

Abstract
PhIP carcinogenesis is initiated by N2-hydroxylation, mediated by several cytochromes P450, including CYP1A1. However, the role of CYP1A1 in PhIP metabolic activation in vivo is unclear. In this study, Cyp1a1-null and wild-type (WT) mice were used to investigate the potential role of CYP1A1 in PhIP metabolic activation in vivo. PhIP N2-hydroxylation was actively catalyzed by lung homogenates of WT mice, at a rate of 14.9 ± 5.0 pmol/min/g tissue, but < 1 pmol/min/g tissue in stomach and small intestine, and almost undetectable in mammary gland and colon. PhIP N2-hydroxylation catalyzed by lung homogenates of Cyp1a1-null mice was ~10-fold lower than that of WT mice. In contrast, PhIP N2-hydroxylation activity in lung homogenates of Cyp1a2-null versus WT mice was not decreased. Pretreatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) increased lung Cyp1a1 mRNA and lung homogenate PhIP N2-hydroxylase activity ~50-fold in WT mice, where the activity was substantially inhibited (70%) by monoclonal antibodies against CYP1A1. In vivo, 30 min after oral treatment with PhIP, PhIP levels in lung were similar to those in liver. After a single dose of 0.1 mg/kg [14C]PhIP, lung PhIP-DNA adduct levels in Cyp1a1-null mice, but not in Cyp1a2-null mice, were significantly lower (P=0.0028) than in WT mice. These results reveal that mouse lung has basal and inducible PhIP N2-hydroxylase activity predominantly catalyzed by CYP1A1. Because of the high inducibility of human CYP1A1, especially in cigarette smokers, the role of lung CYP1A1 in PhIP carcinogenesis should be considered.
Abbreviations: 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP); N2-hydroxy-PhIP (N-OH-PhIP); 4′-hydroxy-PhIP (4-OH-PhIP); 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD); cytochrome P450 (CYP)
Introduction
2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), the most abundant dietary heterocyclic amine, is carcinogenic in multiple organs and in numerous species. PhIP carcinogenesis is initiated by PhIP N2-hydroxylation [1], which has been reported to be primarily catalyzed by CYP1A2 [2–4]. However, recent studies raise the possibility of the CYP1A2-independent pathways for PhIP metabolic activation. In humans, excretion of the glucuronide conjugate of N2-hydroxy PhIP (N-OH-PhIP) in urine shows a low correlation with CYP1A2 activity [5]. In a mouse carcinogen bioassay with PhIP, the incidence of lymphomas and hepatocellular adenomas is higher in Cyp1a2-null mice than WT mice [6]. Moreover, PhIP-DNA adducts are detectable in mammary gland and colon in Cyp1a2-null mice [7].
Multiple in vitro studies have revealed that CYP1A1 catalyzes PhIP metabolic activation [8–11]. cDNA-expressed human CYP1A2, CYP1A1 and CYP1B1, respectively catalyze N-OH-PhIP formation with maximal velocities (Vmax) of 90, 16 and 0.2 nmol/min/nmol P450, and apparent Km values of 79, 5.1 and 4.5 μM [12]. Thus, although CYP1A2 exhibited the highest Vmax for N-OH-PhIP production, the catalytic efficiency (Vmax/Km) of CYP1A1 is 2.75-fold higher than that of CYP1A2. With CYP1B1, PhIP N2-hydroxylation activity is 80-fold lower than that of CYP1A1 and formation of the non-mutagenic metabolite 4-OH-PhIP is favored ~2-fold over that of the promutagenic metabolite N-OH-PhIP [12,13]. Given the high catalytic efficiency of CYP1A1 for PhIP metabolic activation, it is important to establish the potential role of extrahepatic CYP1A1 in PhIP metabolism and carcinogenesis. Such studies are particularly important because PhIP induces tumors in several extrahepatic organs. In the current study, PhIP N2-hydroxylase activity was detected in extrahepatic organs including lung, stomach, small intestine, colon, and mammary gland. Cyp1a2-null mice were used to examine CYP1A2-independent PhIP metabolic pathways and Cyp1a1-null mice were used to assess the potential role of CYP1A1 in PhIP metabolic activation.
