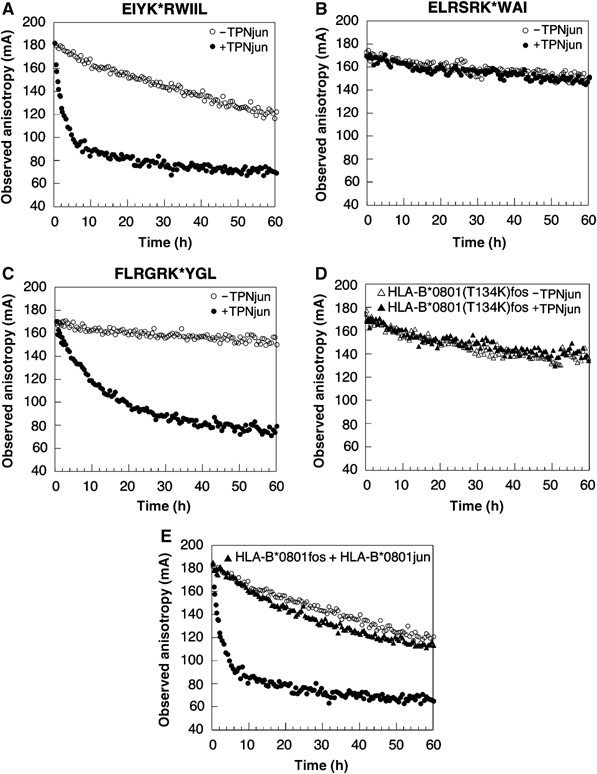
Effect of TPNjun on the kinetics of peptide dissociation. (A) Dissociation of EIYK*RWIIL was monitored as described in Figure 1B: HLA-B*0801fos alone (40 nM) (○) and in the presence of TPNjun (400 nM) (•). Data points were collected at various time points over a period of 166 h (−TPN) or continuously for 60 h (+TPN). (B) As described in Figure 1B for ELRSRK*WAI: HLA-B*0801fos alone (40 nM) (○) and in the presence of TPNjun (400 nM) (•). Data points were collected at various time points over a period of 576 h. (C) As described in Figure 1B for FLRGRK*YGL: HLA-B*0801fos alone (40 nM) (○) and in the presence of TPNjun (400 nM) (•). Data points were collected at various time points over a period of 546 h (−TPN) or continuously for 89 h (+TPN). (D) Dissociation of FLRGRK*YGL from HLA-B*0801(T134K)fos (40 nM) in the absence (▵) or presence (▴) of TPNjun (400 nM) was monitored as described in Figure 1B. Data points were collected continuously for 60 h. (E) Dissociation of EIYK*RWIIL from HLA-B*0801fos (40 nM) in the presence of HLA-B*0801jun/ELRSRKWAI (400 nM) (▴) was monitored as described in Figure 1B. Data points were collected continuously for 60 h. The decay curves shown in (A) are included for comparison purposes.
Table 1.
Kinetics of dissociation
| −TPNjun | +TPNjun | |||||
|---|---|---|---|---|---|---|
| Peptidesa | Phase | Amplitude | τ (h) | Amplitude | τ (h) | R.E.F.b |
| EIYK*RWIIL | 1 | 8.9±0.4 | 2.4±0.8 | 109.3±1.1 | 1.4±0.1 | 78 |
| 2 | 118.2±0.9 | 109±2 | ||||
| ELRSRK*WAI | 1 | 3.2±0.3 | 3.2±0.8 | 6.4±1.5 | 2.4±0.6 | − |
| 2 | 109.5±0.7 | 3210±107 | 112.3±0.7 | 2750±314 | ||
| FLRGRK*YGL | 1 | 8.3±1.7 | 19.3±5.8 | 113.3±1.1 | 9.6±0.5 | 104 |
| 2 | 100.8±2.2 | 1000±53 | ||||
| GLRSRK*WAV | 1 | 8.2±0.7 | 19.3±3.9 | 94.3±1.2 | 4.8±0.2 | 48 |
| 2 | 88.1±0.7 | 231±6 | ||||
| GLRSRK*WAI | 1 | 10.2±2.1 | 4.8±0.9 | 119.5±1.6 | 3.2±0.1 | 42 |
| 2 | 112.8±2.3 | 133±4 | ||||
| ELRSRK*WAV | 1 | 11.2±1.4 | 12.0±2.8 | 10.5±1.9 | 5.7±0.8 | − |
| 2 | 100.7±1.7 | 2940±340 | 106.6±1.3 | 2410±300 | ||
| DPKVKK*WPL | 1 | 10.3±1.4 | 19.3±7.3 | 12.6±1.4 | 34.4±8.7 | − |
| 2 | 115.9±1.2 | 1280±80 | 113.1±1.6 | 1480±80 | ||
| Ndel | 1 | 4.9±0.6 | 1.0±0.3 | 103.0±1.2 | 0.60±0.04 | 47 |
| 2 | 89.5±0.4 | 28.3±0.8 | ||||
| Cdel | 1 | 50.8±2.3 | 0.80±0.05c | 119.9±1.4 | 0.40±0.01c | 17 |
| 2 | 69.2±0.2 | 6.8±0.3c | ||||
| a K* denotes fluorescein-derivatized lysine. | ||||||
| b Rate enhancement factor=τ(intrinsic; slow phase)/τ(TPNjun). | ||||||
| c In units of minutes. | ||||||
These studies were extended by examining the effect of TPNjun on the dissociation rates of two peptides with higher intrinsic kinetics stabilities: the ELRSRK*WAI peptide, derived from the nucleoprotein of influenza A (DiBrino et al, 1994), and FLRGRK*YGL. The anisotropy decay profiles obtained in the absence of TPNjun (Figures 5B and C, respectively) were best fitted to two phases (Table I): for ELRSRK*WAI, a fast phase with τ1=3.2 h (3% of the reaction) and a slower phase with τ2=3210 h (97% of the reaction); and for FLRGRK*YGL, a fast phase with τ1=19.3 h (7% of the reaction) and a slower phase with τ2=1000 h (93% of the reaction). In the presence of TPNjun, the anisotropy decay curve of ELRSRK*WAI (Figure 5B) was essentially unchanged and best described by biphasic kinetics with parameters similar to those obtained in the absence of TPNjun (Table I): a fast phase with τ1=2.4 h (5% of the reaction) and a slower phase with τ2=2750 h (95% of the reaction). In contrast, TPNjun converted the dissociation pattern of FLRGRK*YGL (Figure 5C) to monophasic with τ=9.6 h (Table I). From these data, it was determined that TPNjun accelerated the dissociation of FLRGRK*YGL by 104-fold. Thus, the effect of TPNjun toward FLRGRK*YGL is considerably more similar to its effect on the short-lived EIYK*RWIIL peptide than on ELRSRK*WAI. This lack of direct correlation between the intrinsic dissociation half-life of peptides and the magnitude of the rate enhancement factors was further examined with other peptides (Table I). We identified the GLRSRK*WAI peptide as having a similar intrinsic kinetics stability as EIYK*RWIIL, but yet displaying a TPNjun sensitivity similar to that of the longer-lived GLRSRK*WAV peptide. Thus, results from dissociation experiments show that (1) peptides are differentially sensitive to the action of TPNjun and (2) the magnitude of TPN effect cannot be predicted simply from the intrinsic dissociation half-life of peptides.
