

Abstract
Rho GTPases regulate multiple signal transduction pathways that influence many aspects of cell behaviour, including migration, morphology, polarity and cell cycle. Through their ability to control the assembly and organization of the actin and microtubule cytoskeletons, Rho and Cdc42 make several key contributions during the mitotic phase of the cell cycle, including spindle assembly, spindle positioning, cleavage furrow contraction and abscission. We now report that PRK2/PKN2, a Ser/Thr kinase and Rho/Rac effector protein, is an essential regulator of both entry into mitosis and exit from cytokinesis in HeLa S3 cells. PRK2 is required for abscission of the midbody at the end of the cell division cycle and for phosphorylation and activation of Cdc25B, the phosphatase required for activation of mitotic cyclin/Cdk1 complexes at the G2/M transition. This reveals an additional step in the mammalian cell cycle controlled by Rho GTPases.
Keywords: cell cycle, Cdc25B, Cdk1, PRK, Rho GTPase
Introduction
Members of the Rho family of small GTPases control many important cellular activities, including the organization of the actin and microtubule cytoskeletons, cell–cell and cell–matrix adhesion, polarity, gene expression and cell cycle progression (Jaffe and Hall, 2005). Guanine nucleotide exchange factors (GEFs) stimulate the conversion of these molecular switches from an inactive, GDP-bound conformation to an active, GTP-bound form, which then interacts with target proteins (effectors) to mediate downstream signaling. Some 85 GEFs for the Rho family have been identified in the human genome, and over 60 effectors have been isolated to date for the three best-characterized members of the family, Rho, Rac and Cdc42 (Schmidt and Hall, 2002; Jaffe and Hall, 2005). With such a complexity of cellular functions and interacting proteins, it has proven challenging to dissect individual Rho GTPase signaling pathways and identify the specific GEFs and effectors involved. Attempts to manipulate Rho GTPase function by ectopic expression of Rho GTPases, or their GEFs and effectors, often results in complex phenotypes owing to activation of several pathways, while dominant-negative Rho GTPases can potentially interfere with closely related family members that share the same GEF.
In the cell cycle, Rho, Rac and Cdc42 have each been shown to promote G1/S progression in a variety of cell types, but their exact roles have been difficult to pin down (Yamamoto et al, 1993; Olson et al, 1995). Numerous activities have been reported, including regulation of cyclin D, p21Waf1 and p27Kip1 expression at both the transcriptional and translation levels (Olson et al, 1998; Mettouchi et al, 2001; Welsh et al, 2001; Coleman et al, 2004). Multiple roles for Rho GTPases have been described during mitosis and cytokinesis. A requirement for Rho during cytokinesis was first reported in sand dollar eggs in 1993 (Mabuchi et al, 1993). Rho also promotes mitotic cell rounding in HeLa cells and Cdc42 is required for asymmetric positioning of spindles during zygotic division in Caenorhabditis elegans (Gotta et al, 2001; Maddox and Burridge, 2003). In addition, there is now a well-established requirement for a Rho family GTPase in kinetochore–spindle attachment. In HeLa cells, Cdc42 promotes the attachment of microtubules to the kinetochore, whereas in Rat-2 fibroblasts, kinetochore attachment is Rho dependent (Yasuda et al, 2004; Bakal et al, 2005). In HeLa cells, the Cdc42 effector is mDia3 and the exchange factor is Ect2 (Yasuda et al, 2004; Oceguera-Yanez et al, 2005). In Rat-2 cells, the target of Rho is mDia1 and the exchange factor is Lfc (Bakal et al, 2005).
The mechanism by which Rho regulates the contractile ring during cytokinesis has been studied widely in mammalian cells, Drosophila and C. elegans. Rho is required for the assembly and ingression of the actomyosin filament ring and is activated during mitosis by Ect2 in mammalian cells and by its orthologues Pebble in Drosophila and RhoGEF ect-2 in C. elegans (Tatsumoto et al, 1999; Shandala et al, 2004; Glotzer, 2005; Kamijo et al, 2006). Ect2 is phosphorylated during mitosis by Cdk1 and the Polo-like kinase Plk1, and then activates Rho at the equatorial region and the cleavage furrow (Kamijo et al, 2006; Niiya et al, 2006; Nishimura and Yonemura, 2006). Activated Rho recruits two effector kinases, citron kinase and Rho kinase, and probably other effectors such as Diaphanous-related proteins, to assemble the contractile ring, mediate cleavage furrow ingression and promote abscission (Madaule et al, 1998; Kosako et al, 2000; Eda et al, 2001; Piekny et al, 2005; Gruneberg et al, 2006). Citron kinase is essential for the development of Drosophila, and in HeLa cells, siRNA-mediated depletion of citron kinase, but not Rho kinase, results in the formation of multinucleated cells (Shandala et al, 2004; Kamijo et al, 2006). However, a citron kinase knockout mouse shows a relatively mild cytokinesis phenotype, and it is likely that there is redundancy in the Rho effector kinases that mediate filament contraction (Di Cunto et al, 2000).
Given the multiple roles of individual Rho GTPases in the cell cycle, we have been examining the role of individual effectors using siRNAi. Here, we report the identification of the Rho effector protein kinase C-related kinase (PRK2/PKN2) as a new regulator of both G2/M progression and exit from cytokinesis.
Results
siRNA-mediated targeting of PRK2 causes inhibition of cytokinesis
To assess the role of PRK/PKN during the cell cycle, double-stranded siRNAs targeting each of the three isoforms (PRK1,2,3/PKN1,2,3) were transfected separately into unsynchronized HeLa S3 cells and the cells were analysed for DNA content by FACS 72 h later. siRNA for PRK2 (PRK2 A) resulted in an increase in the percentage of ⩾4n cells compared with control cells (Figure 1A). siRNA against PRK1/PKN (Figure 1A) or PRK3/PKN3 (data not shown) had no effect on cell cycle progression in these cells.
Figure 1.

Depletion of PRK2 from HeLa S3 cells leads to the formation of binucleated cells. (A) Unsynchronized control, PRK2 A and PRK1 siRNA-transfected cells were subjected to FACS analysis and the percentage of ⩾4n DNA cells was determined. The averages±standard deviation for at least two independent experiments are shown. (B) Unsynchronized control, PRK2 A, PRK2 B and PRK1 siRNA-transfected cells were fixed after 72 h and stained with Hoechst. PRK2 A and PRK2 B siRNA-transfected cells result in the accumulation of binucleated cells (arrowheads). (C) Unsynchronized control, PRK2 A, PRK2 B and PRK1 siRNA-transfected cells were fixed after 72 h and stained with Hoechst. The percentage of binucleated cells was determined. The averages of at least three independent experiments±standard deviation are shown. (D) Unsynchronized control, PRK2 A, PRK2 B and PRK1 siRNA-transfected cells were lysed after 72 h and cell extracts were analysed by Western blotting using anti-PRK2, anti-PRK1 and anti-tubulin antibodies. (E) Control and myc PRK2-transfected cells were fixed after 72 h and stained with anti-myc antibody and Hoechst. Overexpression of myc PRK2 results in the accumulation of binucleated cells (arrowheads). (F) Control and myc PRK2-transfected cells were fixed after 72 h, stained with anti-myc antibody and Hoechst, and the percentage of binucleated cells determined. The averages of at least three independent experiments±standard deviation are shown. (G) PRK1 protein is expressed at low levels in HeLa S3 cells. Recombinant Gst-PRK1 and Gst-PRK2 proteins (5–50 ng) together with total cell extract (10+20 μl) were analysed by SDS–PAGE and Western blotting using anti-PRK1 and anti-PRK2 antibodies.