Materials and Methods
Chemicals and reagents
PhIP and N-OH-PhIP were obtained from the National Cancer Institute Chemical Carcinogen Reference Standard Repository at the Midwest Research Institute (Kansas City, MO). [14C]PhIP, > 95% radio-pure, was purchased from Toronto Research Chemicals Inc. (Ontario, Canada). 4′-Hydroxy-PhIP (4-OH-PhIP) was provided by Dr. M. Nagao (National Cancer Research Center, Tokyo, Japan). NADPH and 6-chloromelatonin were purchased from Sigma-Aldrich (St. Louis, MO). Monoclonal antibodies against CYP1A1 (mAb 1-7-1) were used to study CYP1A1 inhibition [14,15]. High-performance liquid chromatography solvents and other chemicals were of the highest grade commercially available. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) was obtained from Dr. Steven Safe at Texas A&M University.
Animals and treatments
Cyp1a1-null mice [16], Cyp1a2-null mice [17], and their corresponding wild-type (WT) mice, female, 2–4 months old, were maintained under a standard 12 h light/12 h dark cycle with water and chow provided ad libitum. Handling was in accordance with animal study protocols approved by the National Cancer Institute Animal Care and Use Committee. For CYP1A1 induction, WT and Cyp1a1-null mice were injected i.p. with 10 μg/kg TCDD and sacrificed 72 h after dosing. TheTCDD dose and time was chosen were based on earlier published studies showing maximal induction of CYP1A1 [18]. For the in vivo PhIP distribution, WT mice were treated by oral gavage with 40 mg/kg PhIP. 30 min after PhIP treatment, mice were killed by CO2 asphyxiation; lung, liver, mammary gland and colon were collected and frozen at −80 °C for further analysis. For PhIP-DNA adducts detection, Cyp1a1-null mice, Cyp1a2-null mice, and WT mice were treated orally with 100 μg/kg of [14C]PhIP (6.3 μCi/kg). The mice were sacrificed 18 h after administration, and tissues were collected and frozen at −80 °C for further analysis.
PhIP metabolism in extrahepatic organs
PhIP metabolism in extrahepatic organs was detected in vitro using tissue homogenates. To prepare homogenates, each organ was washed with 20 mM phosphate-buffered saline (PBS), pH 7.4, and homogenized (500 μl PBS/100 mg tissue) by a motor-driven Teflon-tipped pestle. PhIP metabolism in homogenates (20 mg tissue) were assayed in a final volume of 400 μl, containing 20 mM PBS, pH 7.4, 10 μM PhIP and 1 mM NADPH. To assess the role of CYP1A1 in PhIP metabolism, monoclonal antibodies against CYP1A1 (mAb 1-7-1) were pre-incubated for 5 min with lung homogenates at 37 °C. Reactions were initiated by the addition of NADPH and terminated 10 min later by the addition of 1.0 ml ethyl acetate and 1.0 ml methyl tert-butyl ether. 6-Chloromelatonin (5 μl of 100 μM stock solution) was added as an internal standard. Samples were centrifuged at 3000 rpm for 5 min at 4 °C. The organic layer was then transferred to a new tube, dried with N2 and reconstituted in 100 μl of 70% methanol and 30% H2O containing 0.1% formic acid. All reactions were performed in duplicate. PhIP metabolites were detected by LC-MS/MS.
PhIP tissue distribution
PhIP in various tissues was extracted 30 min after a single oral dose of PhIP administered to WT mice. Briefly, 100 mg of each tissue was homogenized in 500 μl PBS, and 300 μl of each homogenate was extracted with a mixture of 1.0 ml ethyl acetate and 1.0 ml methyl tert-butyl ether. 6-Chloromelatonin (5 μl of 100 μM solution) was added as an internal standard. The extracted samples were dried with N2 and reconstituted in 100 μl of 70% methanol and 30% H2O containing 0.1% formic acid. 6 μl of each sample was used for LC-MS/MS analysis.
Identification and quantification of PhIP and PhIP metabolites by LC-MS/MS
PhIP metabolites were detected by LC-MS/MS, as described previously [4]. Briefly, LC-MS/MS analysis was performed using a PE SCIEX API 2000 ESI triple quadrupole mass spectrometer (PerkinElmer/ABI, Foster City, CA). A Luna C18 50 mm x 4.6 mm i.d. column (Phenomenex, Torrance, CA) was used to separate PhIP metabolites. The flow rate through the column at ambient temperature was 0.25 ml/min with 70% methanol and 30% H2O containing 0.1% formic acid. The mass spectrometer was equipped with a turbo ion spray source and run in the positive ion mode. The turbo ion spray temperature was maintained at 350 °C and a voltage of 4.8 kV was applied to the sprayer needle. N2 was used as the turbo ion spray and nebulizing gas. Identification and quantification of PhIP metabolites and the internal standard were accomplished by multiple reactions monitoring (MRM) with the transitions m/z 225.2/210.2 for PhIP, 241.2/223.2 for N-OH-PhIP, 241.2/226.2 for 4-OH-PhIP, and 267.0/208.4 for 6-Chloromelatonin.