To demonstrate that the kinetics effects observed in the tethered complexes arise from native interactions between TPN and HLA-B*0801, we monitored peptide release from HLA-B*0801fos carrying a T134K mutation in the absence or presence of TPNjun. Previous studies have reported that a T134K mutation in HLA-A2 and H-2Ld abrogated interaction with class I assembly proteins (Peace-Brewer et al, 1996; Lewis and Elliott, 1998; Yu et al, 1999). Results show that the anisotropy decay curve corresponding to dissociation of FLRGRK*YGL from HLA-B*0801(T134K)fos in the presence of TPNjun was identical to the profile obtained in the absence of TPNjun (Figure 5D). This is consistent with TPN losing its ability to associate with HLA-B*0801(T134K) in the zippered complex. The mutations of MHC residues E128A and E222K, known to abolish association with class I assembly proteins (Suh et al, 1999; Paquet and Williams, 2002), were similarly tested and found to also inhibit the function of TPNjun (data not shown). Thus, these control experiments provide strong evidence that the effects of TPNjun on HLA-B*0801fos (Table I) result from specific interactions between TPN and HLA-B*0801. Finally, to demonstrate that the Jun/Fos linkage has no influence on interaction between TPN and HLA-B*0801, we monitored the release of EIYK*RWIIL from HLA-B*0801fos tethered to HLA-B*0801jun/ELRSRKWAI. Results show that the anisotropy decay curve corresponding to the release of EIYK*RWIIL in the presence of HLA-B*0801jun/ELRSRKWAI (Figure 5E) is markedly different from the profile obtained in the presence of TPNjun (panel A and reproduced in panel E). The decay curve is, instead, similar to that of HLA-B*0801fos/EIYK*RWIIL alone. This convincingly shows that the mere tethering of HLA-B*0801fos/EIYK*RWIIL to a Jun-fused protein has no consequence on the release of bound peptides. Thus, kinetics results obtained with TPNjun/HLA-B*0801fos complexes (Table I) are not artifacts of the Jun/Fos zipper.
Influence of TPNjun on peptide association kinetics
To examine if TPNjun also influenced the kinetics of peptide association onto peptide-deficient HLA-B*0801fos molecules, we monitored the binding of two relatively long-lived peptides: ELRSRK*WAI and FLRGRK*YGL (Figure 6). The anisotropy profiles of ELRSRK*WAI and FLRGRK*YGL obtained in the absence of TPNjun (Figures 6A and B, respectively) were fitted to monophasic kinetics (Table II): for ELRSRK*WAI, τ=1019 s with an amplitude of 54; and for FLRGRK*YGL, τ=447 s with an amplitude of 64. In the presence of TPNjun (Figure 6), the anisotropy profiles were best described by monophasic kinetics (Table II): for ELRSRK*WAI, τ=642 s with an amplitude of 81; and for FLRGRK*YGL, τ=322 s with an amplitude of 76. Thus, results show that TPNjun increased the rates of association of both peptides. Results also show that the amplitudes of the binding reactions were increased in the presence of TPNjun (Table II): for ELRSRK*WAI, the amplitude increased from 54 to 81 (57% increase); and for FLRGRK*YGL, the amplitude increased from 64 to 76 (19% increase). These effects are consistent with TPNjun increasing the concentration of active peptide-deficient HLA-B*0801fos molecules in solution.
Figure 6.
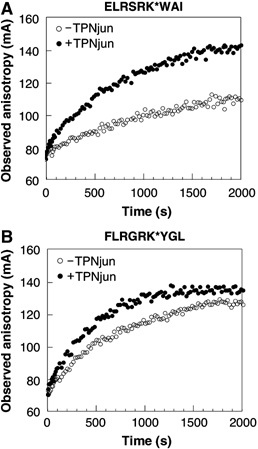
Effect of TPNjun on the kinetics of peptide association. (A) ELRSRK*WAI (160 nM) was mixed automatically with an equal volume of peptide-deficient HLA-B*0801fos (800 nM) (○) together with a 100-fold molar excess of β2m in 20 mM Hepes and 150 mM NaCl (pH 7.5). The time course of the reaction was monitored at 20°C. The binding of ELRSRK*WAI to peptide-deficient molecules in the presence of TPNjun (600 nM) (•) is shown. (B) As described in (A) for FLRGRK*YGL: peptide-deficient HLA-B*0801fos alone (800 nM) (○) and in the presence of TPNjun (600 nM) (•). For (A) and (B), data points were collected for 2000 s.
Table 2.
Kinetics of association
| −TPNjun | +TPNjun | |||
|---|---|---|---|---|
| Peptidesa | Amplitude | τ (s) | Amplitude | τ (s) |
| A | 53.9±1.7 | 1019±45 | 80.5±2.9 | 642±48 |
| B | 64.3±2.3 | 447±12 | 76.2±1.2 | 322±13 |
| a A=ELRSRK*WAI and B=FLRGRK*YGL, K* denotes fluorescein-derivatized lysine. | ||||
Do conserved hydrogen bonds at the N- and C-terminal ends of the binding groove play a role in the mechanism of action of TPNjun?