Thirty-two per cent of cells transfected with PRK2 A siRNA accumulate as binucleated cells (Figure 1B), compared with less than 5% of control or PRK1 siRNA-transfected cells (Figure 1C). A second siRNA against PRK2 (PRK2 B) also showed an accumulation of binucleated cells (Figure 1B and C). Western blot analysis of whole-cell lysates of siRNA-transfected cells verified that these siRNAs specifically reduced PRK2 and PRK1 protein levels, and that PRK2 A was somewhat more effective than PRK2 B (Figure 1D). Attempts to rescue the siRNA-induced effect using a full-length PRK2 construct failed as ectopic expression of PRK2 alone induced a cytokinesis defect (Figure 1E and F).
PRK1 and PRK3 are expressed in HeLa S3 cells and are closely related to PRK2, and so it was surprising that they could not compensate for the loss of PRK2 after siRNA expression (Palmer et al, 1995). However, standardization of the PRK1 and PRK2 monoclonal antibodies on Western blots using recombinant PRK1 and PRK2 protein revealed that HeLa S3 cells contain at least 10-fold more PRK2 than PRK1 (Figure 1G). It remains a possibility, therefore, that PRK1 and PRK3 have roles similar to PRK2 in cell cycle (also see below).
PRK1 and PRK2 localize to the cleavage furrow and the midbody during cytokinesis
To examine the subcellular localization of PRK, unsynchronized HeLa S3 cells were fixed and stained for endogenous PRK1 and PRK2 proteins. During interphase, endogenous PRK2 showed a diffuse distribution in the cytosol (Figure 2A), which did not change during prophase, metaphase or anaphase. During telephase, PRK2 accumulated at the cleavage furrow and finally became concentrated around the midbody in cytokinesis. Midbody staining persisted until late cytokinesis. The cytoplasmic, cleavage furrow and midbody staining all disappeared in the PRK2 siRNA-transfected cells (Figure 2B). Similar, but weaker, staining was seen for endogenous PRK1 and for overexpressed epitope-tagged full-length PRK1 (not shown).
Figure 2.
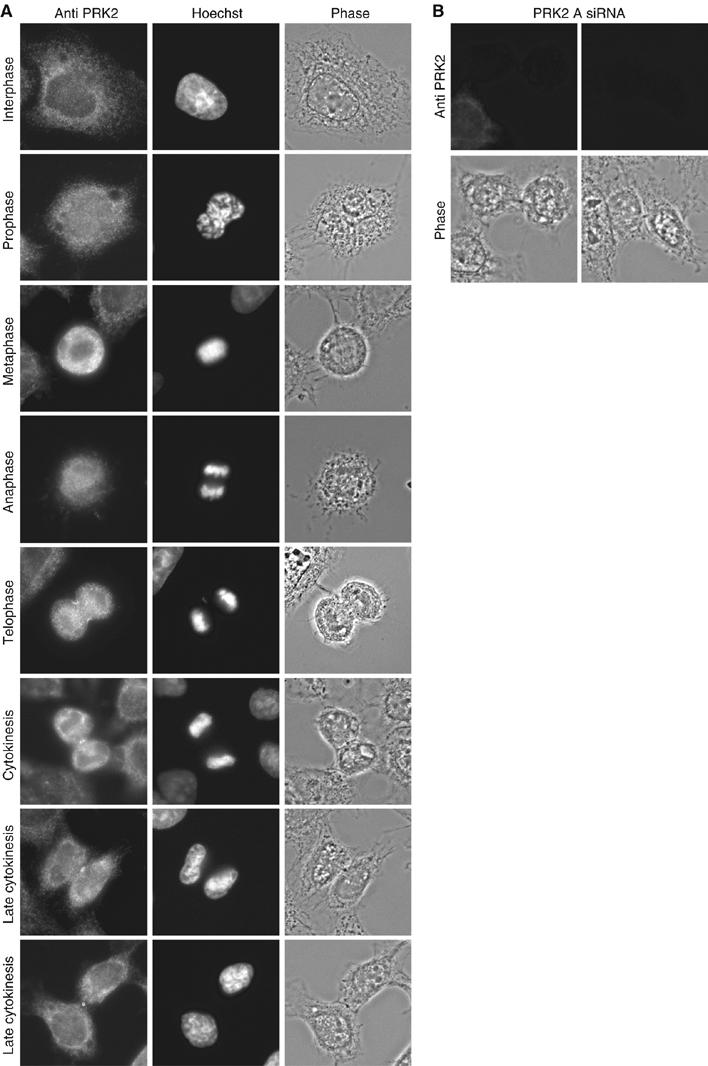
Subcellular localization of endogenous PRK2 during the cell cycle. (A) HeLa S3 cells were fixed and stained with anti-PRK2 antibody and Hoechst. (B) PRK2 siRNA-transfected cells were fixed and stained with anti-PRK2 antibody.
PRK2-depleted cells show a late cytokinesis defect
Activation of Rho by the GEF Ect2 has previously been shown to control cleavage furrow formation during cytokinesis and siRNA-mediated depletion of Ect2 results in the formation of multinucleated cells (Kamijo et al, 2006). To examine if the cytokinesis defect in PRK2-depleted cells was also at the level of cleavage furrow formation, cell division was visualized in PRK2- and Ect2-depleted cells by video time-lapse imaging (Figure 3A–D). Like control cells, PRK2 siRNA-transfected cells round up normally and display normal cleavage furrow ingression and midbody formation (compare Figure 3A and B). However, PRK2-depleted cells fail to undergo abscission and the two daughter cells fuse back together resulting in the formation of binucleated cells. In contrast, in the majority of Ect2-depleted cells, the cleavage furrow does not fully ingress, as has been previously reported (Figure 3C) (Kamijo et al, 2006). We did, however, find that about 15% of Ect2-depleted cells also showed a late cytokinesis defect, similar to those observed in the PRK2 siRNA-transfected cells (Figure 3D). This has also been described by others previously, and suggests that Ect2 has two functions during cytokinesis, controlling cleavage furrow formation as well as cell abscission (Kamijo et al, 2006). Interestingly, in those Ect2-depleted cells that were able to ingress a cleavage furrow and form a midbody, PRK2 no longer localized to the midbody (Figure 3E). These results suggest that PRK2 is required for cell abscission during cytokinesis and that PRK2 localization to the midbody is dependent on Ect2.
Figure 3abc.

PRK2-depleted cells have an abscission defect. Live-cell imaging of control cells (A); PRK2 A siRNA-transfected cells (B); and Ect2 siRNA-transfected cells (C, D).
Figure 3de.