Because peptides have in common a NH2- and COOH-terminal group, it is possible that conserved hydrogen bonds at the N- and C-terminal ends of the groove may be an important feature of a TPN-assisted mechanism of peptide selection. This notion is reinforced by the critical contribution these conserved hydrogen bonds make to the overall stability of class I/peptide molecules (Bouvier and Wiley, 1994). To examine this question, we designed peptides in which their NH2- and COOH-terminal groups were individually substituted by a CH3 group as a way to abolish their hydrogen-bonding potential through these groups (referred to as Ndel and Cdel, respectively). Ndel and Cdel are modified from the control GLRSRK*WAV peptide, which in turn is derived from ELRSRK*WAI (see Materials and methods).
Anisotropy decay curves for GLRSRK*WAV, Ndel, and Cdel, were obtained in the absence or presence of TPNjun (Table I). Results show that in the absence of TPNjun, a biphasic pattern best described the decay profiles. As expected, the intrinsic kinetics stability of GLRSRK*WAV (τ2=231 h) is considerably higher than that of Ndel (τ2=28.3 h) and Cdel (τ2=6.8 min). Results in Table I also show that TPNjun converted the biphasic kinetics of these peptides into a monophasic pattern: GLRSRK*WAV (τ=4.8 h), Ndel (τ=0.6 h), and Cdel (τ=0.4 min) with rate enhancement factors of 48, 47, and 17, respectively (Table I). It is noteworthy that GLRSRK*WAV and Ndel are both similarly sensitive to the action of TPNjun in spite of their considerably different intrinsic dissociation half-lives. This suggests that TPNjun disrupted the same class I/peptide interactions to release GLRSRK*WAV and Ndel from HLA-B*0801fos. On the other hand, TPNjun accelerated the release of Cdel by only 17-fold (Table I). This suggests that dissociation of Cdel from HLA-B*0801fos was less dependent on TPNjun than the control GLRSRK*WAV peptide. In the context of our findings with Ndel, the data thus indicate that disruption of conserved hydrogen bonds at the C-terminal end of the groove plays a role in the mechanism of action of TPNjun. A role for hydrogen bonds at the C-terminal end of the groove in the function of TPN has been hypothesized previously (Wright et al, 2004).
A role for peptide sequence-dependent class I interactions in the mechanism of action of TPNjun
That the release of Cdel was nonetheless accelerated by 17-fold by TPNjun (Table I) suggests that disruption of peptide sequence-dependent class I interactions plays a role in the mechanism of peptide selection under the action of TPNjun. A similar observation can also be made by comparing kinetics parameters of the TPjun-insensitive ELRSRK*WAI peptide with those of the closely related TPjun-sensitive GLRSRK*WAV peptide (REF=48) (Table I); both peptides differ at P1 and P9 only, but yet exhibit significantly different sensitivity to TPNjun. To examine this point, we used a mutational approach in which ELRSRK*WAI was mutated at P1 to Gly (GLRSRK*WAI) and at P9 to Val (ELRSRK*WAV). The release of mutant peptides from HLA-B*0801fos was monitored as described above. Results (Table I) show that GLRSRK*WAI is sensitive to TPNjun (R.E.F.=42), but ELRSRK*WAV is insensitive to TPNjun. Thus, an EP1G mutation in ELRSRK*WAI rendered the peptide sensitive to TPNjun, but the more conservative IP9V mutation did not alter sensitivity to TPNjun. The differential TPN sensitivity of the closely related ELRSRK*WAI and GLRSRK*WAV peptides is therefore due to differences at the P1 rather than the P9 position. On the basis of these results, and those with Ndel and Cdel, we suggest that class I/peptide interactions along the entire length of the groove are likely to influence sensitivity to TPNjun.
A direct TPNjun-assisted peptide exchange assay
As a ‘proof-of-principle' that our findings are likely to have consequences on the peptide repertoire, we examined the effects of TPNjun on the dose-dependent binding of the TPNjun-sensitive EIYKRWIIL peptide to HLA-B*0801fos in the presence of the TPNjun-insensitive ELRSRK*WAI peptide. As the dissociation of EIYK*RWIIL from HLA-B*0801fos molecules was accelerated in the presence of TPNjun (Table I), this suggests that it should be more difficult for EIYKRWIIL to compete for binding against ELRSRK*WAI in the presence of TPNjun. The IC50 of EIYKRWIIL competing against ELRSRK*WAI should therefore increase in the presence of TPNjun. First, results (Table III) show that the IC50 of the control ELRSRKWAI peptide decreased from 0.18 to 0.09 μM in the presence of TPNjun (two-fold decrease). An explanation for this result may be that the binding of fluorescent ELRSRK*WAI to HLA-B*0801fos leads to molecules that are somewhat less kinetically stable relative to those formed with nontagged ELRSRKWAI. On the other hand, the IC50 of EIYKRWIIL increased from 0.19 to 1.13 μM (six-fold increase), consistent with EIYKRWIIL being a weaker competitor in the presence of TPNjun. This experiment, therefore, shows that TPNjun diminished the propensity (by 12-fold) of a TPNjun-sensitive peptide to occupy the groove of HLA-B*0801fos molecules. An implication of these results is that similar TPN-mediated effects in the ER are likely to influence the repertoire of peptides presented by class I molecules at the cell surface.
Table 3.
Effects of TPNjun on IC50
| IC50 (μM) | ||
|---|---|---|
| Peptides | −TPNjun | + TPNjun |
| ELRSRKWAI | 0.18±0.04 | 0.09±0.03 |
| EIYKRWIIL | 0.19±0.02 | 1.13±0.30 |
Discussion
Here we examined the molecular basis of TPN-mediated effects by undertaking an analysis of interactions in a soluble TPNjun/HLA-B*0801fos complex.
Mechanism of class I assembly/disassembly
Dissociation experiments carried out in the absence of TPNjun revealed that peptides obeyed biphasic kinetics. Biphasic behavior was previously interpreted to reflect peptide dissociation from two conformational states of peptide-filled molecules (equilibrium A) (Gakamsky et al, 2000): an open conformation ([HC·β2m·p]open) associated with the fast dissociation phase and a closed conformation ([HC·β2m·p]closed) associated with the slow dissociation phase.
Scheme I 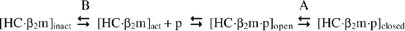 An examination of the relative amplitudes of the fast and slow phases (Table I) indicates that Cdel is the only peptide for which dissociation occurred from the open form of HLA-B*0801fos to a significant degree (τ1=0.8 min; 42% of the reaction). This means that the inability of Cdel to form conserved hydrogen bonds at the C-terminal end of the groove significantly hampered HLA-B*0801fos molecules to mature into a closed conformation. In contrast, the inability of Ndel to mediate conserved hydrogen bonds at the N-terminal end of the groove had essentially no effect on the propensity of HLA-B*0801fos to mature into a closed conformation, as evidenced by the second phase representing 95% of the reaction. An implication of these results is that the conformational equilibrium between peptide-filled, open and peptide-filled, closed (equilibrium A) is strongly dependent on the breaking/making of conserved hydrogen bonds at the C-terminal end of the groove. Importantly, that Cdel dissociated from somewhat more stable HLA-B*0801fos molecules (τ2=6.8 min; 58% of the reaction) suggests that class I/peptide interactions other than the conserved C-terminal hydrogen bonds influence equilibrium A. Such interactions are likely to include peptide sequence-dependent class I interactions along the groove, as presented above with mutant peptides.
An examination of the relative amplitudes of the fast and slow phases (Table I) indicates that Cdel is the only peptide for which dissociation occurred from the open form of HLA-B*0801fos to a significant degree (τ1=0.8 min; 42% of the reaction). This means that the inability of Cdel to form conserved hydrogen bonds at the C-terminal end of the groove significantly hampered HLA-B*0801fos molecules to mature into a closed conformation. In contrast, the inability of Ndel to mediate conserved hydrogen bonds at the N-terminal end of the groove had essentially no effect on the propensity of HLA-B*0801fos to mature into a closed conformation, as evidenced by the second phase representing 95% of the reaction. An implication of these results is that the conformational equilibrium between peptide-filled, open and peptide-filled, closed (equilibrium A) is strongly dependent on the breaking/making of conserved hydrogen bonds at the C-terminal end of the groove. Importantly, that Cdel dissociated from somewhat more stable HLA-B*0801fos molecules (τ2=6.8 min; 58% of the reaction) suggests that class I/peptide interactions other than the conserved C-terminal hydrogen bonds influence equilibrium A. Such interactions are likely to include peptide sequence-dependent class I interactions along the groove, as presented above with mutant peptides.
Previous studies showed that peptide-deficient class I molecules undergo an equilibrium between an inactive and active state (equilibrium B) that involves conformational changes (Gakamsky et al, 2000). The inactive state of peptide-deficient molecules is likely to represent molecules in which the binding groove is closed due to interactions between MHC residues and bound water molecules; crystallographic structures of class I/peptide molecules have indeed revealed bound water molecules in the groove (Ogata and Wodak, 2002). In that sense, equilibrium B between peptide-deficient, closed and peptide-deficient, open conformations is akin to equilibrium A between peptide-filled, closed and peptide-filled, open conformations, as pointed to previously (Khan et al, 2000).
TPNjun increases the rate of peptide dissociation by conformationally disrupting the binding groove of class I molecules
Results from kinetics dissociation experiments (Table I) revealed that TPNjun converted the dissociation process from biphasic to monophasic for all TPNjun-sensitive peptides. This indicates that TPNjun generated a conformationally homogeneous population of peptide-filled HLA-B*0801fos molecules in solution. Results also show that TPNjun increased the dissociation rates of all TPNjun-sensitive peptides (Table I). Importantly, the magnitude of peptide dissociation rates obtained in the presence of TPNjun are considerably more similar to the intrinsic rates of the open (τ1) than the closed (τ2) forms of HLA-B*0801fos. This suggests that peptide-filled class I molecules associated with TPNjun adopt an open, rather than a closed, conformation. Equally important, because TPNjun-assisted peptide dissociation rates are slightly faster than the intrinsic rates of the open form of HLA-B*0801fos (Table I), it can be concluded that peptide-filled class I molecules associated with TPNjun possess an even more widely open binding groove. Taken together, it is the ability of TPNjun to form a complex with the open conformation of peptide-filled molecules, and to widen the binding groove, that causes bound peptides to dissociate faster in the presence of TPNjun. Our data suggest that these effects are likely to be directly mediated through disruption of the C-terminally localized conserved hydrogen bonds in the groove, but also involve peptide sequence-dependent class I interactions along the groove.
TPNjun increases the rate of peptide association and acts as a chaperone
Results from kinetics association experiments revealed that TPNjun increased the ratio of active-to-inactive peptide-deficient molecules in solution (Table I). Although we did not investigate how TPNjun affected the conversion between the inactive and active states (forward or reverse reaction), it is reasonable to suggest that TPNjun associates with the open (active) conformation of peptide-deficient molecules in a way that is similar to its association with the peptide-filled, open conformation. This association is likely to prevent class I molecules to become inactive, much like the effect exerted by chaperones on conformationally immature proteins. Consistent with this, previous studies have suggested that TPN stabilizes immature class I molecules in the ER (Ortmann et al, 1997; Schoenhals et al, 1999; Barnden et al, 2000). It is also reasonable to propose that the association of TPNjun with peptide-deficient, open class I molecules causes a widening of the binding groove, as discussed above for peptide-filled, open molecules. Therefore, it is the effect of TPNjun on stabilizing peptide-deficient, open class I molecules together with its ability to widen the groove of class I molecules that causes peptides to bind more rapidly in the presence of TPNjun (note that a TPNjun-mediated increase in the ratio of active-to-inactive peptide-deficient molecules also contributes to increasing the rate of peptide association, under pseudo-first-order conditions).