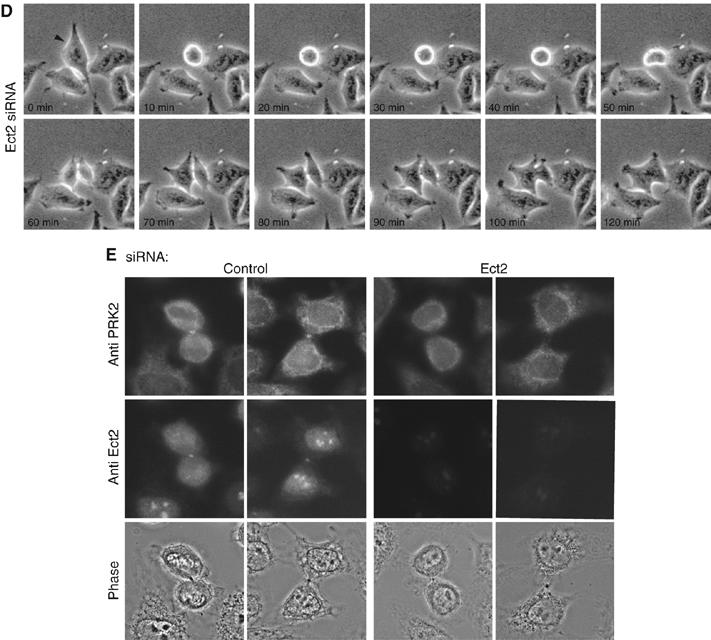
Images were taken every 10 min and representative frames are shown. The experiment was repeated five times. For PRK2 siRNA, 85% of binucleate cells arose from a late cytokinesis defect at abscission and 15% arose from an early cleavage furrow defect. For Ect2 siRNA, 17% of binucleate cells arose from an abscission defect and 83% arose from a cleavage furrow defect. (E) PRK2 localization is dependent on Ect2. Control or Ect2 siRNA-transfected cells were fixed after 48 h and visualized with anti-PRK2 and anti-Ect2 antibodies.
PRK2-depleted cells are delayed in G2/M progression
In the course of analysing PRK2- and Ect2-depleted cells, we noticed that the majority of Ect2 siRNA-transfected cells were multinucleated 72 h after transfection, whereas PRK2 siRNA-transfected cells remained binucleated during this time period (Figure 4A). This suggests that cell cycle progression is disturbed specifically in PRK2-knockdown cells. Using video time-lapse imaging of control, PRK2- and Ect2 siRNA-treated cells, we find that the cell cycle length of PRK2-depleted binucleated cells is almost twice as long (36 h) as that of control cells or Ect2-depleted binucleated cells (21 h) (Figure 4B). Furthermore, some 30% of PRK2-depleted binucleated cells underwent apoptosis without further dividing (not shown). We conclude that PRK2 is required for cell cycle progression as well as cytokinesis.
Figure 4.

PRK2 depletion delays cell cycle progression. (A) Control, PRK2 A siRNA- and Ect2 siRNA-transfected cells were fixed after 72 h and visualized with Hoechst. PRK2-depleted cells are binucleated, whereas Ect2-depleted cells are multinucleated. (B) Control, PRK2 A siRNA- and Ect2 siRNA-transfected cells were observed by time-lapse imaging, and cell cycle lengths, that is, the time between two consecutive mitosis, were calculated. For PRK2 A siRNA- and Ect2 siRNA-transfected cells, the cell cycle duration was determined for cells that entered mitosis and then failed cytokinesis. Average cell cycle lengths±standard deviation for 20–30 cells from independent experiments are shown. (C) Control cells and PRK2 A siRNA-transfected cells were synchronized with a double thymidine block and released into fresh medium containing BrdU. Cells were fixed after 6 h and stained with anti-BrdU antibody, and the percentage of cells with incorporated BrdU was determined. The average percentages from four independent experiments±standard deviation are shown. (D) PRK2-depleted cells show a delay in G2/M progression. Control cells and PRK2 A siRNA-transfected cells were synchronized with a double thymidine block and released into fresh medium. Whole cell extracts prepared at the indicated time points were analysed by SDS–PAGE and Western blotting using anti-PRK2, anti-Cdc25B, anti-P-Cdk1, anti-Cdk1, anti-cyclin A, anti-cyclin B1, anti-P-Histone3 and anti-tubulin antibodies.
To investigate this further, control and PRK2 siRNA-transfected cells were synchronized at the G1/S boundary with a double thymidine block. Cells were released into fresh medium containing BrdU, fixed 6 h after release and stained with anti-BrdU antibody as a read-out for DNA synthesis. Both control cells and PRK2 siRNA-transfected cells showed BrdU incorporation in >85% of cells, indicating that progression through S phase is not dependent on PRK2 (Figure 4C). To monitor the timing of mitosis, thymidine-synchronized control and PRK2 siRNA-transfected cells were observed under the microscope at different time points after release. Control cells started to round up for mitosis 10 h post-release, whereas the majority of PRK2-depleted cells started to enter mitosis only after 14 h (not shown), suggesting that PRK2 is required for entry into mitosis.
To identify the molecular consequences of PRK2 depletion during G2/M progression, synchronized PRK2 siRNA-transfected cells were harvested at the indicated times and lysates analysed on Western blots (Figure 4D). The levels of phosphorylated Histone 3 (P-Histone3), which accumulates in prophase, and the degradation of cyclin A and cyclin B1, which occurs during prometaphase and metaphase, respectively, were monitored. In control cells, maximal accumulation of P-Histone3 occurs 10 h after release from the thymidine block (Figures 4D and 6A). In contrast, in PRK2-depleted cells, P-Histone3 is maximal only after 14 h. Similarly, the degradation of cyclin A and cyclin B1 was significantly delayed in PRK2 siRNA-transfected cells compared with control cells. Together, these data suggest that PRK2 is required for G2/M progression.
Figure 6.

PRK2 is phosphorylated during mitosis in a Rho GTPase- and Cdk1-dependent manner. (A) HeLa S3 cells were synchronized with a double thymidine block and released into fresh medium. Whole cell extracts were prepared at the indicated time points and analysed by SDS–PAGE and Western blotting using anti-PRK2, anti-PRK1, anti-Ect2, anti-Cdc25B, anti-P-Cdk1, anti-Cdk1, anti-cyclin A, anti-cyclin B1, anti-P-Histone3 and anti-tubulin antibodies. PRK proteins are modified during mitosis. (B) Endogenous PRK2 was precipitated from lysates of cells synchronized with a double thymidine block and released for 0 or 10 h. PRK2 precipitates from the 10 h samples were either left untreated or incubated with or without λ phosphatase for 30 min at 30°C. Samples were subjected to SDS–PAGE and Western analysis with anti-PRK2 antibody. (C) HeLa S3 cells were synchronized with a double thymidine block and released into fresh medium. After 6 h, Toxin B was added at the indicated concentrations and whole cell extracts were prepared 10 h after release from the thymidine block and analysed by SDS–PAGE and Western blotting using anti-PRK2 and anti-tubulin antibodies. (D) Control and Ect2 siRNA-transfected cells were synchronized with a double thymidine block and released into fresh medium. Whole cell extracts were prepared at the indicated time points and analysed by SDS–PAGE and Western blotting using anti-Ect2, anti-PRK2, anti-cyclin A, anti-P-Histone3 and anti-tubulin antibodies. (E) HeLa S3 cells were synchronized with a double thymidine block and released into fresh medium. Roscovitine (25 μM) was added after 8 h and whole cell extracts were prepared 10 h after release from the thymidine block and analysed by SDS–PAGE and Western blotting using anti-PRK2 and anti-tubulin antibodies. (F) Control and Cdc25B siRNA-transfected cells were synchronized with a double thymidine block and released into fresh medium. Whole cell extracts were prepared at the indicated time points and analysed by SDS–PAGE and Western blotting using anti-Cdc25B, anti-PRK2, anti-P-Cdk1, anti-cyclin A, anti-P-Histone3 and anti-tubulin antibodies.
Cyclin B1/Cdk1 and cyclin A/Cdk1 are the main cyclin/Cdk complexes activated at the G2/M transition, and this requires dephosphorylation of Cdk1 at Tyr-15 (Gould and Nurse, 1989). In control HeLa S3 cells, dephosphorylation of Cdk1 takes place approximately 10–12 h after being released from the thymidine block (Figures 4D and 6A), indicative of Cdk1 activation. In PRK2-depleted cells, phosphorylated, inactivated Cdk1 persists for more than 14 h post-thymidine release. Dephosphorylation of Cdk1 is mediated by the Cdc25 family of protein phosphatases, which themselves are activated at the G2/M boundary by phosphorylation (Karlsson-Rosenthal and Millar, 2006). Western blot analysis of synchronized PRK2-depleted cells revealed that Cdc25B phosphorylation (seen as a supershift in gel mobility) was severely delayed compared with control cells (Figure 4D). We conclude that PRK2 is required for phosphorylation and activation of Cdc25B at the G2/M transition.
siRNA-mediated depletion of Cdc25B induces a G2/M delay and a cytokinesis defect
To establish if defective Cdc25B activation seen in PRK2-depleted cells is linked to the defect in cytokinesis and the increased cell cycle length, an siRNA targeting Cdc25B was transfected into unsynchronized HeLa S3 cells and cells were analysed 72 h post-transfection by Western blotting and immunofluorescence. Figure 5A shows that Cdc25B was efficiently and specifically depleted in siRNA-transfected cells. Depletion of Cdc25B resulted in approximately 33% binucleated cells, similar to that observed in PRK2 siRNA-transfected cells and in contrast to the multinucleated phenotype of Ect2 cells (Figure 5B and C). Video time-lapse imaging of Cdc25B-depleted binucleated cells revealed a severe increase in the duration of their cell cycle compared with control cells (Figure 5D). In addition, around 30% of Cdc25B-depleted binucleated cells underwent apoptosis without further dividing (not shown). As previously published, synchronized Cdc25B siRNA-transfected cells showed a delay in G2/M progression, as judged by a delay in Cdk1 dephosphorylation, cyclin A degradation and P-Histone3 accumulation (Figure 6F) (Lindqvist et al, 2005).
Figure 5.

Cdc25B depletion causes a cell cycle delay and a cytokinesis defect. (A) Unsynchronized control and Cdc25B siRNA-transfected cells were lysed after 72 h and whole cell extracts were subjected to SDS–PAGE and Western analysis using anti-Cdc25B and anti-PRK2 antibodies. (B) Unsynchronized control and Cdc25B siRNA-transfected cells were fixed after 72 h and visualized with Hoechst. Cdc25B-depleted cells are binucleated. (C) Unsynchronized control, Cdc25B siRNA- and PRK2 siRNA-transfected cells were fixed after 72 h and visualized with Hoechst. The percentages of binucleated cells were determined. The averages of at least three independent experiments±standard deviation are shown. (D) Control, Cdc25B siRNA- and PRK2 siRNA-transfected cells were observed by time-lapse imaging, and the cell cycle lengths, that is, the time between two consecutive mitosis, were calculated. For Cdc25B siRNA- and PRK2 siRNA-transfected cells, the cell cycle duration was determined for cells that entered mitosis and then failed cytokinesis. Average cell cycle lengths±standard deviation are shown.
Regulation of PRK2 during mitosis
PRK proteins are activated by Rho.GTP, which disrupts an autoinhibitory, intramolecular interaction, allowing PRK phosphorylation and further activation (Mukai, 2003). To examine whether PRK2 is phosphorylated during G2/M progression, cell extracts from synchronized HeLa S3 cells were harvested over the course of 22 h after release from thymidine block and analysed on Western blots (Figure 6A). Eight to twelve hours after thymidine release, a second, slower migrating form of PRK2 was detected, which coincided with the appearance of phosphorylated Cdc25B, dephosphorylation of Cdk1, degradation of cyclin A and B1, as well as accumulation of P-Histone3. The pattern of PRK2 modification is similar to that of Ect2 phosphorylation (Tatsumoto et al, 1999). Treatment of immunoprecipitated PRK2 with λ phosphatase confirmed that the new form of PRK2 is due to phosphorylation (Figure 6B). PRK1 is also modified similar to PRK2, suggesting that both PRK proteins have likely redundant functions in the control of G2/M progression (Figure 6A).
To investigate if Rho GTPases are involved, synchronized HeLa S3 cells were released from thymidine block, treated with the Rho GTPase inhibitor toxin B after 6 h, harvested after 10 h and examined for phosphorylation of PRK2. ToxinB blocked PRK2 phosphorylation in a dose-dependent manner (Figure 6C), and caused a mitotic delay in Cdc25B phosphorylation (data not shown), suggesting that Rho GTPases are required for phosphorylation and activation of PRK2 at G2/M. As PRK2 localization at the cleavage furrow and midbody is dependent on Ect2 (Figure 3E), we examined if Ect2 is required for Rho GTPase-dependent PRK2 phosphorylation at G2/M. After synchronization and release of Ect2 siRNA-transfected cells, PRK2 phosphorylation was identical to control cells (Figure 6D). We conclude that Ect2 is required to localize PRK2 during cytokinesis, but is not required for phosphorylation and activation of PRK2 at the beginning of mitosis.
Finally, PRK2 has a Cdk phosphorylation consensus site at the end of the PRK2 kinase domain, which is conserved across species as well as in PRK1/PKN. To investigate whether Cdk mediates phosphorylation of PRK2, cells were synchronized with thymidine, released, treated with the Cdk inhibitor roscovitine after 8 h (after cells had progressed through S phase) and harvested after 10 h. Unlike control cells, PRK2 was no longer phosphorylated in the presence of roscovitine, suggesting that PRK2 phosphorylation is downstream of Cdk1 (Figure 6E). In agreement with this, PRK2 phosphorylation was also blocked in synchronized Cdc25B siRNA-transfected cells (Figure 6F).
Discussion
Rho GTPases have been implicated in a variety of steps during the cell cycle, including progression through the G1 phase, spindle assembly and positioning in mitosis, and contractile ring formation during cytokinesis (Jaffe and Hall, 2005). Using siRNA, we have identified PRK2/PKN2, a Ser/Thr kinase and Rho GTPases target protein, as a new regulator of both G2/M progression and cytokinesis.