A TPN-assisted mechanism of peptide selection
We propose that the assembly/disassembly mechanism of class I/peptide molecules in the presence of full-length TPN is altered as follows:
Scheme II 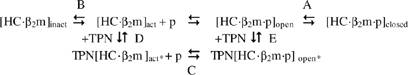 where ‘act*' and ‘open*' denote a more open binding groove relative to ‘act' and ‘open'.
where ‘act*' and ‘open*' denote a more open binding groove relative to ‘act' and ‘open'.
TPN can associate at two distinct stages: with the peptide-deficient, open (active) conformation and with the peptide-filled, open conformation as defined by equilibria D and E, respectively. The equilibrium C between TPN-stabilized open forms of class I molecules is established through the rapid association/dissociation of peptides.
How may this mechanism explain the ability of TPN to influence the cell-surface presentation of antigenic peptides? First, we suggest that the ability of TPN to widen the binding groove of class I molecules in the TPN[HC·β2m]act* complex is likely to diminish the specificity of the groove toward peptides. This means that a greater variety of peptides in terms of intrinsic dissociation half-lives, sequences, lengths, etc. can be captured in the initial binding step. To progress along the assembly pathway, bound candidate peptides establish sequence-independent and sequence-dependent interactions with class I molecules. This dynamic process is accompanied by conformational changes in various regions of the groove. It is such peptide-induced conformational changes that disengage TPN from the initial TPN[HC·β2m·p]open* complex. These peptide-induced maturation events shift the equilibria E and A toward the closed form of peptide-filled molecules. In contrast, bound candidate peptides in the initial TPN[HC·β2m·p]open* complex that are unable to induce maturing conformational changes in the binding groove of class I molecules, owing to their inability to establish energetically stabilizing interactions with MHC residues, will not as easily disengage TPN. Such peptides will eventually dissociate from the TPN[HC·β2m·p]open* complex allowing other peptides to be loaded into the groove. This dynamic peptide selection process under the action of TPN will continue until a peptide capable of conformationally disengaging TPN is captured. Thus, we suggest that TPN stabilizes the open forms of class I molecules and serves as an energy barrier that bound candidate peptides need to overcome for the productive assembly of mature class I/peptide molecules.
That alleles such as HLA-B*2705 and HLA-B*4405 show reduced TPN dependencies (Peh et al, 1998; Williams et al, 2002; Park et al, 2003; Zernich et al, 2004) implies that these alleles can mature through a pathway that bypasses equilibria D and E (Scheme II). This in turn implies that the open forms of HLA-B*2705 and HLA-B*4405 molecules possess a binding groove that is intrinsically more stable relative to the more TPN-dependent alleles. An increased stability could be achieved, for example, through increased intrinsic conformational maturity in the groove. As a consequence, it is expected that the open forms of peptide-deficient HLA-B*2705 and HLA-B*4405 molecules show more specificity in the initial capture of peptides relative to TPN-dependent alleles, which we suggested above to be more permissive in their initial selection of peptides. In other words, it is possible that allele-specific TPN dependencies may be rooted in differences in intrinsic conformational maturity. Although this remains to be examined experimentally, a role for MHC residues 114 and 116 (Williams et al, 2002; Park et al, 2003; Zernich et al, 2004) may be of consideration in influencing the intrinsic conformational maturity of class I binding groove.
Conclusions
TPN exerts two critical functions in the class I assembly complex
Data presented here are consistent with TPN exerting two critical functions in the class I assembly complex: (1) TPN permits the initial binding of a more diversified pool of candidate peptides; and (2) the presence of TPN in the initial assembly complex ensures that bound candidate peptides that cannot meet a certain minimum-energy threshold, as needed to conformationally disengage TPN, are excluded from the repertoire. The second function of TPN dampens the more permissive effects of the first function. The net outcome of a TPN-assisted mechanism of peptide selection is to maximize at any point in time, and according to the pool of peptides available in the ER, the diversity of minimally stable class I/peptide molecules presented at the cell surface. This effect in turn permits individuals to recruit a broader range of CD8+ T cells for protective immune responses against pathogens. Under such a selection process, class I/peptide molecules with higher or lower overall kinetics stability, relative to molecules formed in the absence of TPN, can be presented at the cell surface, as pointed to previously (Zarling et al, 2003). Overall, the presentation of antigenic peptides to CD8+ T cells under the action of TPN is the result of a mechanistically complicated selection process that cannot be directly correlated with the intrinsic dissociation half-life of peptides. This is reminiscent of the way in which HLA-DM influences peptide selection by class II molecules (Chou and Sadegh-Nasseri, 2000; Belmares et al, 2002).
Materials and methods
Expression and purification of TPNjun
The cDNA encoding the ER-lumenal domain (residues 1–392) of TPN was generated by PCR from a previously described plasmid (Chen et al, 2002) using as forward primer (5′-GGATCCTATAAATATGAAGTCCCTGTCTCTG-3′) and as reverse primer (5′-CGCGGAACCAGACCGCCGGAACCTCCGTCCTCAAGGGAGGG-3′) encoding a short linker (GGSGG; in bold). The cDNA encoding the Jun peptide was obtained by PCR from a previously described plasmid (Kalandadze et al, 1996) (a gift of Dr K Wucherpfennig) using as forward primer (5′-GGCGGTCTGGTTCCGCGTGGCTCGCGCATCGCCCGGCTCGAG-3′) encoding a thrombin site (LVPRGS; in bold) and as reverse primer (5′-GTCGACCTAGTGGTGATGGTGATGATGGTTCATGACTTT-3′) including a (His)6 tag sequence (in bold). The cDNAs encoding the engineered ER-lumenal domain of TPN and Jun peptide were linked together through an overlapping region (italicized) to generate the complete cDNA of TPNjun, which was then ligated into the pFastBac-1 vector (Invitrogen). Production of the TPNjun baculovirus expression vector and purification of the recombinant protein were carried out as described previously (Chen et al, 2002).