Video time-lapse imaging revealed that siRNA-mediated depletion of PRK2 in HeLa S3 cells caused a late-stage cytokinesis defect. These cells showed normal cleavage furrow ingression and midbody formation, but failed to undergo abscission and eventually fused back together to form binucleated cells. In agreement with a role for PRK2 in abscission, endogenous PRK2 (and the closely related family member PRK1) localizes to the cleavage furrow and then the midbody during cytokinesis. We have also obtained evidence that this localization is dependent on the exchange factor Ect2. Ect2, acting through Rho, is known to be required for formation of the contractile ring, and HeLa cells depleted of Ect2 with siRNA fail to undergo cleavage furrow ingression (Yuce et al, 2005; Kamijo et al, 2006). Interestingly, and as first observed by others, we found that a small percentage of Ect2-depleted cells do form a contractile ring and a midbody, but then fail to undergo abscission, presumably due to variations in the efficiency of Ect2 depletion in the experiment (Kamijo et al, 2006). In those cells, however, PRK2 no longer localizes to the midbody. From this we conclude that PRK2 is required for abscission and that its localization to the midbody requires Ect2 and perhaps Rho activity. Thus, Ect2 and Rho control at least two distinct functions during cytokinesis: (i) contractile ring formation and cleavage furrow ingression through Rho kinase and formins (Piekny et al, 2005) and (ii) cell abscission through citron kinase and PRK/PKN family kinases (Figure 7) (Gruneberg et al, 2006). We have also found a similar effect of PRK2 siRNA in the human glioma cell line U373 under conditions where Ect2 siRNA produces primarily multinucleated cells, loss of PRK2 induces binucleated cells (Supplementary Figure S1). The biochemical role of Rho and PRK/PKN proteins in abscission, a process that requires delivery of membrane vesicles and subsequent fusion events to remodel the membrane surrounding the two daughter cells, is not known (Schweitzer and D'Souza-Schorey, 2004). PRK2 has been described to interact with a protein tyrosine phosphatase, PTP-BL, which localizes to the cleavage furrow and midbody, and has been implicated in cytokinesis, and PKN/PRK1 binds to CG-NAP, a scaffold protein localized to the Golgi apparatus, the centrosome and midbody during cytokinesis (Takahashi et al, 1999; Gross et al, 2001; Herrmann et al, 2003). Whether these proteins are involved in linking PRK proteins to the abscission machinery remains to be seen.
Figure 7.

Dual role of Rho GTPases and PRK2 during mitosis. See Discussion for further details.
In addition to controlling exit from cytokinesis, PRK2 is required for entry into mitosis. Through the analysis of known players in G2/M progression, we find that PRK2 depletion leads to a significant delay in numerous activities associated with progression through mitosis including activation of Cdk1, degradation of cyclin A and cyclin B1 and accumulation of P-Histone3. A key event for initiating mitosis is the dephosphorylation and activation of Cdk1 by members of the Cdc25 family of protein phosphatases, which are themselves activated by phosphorylation (Karlsson-Rosenthal and Millar, 2006). In PRK2-depleted cells, Cdc25B phosphorylation is delayed, and we conclude that PRK2 activity is required for phosphorylation of Cdc25B at the G2/M transition. In agreement with this, and as previously described, siRNA-mediated depletion of Cdc25B phenocopies the effects of PRK2-depletion, causing a similar delay in G2/M progression. Interestingly, Cdc25B depletion also leads to a similar defect in cytokinesis and increased cell cycle duration as PRK2 depletion.
Cdc25 phosphatases play a crucial role in the activation of mitotic cyclin/Cdks at the onset of mitosis (Karlsson-Rosenthal and Millar, 2006). Mammalian cells have three Cdc25 isoforms, Cdc25A, Cdc25B and Cdc25C, and each have been implicated in the control of mitotic entry. Antibodies or dominant-negative versions of Cdc25B and Cdc25C block G2/M progression (Millar et al, 1991; Gabrielli et al, 1996; Lammer et al, 1998), although more recent studies employing siRNA suggest that Cdc25A and Cdc25B, but not Cdc25C, are crucial for mitotic entry (Mailand et al, 2002; Lindqvist et al, 2005). Cdc25B has been shown to be specifically required for the initial activation of cyclinB/Cdk1 at the centrosome, which triggers G2/M progression and precedes the activation of nuclear mitotic events (De Souza et al, 2000; Jackman et al, 2003; Lindqvist et al, 2005). On the other hand, Cdc25B−/− Cdc25C−/− mice develop normally, suggesting that Cdc25A can fulfill all Cdc25 protein functions (Ferguson et al, 2005). Some of these discrepancies are likely due to overlapping, if not redundant activities and cell type/species differences.
The regulation of Cdc25 phosphatase activity occurs at the level of protein expression, subcellular localization and phosphorylation (Karlsson-Rosenthal and Millar, 2006). Cdc25 proteins are substrates for many kinases, which can either activate or inhibit catalytic activity depending on the site of phosphorylation. In response to environmental stress or DNA damage, a G2/M checkpoint is induced and Cdc25 proteins are phosphorylated and inhibited by a number of kinases including Chk1, Chk2 and MAPKAPK-2, thus preventing cyclin/Cdk1 activation and cell cycle progression (Karlsson-Rosenthal and Millar, 2006). During normal cell cycle progression, other kinases, including Aurora-A, CK2, Cdk2 and Cdk1, have been implicated in phosphorylation and activation of Cdc25 (Karlsson-Rosenthal and Millar, 2006). Phosphorylation of Cdc25B by Aurora-A on Ser353 correlates with its centrosomal localization and cyclinB1/Cdk1 activation, although the in vitro catalytic activity of Cdc25B is not affected by Aurora-A phosphorylation (Dutertre et al, 2004; Cazales et al, 2005). siRNA depletion of Aurora-A in HeLa cells inhibits activation of cyclinB1/Cdk1 at the centrosome and thus entry into mitosis, and in addition, results in the formation of binucleated cells and increased apoptosis, similar to what we observe in PRK2- and Cdc25B-depleted cells (Hirota et al, 2003; Du and Hannon, 2004; Dutertre et al, 2004). Currently, we can only speculate how PRK2 contributes to Cdc25B activation, but given the similarity of some of the phenotypes of PRK2- and Aurora-A-depleted cells, it is possible that PRK2 lies upstream of Aurora-A kinase in the activation of Cdc25B. Alternatively, PRK2 might act parallel to Aurora-A and directly phosphorylate Cdc25B to stimulate its catalytic activity. Consistent with the latter hypothesis, we have shown that recombinant PRK1 and PRK2 are both able to phosphorylate CDC25B in vitro (Supplementary Figure S2). However, it remains to be determined whether this mechanism is responsible for PRK2-dependent CDC25B activation in the cell. Interestingly, PKN/PRK1 has been reported to interact directly with and phosphorylate Cdc25C in vitro, although in this case, phosphorylation seems to inhibit Cdc25C activity and so the relevance of these in vitro kinase assays to the work described here is unclear (Misaki et al, 2001).
A final question relates to how PRK2 is itself regulated during mitosis. Previous work has shown that the PRK/PKN family of proteins (PRK1, PRK2 and PRK3) can be activated through binding of active Rho at the N-terminus to disrupt an intra-molecular, autoinhibitory interaction (Mukai, 2003). Subsequent phosphorylation can further activate the kinase. We have shown here that PRK2 (and PRK1) is phosphorylated during mitosis and that this is dependent on Rho GTPases, Cdc25B and cyclin/Cdk1 activity (Figure 7). It seems likely, therefore, that a positive feedback loop is operating here, where PRK2 initially activates Cdc25B and cyclin/Cdk1, which then phosphorylate PRK2 to amplify its activity (Figure 7). In agreement with this, PRK2 phosphorylation is inhibited by roscovitine, a Cdk inhibitor. At present, we do not know which Rho GTPase is involved in PRK2 activation at G2/M, although it is thought to be mainly an effector for Rho and so this is the most likely candidate. However, PRK/PKN family members also have N-terminally located Rac binding sites and so we cannot rule out a role for other Rho family GTPases (Vincent and Settleman, 1997; Flynn et al, 1998). Interestingly, although Ect2 is the GEF acting upstream of Rho in controlling contractile ring formation and, as we show here, in localizing PRK2 to the midbody during abscission, the activation and phosphorylation of PRK2 at G2/M are not Ect2-dependent. We are currently trying to identify the GEF involved.
In conclusion, we describe here two new activities controlled by Rho GTPases and the PRK/PKN kinase family that are required for progression through the mitotic phase of the cell cycle. PRK2 is required for activation of Cdc25B to allow entry into mitosis, and for abscission to allow exit from cytokinesis into G1 phase.
Materials and methods
Plasmids, siRNAs, antibodies and reagents
Plasmid expressing full-length myc-tagged PRK2 was obtained from H Mellor (University of Bristol, UK). Gst-PRK1 and Gst-PRK2 proteins were from Invitrogen. PRK2 A siRNA was from Ambion (siRNA #781), Ect2 siRNA (5′-GAUAAAGGAUGAUCUUGAAUU-3′), PRK2 B siRNA (5′-GGAGCGCUCUGAUGGACAAUU-3′), PRK1 siRNA (Dharmacon Smartpool) and Cdc25B siRNA were from Dharmacon. Sequence for Cdc25B siRNA, 5′-UCCUCCCUGUCGUCUGAAUUU-3′, was previously published (Lindqvist et al, 2004). Antibodies were obtained as follows: monoclonal anti-PRK1 and anti-PRK2 antibodies from BD Transduction Laboratories, anti-Ect2 antibodies from H Sillje and E Nigg (MPI, Munich, Germany) and Santa Cruz (C-20), anti-myc 9E10 from S Moss (University College London, UK), anti-P-Histone3 from Upstate, anti-cyclin A (H-432), cyclin B1 (GNSI) and Cdc25B (C-20) from Santa Cruz, anti-Cdk1 and P-Cdk1 from Cell Signaling, anti-BrdU from Roche Diagnostics and anti-tubulin from Sigma. λ phosphatase was obtained from New England Biolabs and thymidine, BrdU and roscovitine were from Sigma. Hoechst 33342 was purchased from Molecular Probes and Toxin B from Calbiochem.
Cell culture and transfections
HeLa S3 cells were maintained in DMEM supplemented with 10% FCS and penicillin/streptomycin (100 IU/ml and 100 μg/ml) and incubated at 37°C and 5% CO2. U373 cells were maintained in MEM supplemented with 10% heat-inactivated FCS, 5% non-essential amino acids, 20 mM HEPES and penicillin/streptomycin (100 IU/ml and 100 μg/ml) and incubated at 37°C and 5% CO2. DNA transfections were performed using GeneJuiceTM (Novagen) according to the manufacturer's specifications. Transfection of siRNA was performed using OligofectamineTM (Invitrogen). Briefly, HeLa S3 and U373 cells were seeded in six-well plates at a density of 20% and transfected the next day. For transfections, 3 μl of OligofectamineTM was incubated with 200 μl of Opti-MEM (Invitrogen) for 5 min at room temperature. The mixture was added to 30–60 pmol siRNA and further incubated at room temperature for 15 min before addition to the cells. The medium was replaced the next day.
Preparation of cell extracts
For preparation of whole cell extracts, cells from six-well plates were lysed in 150 μl of cold lysis buffer (20 mM Tris–HCl (pH 7.5), 100 mM NaCl, 1% Triton X-100, 12 mM β-glycerophosphate, 5 mM EGTA, 0.5% deoxycholate, 1 mM DTT, 10 mM NaF, 1 mM Na3VO4, 0.1 mM PMSF, 20 μg/ml aprotinin and leupeptin), mixed with 35 μl of 5 × SDS protein sample buffer, boiled for 3 min, sonicated and analysed by SDS–PAGE and Western blotting. For immunoprecipitation (IP) and λ phosphatase treatment, cells were lysed in 600 μl of cold IP buffer (50 mM Tris–HCl (pH 8), 150 mM NaCl, 1% Nonident P-40, 12 mM β-glycerophosphate, 0.5% deoxycholate, 1 mMT DTT, 10 mM NaF, 1 mM Na3VO4, 0.1 mM PMSF, 20 μg/ml aprotinin and leupeptin). Cell debris was pelleted by centrifugation at 14 000 r.p.m. for 5 min at 4°C and lysates were incubated for 2 h at 4°C with anti-PRK2 antibody and 20 μl protein G-Sepharose. Immunoprecipitates were collected by centrifugation and washed 3 × in IP buffer without phosphatase inhibitors and once with λ phosphatase buffer containing 2 mM MnCl2. Beads were resuspended in 40 μl of λ phosphatase buffer containing 2 mM MnCl2 and 400 U λ phosphatase was added and the reaction incubated at 30°C for 30 min. Immunoprecipitated proteins were eluted from the beads with SDS protein sample buffer and analysed by SDS–PAGE and Western blotting.
Immunofluorescence
HeLa S3 and U373 cells were plated onto glass coverslips and transfected as described above. For immunofluorescence analysis of cells transfected with epitope-tagged plasmids, cells were fixed in 4% paraformaldehyde for 15 min at room temperature and stained for the appropriate epitope tags, as previously described (Nobes and Hall, 1999). For staining of endogenous proteins, cells were fixed in −20°C methanol for 10 min and then stained as described. For staining with anti-BrdU antibody, cells were fixed in 4% paraformaldehyde for 15 min, incubated with 0.5% Triton X-100 in 0.2 M HCl, blocked with 5% BSA in PBS and then stained as described. Fluorescence images were recorded on a CCD camera and processed using Openlab and Adobe Photoshop software.
Cell synchronization
For synchronization experiments, cells were seeded in six-well plates at a density of 20%, transfected as described and immediately after transfection incubated with 2 mM thymidine for 24 h. Cells were washed 3 × with PBS and released into fresh medium for 8 h, followed by a second round of incubation with 2 mM thymidine for 15 h, 3 × washing with PBS and release into fresh medium (t=0 h). Where indicated, BrdU was added after release into fresh medium (t=0 h), and Toxin B or roscovitine was added 6 and 8 h, respectively, after release into fresh medium. Cells were harvested at the indicated time points and whole cell extracts were prepared as described above.
FACS analysis
For FACS analysis, cells were transfected with siRNA as described and harvested after 72 h with trypsin, washed twice in PBS and fixed with −20°C 70% ethanol for 30 min at 4°C. Cells were washed twice with ice-cold PBS, incubated with 6 μg/ml propidium iodide, and 10 μg/ml RNAse for 30 min at room temperature and analysed for DNA content by flow cytometry on a FACSCalibur (Becton Dickinson).