Expression and purification of HLA-B*0801fos heavy chain
The cDNA encoding the ER-lumenal domain of HLA-B*0801 heavy chain (residues 1–275), extended by the linker (GGSGG) at its 3′-end, was generated based on the plasmid pGM-T7 encoding HLA-B*0801 heavy chain (a gift of Dr Y Jones). The cDNA encoding the Fos peptide, extended by a thrombin site at its 5′-end, was generated from a previously described plasmid (Kalandadze et al, 1996) (a gift of Dr K Wucherpfennig). These two cDNAs were linked together to generate the complete cDNA of HLA-B*0801fos heavy chain, which was ligated into the pGM-T7 vector. Mutants of HLA-B*0801fos heavy chains were generated by PCR using the QuickChange site-directed mutagenesis approach (Stratagene). Plasmids harboring the correct DNA sequences were transformed into BL21(DE3)pLysS cells. Protein expression was induced with IPTG. Inclusion bodies of HLA-B*0801fos heavy chain (or mutants) were isolated from cell pellets, washed, and solubilized (Bouvier and Wiley, 1998).
In vitro assembly of peptide-filled and peptide-deficient HLA-B*0801fos molecules
Peptide-filled HLA-B*0801fos molecules were reconstituted as described previously (Bouvier and Wiley, 1998). These molecules were purified on a Superdex 200 HR 10/30 column in 20 mM Tris and 150 mM NaCl (pH 7.5). Peptide-deficient HLA-B*0801fos molecules were assembled from the denaturation of peptide-filled molecules (Bouvier and Wiley, 1998). Peptide-deficient HLA-B*0801fos molecules were purified by gel filtration chromatography; fractions were supplemented with glycerol (to 15%).
Native gel band-shift assay
Samples of TPNjun (or TPN) (10 μg) and HLA-B*0801fos molecules (or HLA-B*0801) (10 μg) (1:1 molar ratio) were incubated on ice in 20 mM Tris, 150 mM NaCl, and 10% glycerol (pH 7.5) for 30 min followed by the addition of native gel loading buffer (50 mM Tris, 0.1% bromophenol blue, 10% glycerol (pH 6.8)). Mixtures were assayed on native PAGE gel (8%) at 4°C in 25 mM Tris and 200 mM glycine (pH 8.3). Proteins were visualized with Coomassie blue staining.
Fluorescence anisotropy
Dissociation kinetics was measured by fluorescence anisotropy using an Aminco-Bowman 2 spectrofluorometer (Thermo Electron Co.). The excitation wavelength was 495 nm and polarization was detected at 524 nm. Association kinetics was measured by fluorescence anisotropy using an SX.18 MV stopped-flow spectrofluorometer (Applied Photophysics). The excitation light was 495 nm and the wavelength for emission was 510 nm. Experiments were carried out at 20°C in 20 mM Hepes and 150 mM NaCl (pH 7.5). Anisotropy data were processed by the software KaleidaGraph 4.0.1 (Synergy Software); plots of residuals were visibly examined.
Dissociation kinetics
Peptide dissociation kinetics experiments were initiated by adding under stirring HLA-B*0801fos molecules (or HLA-B*0801) (40 nM), loaded with a tagged peptide, into the buffer containing nontagged peptide (40 μM) (to block the rebinding of dissociated tagged peptide) and β2m (4 μM) (to avoid complications due to dissociation of β2m) in the absence or presence of TPNjun (or in the presence of TPN or HLA-B*0801jun) (400 nM). Given that the equilibrium dissociation constant, Kd, of Jun/Fos interaction is 54 nM (Kohler and Schepartz, 2001), it is expected that 87% of HLA-B*0801fos molecules are engaged in TPNjun/HLA-B*0801fos complexes in our assay. Also, note that the Kd of Jun/Jun homodimerization is estimated to be ∼600 nM (Kohler and Schepartz, 2001). It is thus expected that less than 42 nM TPNjun/TPNjun dimer forms at a total TPNjun concentration of 400 nM, leaving the majority of TPNjun available for interaction with HLA-B*0801fos. The effect of TPNjun homodimerization on the concentration of heterodimer is accounted for in ftpn (see below) and does not affect the kinetic parameters presented in Tables I and II. For each peptide, 3–5 independent experiments were carried out using different preparations of class I/peptide molecules and TPNjun (or TPN). For experiments carried out in the absence of TPNjun, the observed averaged intrinsic anisotropy data were fitted to an exponential decay function of the form y(t)int=y0 int+∑i(Ai exp(−kit)), where y0 int is the baseline offset, the summation is over the number of phases i, Ai and ki (ki=0.693/τi) are the amplitude and rate constant for phase i, respectively, and t is time. For experiments carried out in the presence of TPNjun, the averaged observed anisotropy data were fitted to an exponential decay function described by the sum of TPNjun-dependent (tpn) and intrinsic dissociation data: y(t)tpn=y0 tpn+ftpn∑i(Ai exp(−kit))+((1−ftpn)(y(t)int−y0 int)), where ftpn represents the fraction of dissociation reaction under the action of TPNjun, y(t)int is the intrinsic decay function of the corresponding class I/peptide molecule as computed above and other parameters are as described above. The dissociation rate enhancement factor is defined as τ(intrinsic; slow phase)/τ(TPNjun).
Association kinetics
Peptide association experiments were initiated by mixing automatically equal volumes (125 μl) from chamber A containing peptide-deficient HLA-B*0801fos molecules (or HLA-B*0801) (active concentration of 800 nM) and β2m (80 μM), and from chamber B containing a tagged peptide (160 nM) with or without TPNjun (or TPN) (1200 nM). Polarized emission signals of 10 experiments were averaged. For experiments carried out in the absence of TPNjun, the averaged observed intrinsic anisotropy data were fitted to an exponential function of the form y(t)int=y0 int+∑i(Ai(1−exp(−kit))), where parameters are as defined above. For experiments carried out in the presence of TPNjun, the averaged observed anisotropy data were fitted to an exponential function described by the sum of TPNjun-dependent (tpn) and intrinsic association data: y(t)tpn=y0 tpn+ftpn∑i(Ai(1−exp(−kit)))+((1−ftpn)(y(t)int−y0int)), where ftpn represents the fraction of dissociation reaction under the action of TPNjun and other parameters are as described above.
Activity of peptide-deficient HLA-B*0801fos molecules
Peptide-deficient HLA-B*0801fos molecules (or HLA-B*0801) (150 nM) were incubated with β2m (15 μM) and different concentrations of ELRSRK*WAI under identical conditions as those of association kinetics experiments. After 20 h, the anisotropy value for each mixture was measured. The observed anisotropy values, r, are described by the equation r=(fbrb)+(ffrf), where fb is the fraction of bound ELRSRK*WAI, rb is the maximum anisotropy value corresponding to bound ELRSRK*WAI, ff is the faction of free ELRSRK*WAI, and rf is the minimum anisotropy value corresponding to free ELRSRK*WAI. The values of rb and rf were determined in separate experiments. As fb+ff=1, the fraction of bound peptides, fb, was calculated from the observed anisotropy value of each reaction. Values of fb were used to determine the concentration of bound, Cb, and free, Cf, peptides. A Schatchard plot, Cb/Cf versus Cb, was produced and fitted to the linear function Cb/Cf=−(1/Kd)Cb+(Cact/Kd), from which the concentration of active peptide-deficient molecules, Cact, and Kd were derived. The activity of peptide-deficient molecules was calculated as % activity=(Cact/CT)·100, where CT, the total concentration of peptide-deficient molecules, was 150 nM.
Determination of IC50 values
Peptide-deficient HLA-B*0801fos molecules (100 nM in the absence of TPNjun and only 30 nM in the presence of TPNjun owing to the effect of TPNjun on increasing the concentration of active peptide-deficient HLA-B*0801fos molecules in solution) were incubated with the control ELRSRK*WAI peptide (100 nM) and different doses of EIYKRWIIL (0–10 μM) together with an excess of β2m (1 μM) in the absence or presence of TPNjun (100 nM). After 20 h, the anisotropy value of each reaction was measured. Inhibition was calculated from observed anisotropy signals in the absence, ro, and presence, r, of the competitor EIYKRWIIL peptide: % inhibition=(ro−r)/(ro−rf)·100, where rf, the minimum anisotropy signal corresponding to free ELRSRK*WAI, was measured separately. The concentration of competitor peptide required to measure 50% inhibition of the binding of ELRSRK*WAI represents IC50.
Peptides
Fluorescein-derivatized lysine residues (K*) were introduced at position 4 or 6 in EIYK*RWIIL, ELRSRK*WAI, FLRGRK*YGL, GLRSRK*WAI, GLRSRK*WAV, DPKVKK*WPL, and ELRSRK*WAV. This involved a YP6K* mutation in ELRSRK*WAI, GLRSRK*WAI, GLRSRK*WAV, and ELRSRK*WAV; a AP6K* mutation in FLRGRK*YGL; and a QP6K in DPKVKK*WPL. Peptides were synthesized and purified at Tufts University. Ndel and Cdel are derived from GLRSRK*WAV in which EP1G and IP9V mutations were introduced to facilitate syntheses: Ndel, (CH3-CH2-CO)-LRSRK*WAV and Cdel, GLRSRK*WA-(NH-CH(CH3)CH(CH3)2) (note that Cdel retains the side chain of Val at P9). Ndel and Cdel were synthesized and purified by AnaSpec.
Acknowledgments
We thank L Cheng and S Wang for excellent technical help. We are thankful to Dr CM Teschke for her generous permission to use fluorescence instruments. This research was funded by NIH/NIAID AI45070 (to MB) and in part by a Predoctoral Fellowship from Boehringer-Ingelheim Pharmaceuticals Inc. (to MC).
References
- Barber LD, Howarth M, Bowness P, Elliott T (2001) The quantity of naturally processed peptides stably bound by HLA-A*0201 is significantly reduced in the absence of tapasin. Tissue Antigens 58: 363–468 [DOI] [PubMed] [Google Scholar]
- Barnden MJ, Purcell AW, Gorman JJ, McCluskey J (2000) Tapasin-mediated retention and optimization of peptide ligands during the assembly of class I molecules. J Immunol 165: 322–330 [DOI] [PubMed] [Google Scholar]
- Belmares MP, Busch R, Wucherpfennig KW, McConnell HM, Mellins ED (2002) Structural factors contributing to DM susceptibility of MHC class II/peptide complexes. J Immunol 169: 5109–5117 [DOI] [PubMed] [Google Scholar]
- Binz AK, Rodriguez RC, Biddison WE, Baker BM (2003) Thermodynamic and kinetic analysis of a peptide-class I MHC interaction highlights the noncovalent nature and conformational dynamics of the class I heterotrimer. Biochemistry 42: 4954–4961 [DOI] [PubMed] [Google Scholar]
- Bouvier M, Wiley DC (1994) Importance of peptide amino and carboxyl termini to the stability of MHC class I molecules. Science 265: 398–402 [DOI] [PubMed] [Google Scholar]
- Bouvier M, Wiley DC (1998) Structural characterization of a soluble and partially folded class I major histocompatibility/β2m heterodimer. Nat Struct Biol 5: 377–382 [DOI] [PubMed] [Google Scholar]
- Burrows SR, Rodda SJ, Suhrbier A, Geysen HM, Moss DJ (1992) The specificity of recognition of a cytotoxic T lymphocyte epitope. Eur J Immunol 22: 191–195 [DOI] [PubMed] [Google Scholar]
- Busch R, Pashine A, Garcia KC, Mellins ED (2002) Stabilization of soluble, low-affinity HLA-DM/HLA-DR1 complexes by leucine zippers. Immunol Methods 263: 111–121 [DOI] [PubMed] [Google Scholar]
- Chen M, Stafford WF, Diedrich G, Khan A, Bouvier M (2002) A characterization of the lumenal region of human tapasin reveals the presence of two structural domains. Biochemistry 41: 14539–14545 [DOI] [PubMed] [Google Scholar]
- Chou C-L, Sadegh-Nasseri S (2000) HLA-DM recognizes the flexible conformation of major histocompatibility complex class II. J Exp Med 192: 1697–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiBrino M, Parker KC, Shiloach J, Turner RV, Tsuchida T, Garfield M, Biddison WE, Coligan JE (1994) Endogenous peptides with distinct amino acid anchor residue motifs bind to HLA-A1 and HLA-B8. J Immunol 152: 620–631 [PubMed] [Google Scholar]