Time-lapse analysis
Time-lapse microscopy was carried out on an Axiovert S100 equipped with a Qimaging Qicam camera. Images were analysed using Openlab and Adobe Photoshop software. Cell cycle length was calculated as the time between entry into mitosis (rounding up of cells) and entry into mitosis of the next division cycle.
In vitro kinase assays
In vitro kinase assays were performed using 250 ng of recombinant, human GST-PRK1/PKN1 or GST-PRK2/PKN2 (Invitrogen) and either 1 μg of glutathione S-transferase (GST; Sigma), 0.5 μg of myelin basic protein (MBP; Sigma) or 1.5 μg of Cdc25B (Biomol) as a substrate in 50 μl of the following assay buffer: 12.5 mM Tris (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.5 mM Na3VO4, 5 mM β-glycerophosphate, 2.5 mM DTT, 0.01% Triton X-100, 50 μM ATP and 5 μCi [γ-32P]ATP (Amersham). Reactions were initiated with the addition of ATP, incubated for 30 min at 30°C and stopped with 25 μl of kinase assay sample buffer: 75% (v/v) 4 × LDS sample buffer (Invitrogen), 0.2 M dithiothreitol, 4% (v/v) β-mercaptoethanol and 5 mM EDTA. Samples were boiled at 100°C for 5 min, resolved by SDS–PAGE and Coomassie stained/destained. The gel was then exposed to a Kodak Phosphor Screen K and analysed using the Personal Molecular Imager System according to the manufacturer's instructions (Bio-Rad).
Supplementary Material
Supplementary Figures S1 and S2
Acknowledgments
We are grateful to Harry Mellor, Herman Sillje and Erich Nigg for reagents, Chris Marshall for help with the RNAi library construction, Derek Davies and Nathalie Signoret for advice on FACS analysis, Volker Stucke for technical advice and members of the laboratory for valuable discussion. The work was generously supported by a programme grant for Cancer Research UK.
References
- Bakal CJ, Finan D, LaRose J, Wells CD, Gish G, Kulkarni S, DeSepulveda P, Wilde A, Rottapel R (2005) The Rho GTP exchange factor Lfc promotes spindle assembly in early mitosis. Proc Natl Acad Sci USA 102: 9529–9534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazales M, Schmitt E, Montembault E, Dozier C, Prigent C, Ducommun B (2005) CDC25B phosphorylation by Aurora-A occurs at the G2/M transition and is inhibited by DNA damage. Cell Cycle 4: 1233–1238 [DOI] [PubMed] [Google Scholar]
- Coleman ML, Marshall CJ, Olson MF (2004) RAS and RHO GTPases in G1-phase cell-cycle regulation. Nat Rev Mol Cell Biol 5: 355–366 [DOI] [PubMed] [Google Scholar]
- De Souza CP, Ellem KA, Gabrielli BG (2000) Centrosomal and cytoplasmic Cdc2/cyclin B1 activation precedes nuclear mitotic events. Exp Cell Res 257: 11–21 [DOI] [PubMed] [Google Scholar]
- Di Cunto F, Imarisio S, Hirsch E, Broccoli V, Bulfone A, Migheli A, Atzori C, Turco E, Triolo R, Dotto GP, Silengo L, Altruda F (2000) Defective neurogenesis in citron kinase knockout mice by altered cytokinesis and massive apoptosis. Neuron 28: 115–127 [DOI] [PubMed] [Google Scholar]
- Du J, Hannon GJ (2004) Suppression of p160ROCK bypasses cell cycle arrest after Aurora-A/STK15 depletion. Proc Natl Acad Sci USA 101: 8975–8980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutertre S, Cazales M, Quaranta M, Froment C, Trabut V, Dozier C, Mirey G, Bouche JP, Theis-Febvre N, Schmitt E, Monsarrat B, Prigent C, Ducommun B (2004) Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2–M transition. J Cell Sci 117: 2523–2531 [DOI] [PubMed] [Google Scholar]
- Eda M, Yonemura S, Kato T, Watanabe N, Ishizaki T, Madaule P, Narumiya S (2001) Rho-dependent transfer of Citron-kinase to the cleavage furrow of dividing cells. J Cell Sci 114: 3273–3284 [DOI] [PubMed] [Google Scholar]
- Ferguson AM, White LS, Donovan PJ, Piwnica-Worms H (2005) Normal cell cycle and checkpoint responses in mice and cells lacking Cdc25B and Cdc25C protein phosphatases. Mol Cell Biol 25: 2853–2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn P, Mellor H, Palmer R, Panayotou G, Parker PJ (1998) Multiple interactions of PRK1 with RhoA. Functional assignment of the Hr1 repeat motif. J Biol Chem 273: 2698–2705 [DOI] [PubMed] [Google Scholar]
- Gabrielli BG, De Souza CP, Tonks ID, Clark JM, Hayward NK, Ellem KA (1996) Cytoplasmic accumulation of cdc25B phosphatase in mitosis triggers centrosomal microtubule nucleation in HeLa cells. J Cell Sci 109 (Part 5): 1081–1093 [DOI] [PubMed] [Google Scholar]
- Glotzer M (2005) The molecular requirements for cytokinesis. Science 307: 1735–1739 [DOI] [PubMed] [Google Scholar]
- Gotta M, Abraham MC, Ahringer J (2001) CDC-42 controls early cell polarity and spindle orientation in C. elegans. Curr Biol 11: 482–488 [DOI] [PubMed] [Google Scholar]
- Gould KL, Nurse P (1989) Tyrosine phosphorylation of the fission yeast cdc2+ protein kinase regulates entry into mitosis. Nature 342: 39–45 [DOI] [PubMed] [Google Scholar]
- Gross C, Heumann R, Erdmann KS (2001) The protein kinase C-related kinase PRK2 interacts with the protein tyrosine phosphatase PTP-BL via a novel PDZ domain binding motif. FEBS Lett 496: 101–104 [DOI] [PubMed] [Google Scholar]
- Gruneberg U, Neef R, Li X, Chan EH, Chalamalasetty RB, Nigg EA, Barr FA (2006) KIF14 and citron kinase act together to promote efficient cytokinesis. J Cell Biol 172: 363–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann L, Dittmar T, Erdmann KS (2003) The protein tyrosine phosphatase PTP-BL associates with the midbody and is involved in the regulation of cytokinesis. Mol Biol Cell 14: 230–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota T, Kunitoku N, Sasayama T, Marumoto T, Zhang D, Nitta M, Hatakeyama K, Saya H (2003) Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell 114: 585–598 [DOI] [PubMed] [Google Scholar]
- Jackman M, Lindon C, Nigg EA, Pines J (2003) Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat Cell Biol 5: 143–148 [DOI] [PubMed] [Google Scholar]
- Jaffe AB, Hall A (2005) Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 21: 247–269 [DOI] [PubMed] [Google Scholar]
- Kamijo K, Ohara N, Abe M, Uchimura T, Hosoya H, Lee JS, Miki T (2006) Dissecting the role of Rho-mediated signaling in contractile ring formation. Mol Biol Cell 17: 43–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson-Rosenthal C, Millar JB (2006) Cdc25: mechanisms of checkpoint inhibition and recovery. Trends Cell Biol 16: 285–292 [DOI] [PubMed] [Google Scholar]