- Gakamsky DM, Davis DM, Strominger JL, Pecht I (2000) Assembly and dissociation of human leukocyte antigen (HLA)-A2 studied by real-time fluorescence resonance energy transfer. Biochemistry 39: 11163–11169 [DOI] [PubMed] [Google Scholar]
- Garbi N, Tan P, Diehl AD, Chambers BJ, Ljunggren HG, Momburg F, Hammerling GJ (2000) Impaired immune responses and altered peptide repertoire in tapasin-deficient mice. Nat Immunol 1: 234–238 [DOI] [PubMed] [Google Scholar]
- Howarth M, Williams A, Tolstrup AB, Elliott T (2004) Tapasin enhances MHC class I peptide presentation according to peptide half-life. Proc Natl Acad Sci USA 101: 11737–117442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalandadze A, Gallenos M, Foncerrada L, Strominger JL, Wucherpfennig KW (1996) Expression of recombinant HLA-DR2 molecules. J Biol Chem 271: 20156–20162 [DOI] [PubMed] [Google Scholar]
- Khan AR, Baker BM, Ghosh P, Biddison WE, Wiley DC (2000) The structure and stability of an HLA-A*0201/octameric tax peptide complex with an empty conserved peptide N-terminal binding site. J Immunol 164: 6398–6405 [DOI] [PubMed] [Google Scholar]
- Kohler JJ, Schepartz A (2001) Kinetic studies of Fos.Jun.DNA complex formation: DNA binding prior to dimerization. Biochemistry 40: 130–142 [DOI] [PubMed] [Google Scholar]
- Lehner PJ, Surman MJ, Cresswell P (1998) Soluble tapasin restores MHC class I expression and function in the tapasin-negative cell line .220. Immunity 8: 221–231 [DOI] [PubMed] [Google Scholar]
- Lewis JW, Elliott T (1998) Evidence for successive peptide binding and quality control stages during MHC class I assembly. Curr Biol 8: 717–720 [DOI] [PubMed] [Google Scholar]
- Ogata K, Wodak SJ (2002) Conserved water molecules in MHC class-I molecules and their putative structural and functional roles. Prot Eng 15: 697–705 [DOI] [PubMed] [Google Scholar]
- Ortmann B, Copeman J, Lehner PJ, Sadasivan B, Herberg JA, Grandea AG, Riddell SR, Tampe R, Spies T, Trowsdale J, Cresswell P (1997) A critical role for tapasin in the assembly and function of multimeric MHC-class I-TAP complexes. Science 277: 1306–1309 [DOI] [PubMed] [Google Scholar]
- O'Shea EK, Rutkowski R, Stafford WF III, Kim PS (1989) Preferential heterodimer formation by isolated leucine zippers from Fos and Jun. Science 254: 646–648 [DOI] [PubMed] [Google Scholar]
- Paquet ME, Williams DB (2002) Mutant MHC class I molecules define interactions between components of the peptide-loading complex. Int Immunol 14: 347–358 [DOI] [PubMed] [Google Scholar]
- Park B, Lee S, Kim E, Ahn K (2003) A single polymorphic residue within the peptide-binding cleft of MHC class I molecules determines spectrum of tapasin dependence. J Immunol 170: 961–968 [DOI] [PubMed] [Google Scholar]
- Peace-Brewer AL, Tussey LG, Matsui M, Li G, Quinn DG, Frelinger JA (1996) A point mutation in HLA-A*0201 results in failure to bind to TAP complex and to present virus-derived peptides to CTL. Immunity 4: 505–514 [DOI] [PubMed] [Google Scholar]
- Peh CA, Burrows SR, Barnden M, Khanna R, Cresswell P, Moss DJ, McCluskey J (1998) HLA-B27-restricted antigen presentation in the absence of tapasin reveals polymorphism in mechanisms of HLA class I peptide loading. Immunity 8: 531–542 [DOI] [PubMed] [Google Scholar]
- Schoenhals GJ, Krishna RM, Grandea AG III, Spies T, Peterson PA, Yang Y, Fruh K (1999) Retention of empty MHC class I molecules by tapasin is essential to reconstitute antigen presentation in invertebrate cells. EMBO J 3: 743–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer S, Doring K, Skipper JCA, Townsend ARM, Cerundolo V (1998) Fast association rates suggest a conformational change to the MHC class I molecules H-2Db upon peptide binding. Biochemistry 37: 3001–3012 [DOI] [PubMed] [Google Scholar]
- Suh WK, Derby MA, Cohen-Doyle MF, Schoenhals GJ, Fruh K, Berzofsky JA, Williams DB (1999) Interaction of murine MHC class I molecules with tapasin and TAP enhances peptide loading and involves the heavy chain α3 domain. J Immunol 162: 1530–1540 [PubMed] [Google Scholar]
- Williams AP, Peh CA, Purcell AW, McCluskey J, Elliott T (2002) Optimization of the MHC class I peptide cargo is dependent on tapasin. Immunity 16: 509–520 [DOI] [PubMed] [Google Scholar]
- Wright CA, Kozik P, Zacharias M, Springer S (2004) Tapasin and other chaperones: models of the MHC class I loading complex. Biol Chem 385: 763–778 [DOI] [PubMed] [Google Scholar]
- Yu YYL, Turnquist HR, Myers NB, Balendiran GK, Hansen TH, Solheim JC (1999) An extensive region of an MHC class I α2 domain loop influences interaction with the assembly complex. J Immunol 163: 4427–4433 [PubMed] [Google Scholar]
- Zarling AL, Luckey CJ, Marto JA, White FM, Brame CJ, Evans AM, Lehner PJ, Cresswell P, Shabanowitz J, Hunt DF, Engelhard VH (2003) Tapasin is a facilitator, not an editor, of class I MHC peptide binding. J Immunol 171: 5287–5295 [DOI] [PubMed] [Google Scholar]
- Zernich D, Purcell AW, MacDonald WA, Kjer-Nielsen L, Ely LK, Laham N, Crockford T, Misfud NA, Bharadwaj M, Chang L, Tait BD, Holdsworth R, Brooks AG, Bottomley SP, Beddoe T, Peh CA, Rossjohn J, McCluskey J (2004) Natural polymorphism controls the pathway of antigen presentation and susceptibility to viral evasion. J Exp Med 200: 13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]