- Kosako H, Yoshida T, Matsumura F, Ishizaki T, Narumiya S, Inagaki M (2000) Rho-kinase/ROCK is involved in cytokinesis through the phosphorylation of myosin light chain and not ezrin/radixin/moesin proteins at the cleavage furrow. Oncogene 19: 6059–6064 [DOI] [PubMed] [Google Scholar]
- Lammer C, Wagerer S, Saffrich R, Mertens D, Ansorge W, Hoffmann I (1998) The cdc25B phosphatase is essential for the G2/M phase transition in human cells. J Cell Sci 111 (Part 16): 2445–2453 [DOI] [PubMed] [Google Scholar]
- Lindqvist A, Kallstrom H, Karlsson Rosenthal C (2004) Characterisation of Cdc25B localisation and nuclear export during the cell cycle and in response to stress. J Cell Sci 117: 4979–4990 [DOI] [PubMed] [Google Scholar]
- Lindqvist A, Kallstrom H, Lundgren A, Barsoum E, Rosenthal CK (2005) Cdc25B cooperates with Cdc25A to induce mitosis but has a unique role in activating cyclin B1-Cdk1 at the centrosome. J Cell Biol 171: 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabuchi I, Hamaguchi Y, Fujimoto H, Morii N, Mishima M, Narumiya S (1993) A rho-like protein is involved in the organisation of the contractile ring in dividing sand dollar eggs. Zygote 1: 325–331 [DOI] [PubMed] [Google Scholar]
- Madaule P, Eda M, Watanabe N, Fujisawa K, Matsuoka T, Bito H, Ishizaki T, Narumiya S (1998) Role of citron kinase as a target of the small GTPase Rho in cytokinesis. Nature 394: 491–494 [DOI] [PubMed] [Google Scholar]
- Maddox AS, Burridge K (2003) RhoA is required for cortical retraction and rigidity during mitotic cell rounding. J Cell Biol 160: 255–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N, Podtelejnikov AV, Groth A, Mann M, Bartek J, Lukas J (2002) Regulation of G(2)/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J 21: 5911–5920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettouchi A, Klein S, Guo W, Lopez-Lago M, Lemichez E, Westwick JK, Giancotti FG (2001) Integrin-specific activation of Rac controls progression through the G(1) phase of the cell cycle. Mol Cell 8: 115–127 [DOI] [PubMed] [Google Scholar]
- Millar JB, Blevitt J, Gerace L, Sadhu K, Featherstone C, Russell P (1991) p55CDC25 is a nuclear protein required for the initiation of mitosis in human cells. Proc Natl Acad Sci USA 88: 10500–10504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misaki K, Mukai H, Yoshinaga C, Oishi K, Isagawa T, Takahashi M, Ohsumi K, Kishimoto T, Ono Y (2001) PKN delays mitotic timing by inhibition of Cdc25C: possible involvement of PKN in the regulation of cell division. Proc Natl Acad Sci USA 98: 125–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai H (2003) The structure and function of PKN, a protein kinase having a catalytic domain homologous to that of PKC. J Biochem (Tokyo) 133: 17–27 [DOI] [PubMed] [Google Scholar]
- Niiya F, Tatsumoto T, Lee KS, Miki T (2006) Phosphorylation of the cytokinesis regulator ECT2 at G2/M phase stimulates association of the mitotic kinase Plk1 and accumulation of GTP-bound RhoA. Oncogene 25: 827–837 [DOI] [PubMed] [Google Scholar]
- Nishimura Y, Yonemura S (2006) Centralspindlin regulates ECT2 and RhoA accumulation at the equatorial cortex during cytokinesis. J Cell Sci 119: 104–114 [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A (1999) Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol 144: 1235–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oceguera-Yanez F, Kimura K, Yasuda S, Higashida C, Kitamura T, Hiraoka Y, Haraguchi T, Narumiya S (2005) Ect2 and MgcRacGAP regulate the activation and function of Cdc42 in mitosis. J Cell Biol 168: 221–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson MF, Ashworth A, Hall A (1995) An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science 269: 1270–1272 [DOI] [PubMed] [Google Scholar]
- Olson MF, Paterson HF, Marshall CJ (1998) Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature 394: 295–299 [DOI] [PubMed] [Google Scholar]
- Palmer RH, Ridden J, Parker PJ (1995) Cloning and expression patterns of two members of a novel protein-kinase-C-related kinase family. Eur J Biochem 227: 344–351 [DOI] [PubMed] [Google Scholar]
- Piekny A, Werner M, Glotzer M (2005) Cytokinesis: welcome to the Rho zone. Trends Cell Biol 15: 651–658 [DOI] [PubMed] [Google Scholar]
- Schmidt A, Hall A (2002) Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev 16: 1587–1609 [DOI] [PubMed] [Google Scholar]
- Schweitzer JK, D'Souza-Schorey C (2004) Finishing the job: cytoskeletal and membrane events bring cytokinesis to an end. Exp Cell Res 295: 1–8 [DOI] [PubMed] [Google Scholar]
- Shandala T, Gregory SL, Dalton HE, Smallhorn M, Saint R (2004) Citron kinase is an essential effector of the Pbl-activated Rho signalling pathway in Drosophila melanogaster. Development 131: 5053–5063 [DOI] [PubMed] [Google Scholar]
- Takahashi M, Shibata H, Shimakawa M, Miyamoto M, Mukai H, Ono Y (1999) Characterization of a novel giant scaffolding protein, CG-NAP, that anchors multiple signaling enzymes to centrosome and the golgi apparatus. J Biol Chem 274: 17267–17274 [DOI] [PubMed] [Google Scholar]
- Tatsumoto T, Xie X, Blumenthal R, Okamoto I, Miki T (1999) Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J Cell Biol 147: 921–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent S, Settleman J (1997) The PRK2 kinase is a potential effector target of both Rho and Rac GTPases and regulates actin cytoskeletal organization. Mol Cell Biol 17: 2247–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh CF, Roovers K, Villanueva J, Liu Y, Schwartz MA, Assoian RK (2001) Timing of cyclin D1 expression within G1 phase is controlled by Rho. Nat Cell Biol 3: 950–957 [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Marui N, Sakai T, Morii N, Kozaki S, Ikai K, Imamura S, Narumiya S (1993) ADP-ribosylation of the rhoA gene product by botulinum C3 exoenzyme causes Swiss 3T3 cells to accumulate in the G1 phase of the cell cycle. Oncogene 8: 1449–1455 [PubMed] [Google Scholar]
- Yasuda S, Oceguera-Yanez F, Kato T, Okamoto M, Yonemura S, Terada Y, Ishizaki T, Narumiya S (2004) Cdc42 and mDia3 regulate microtubule attachment to kinetochores. Nature 428: 767–771 [DOI] [PubMed] [Google Scholar]
- Yuce O, Piekny A, Glotzer M (2005) An ECT2-centralspindlin complex regulates the localization and function of RhoA. J Cell Biol 170: 571–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1 and S2