Lung Cyp1a1 mRNA detection
Relative levels of mouse lung Cyp1a1 RNA were quantified by real time quantitative PCR (qPCR) using SYBR Green I chemistry. Total RNA was isolated from individual mouse lung samples obtained from untreated and TCDD-treated WT and Cyp1a1-null mice, using Trizol reagent (Invitrogen Life Technologies, Carlsbad, CA) and the manufacturer’s protocol for tissue. The isolated RNA was treated with ribonuclease-free deoxyribonuclease followed by reverse transcription using random hexamers and murine leukemia virus reverse transcriptase to yield cDNA using the GeneAmp RNA core kit (Applied Biosystems, Foster City, CA). One μg of total lung RNA was converted to cDNA in a final vol of 20 μl according to the manufacturer’s protocol. Triplicate samples of each qPCR mixture, each containing 4 μl of SYBR Green I PCR master mix (Applied Biosystems), were transferred into separate wells of a 384-well plate and run through 40 cycles on an ABS 7900HT Sequence Detection System (Applied Biosystems) [19]. Samples were initially incubated at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Dissociation curves were generated after each qPCR run to ensure that a single, specific product was amplified. Results were analyzed using the comparative CT (ΔΔCT) method, as described in User Bulletin 2 of the ABI PRISM 7700 Sequence Detection System. Data are graphed as relative RNA values, normalized to the 18S rRNA content of each sample. Similar results were obtained by normalization to β-actin RNA (data not shown). qPCR primers for Cyp1a1 were as follows: forward primer ON-1582 (5′-GGT TAA CCA TGA CCG GGA ACT-3′) and reverse primer ON-1583 (5′-TGC CCA AAC CAA AGA GAG TGA), yielding an amplicon length of 122 nucleotides. The forward primer was designed to span the junction between exons 6 and 7 of the mouse Cyp1a1 gene (NM_009992) to eliminate amplification of any contaminating genomic DNA. Primers for β-actin (forward primer 5′-TCC ATC ATG AAG TGT GAC GTT-3′; reverse primer 5′-TGT GTT GGC ATA GAG GTC TTT ACG-3′) and 18S rRNA (forward primer 5′-CGC CGC TAG AGG TGA AA TC-3′; reverse primer 5′-CCA GTC GGC ATC GTT TAT GG-3′) were as shown. Primer specificity was verified by BLAST analysis.
Lung PhIP-DNA adduct detection
DNA isolation from mouse lung, and sample preparation for the quantification of DNA adduct levels by accelerator mass spectrometry (AMS) has been reported elsewhere [20]. Briefly, lung tissues were homogenized then digested in lysis buffer (4 M urea, 1.0% Triton X-100, 10 mM EDTA, 100 mM NaCl 10 mM DTT, 10 mM Tris-HCl, pH 8.0) containing 0.8 mg/ml proteinase K overnight at 37 °C. Undigested tissue was removed by centrifugation, and the supernatant was treated for 1 h at room temperature with RNase A, (0.5 mg/ml) and RNase T1 (5 μg/ml). DNA was extracted using Qiagen column chromatography (Qiagen, Valencia, CA) according to the manufacturer’s instructions. DNA purity was determined by the A260 nm/A280 nm ratio. A ratio between 1.6–1.8 was considered pure. Pure DNA samples were then submitted for adduct analysis by AMS.
Statistical analysis
All values are expressed as the means ± SD and analyzed by Student’s t test. P<0.05 was regarded as significantly different between the compared groups.
Results
PhIP metabolism in extrahepatic organs
Extrahepatic PhIP metabolism was investigated using tissue homogenates prepared from WT mouse lung, stomach, small intestine, colon and mammary gland. The velocity of PhIP N2-hydroxylation was highest in the lung, 14.9 ± 5.0 pmol/min/g tissue, whereas it was below 1 pmol/min/g tissue in stomach and small intestine, and almost undetectable in mammary gland and colon (Figure 1A). PhIP 4′-hydroxylation, a non-mutagenic metabolic pathway, exhibited similar tissue specificity. PhIP N2-hydroxylation in lung is NADPH- and enzyme(s)-dependent, as N-OH-PhIP was not detected in the absence of NADPH or tissue homogenate (Figure 1B).
Fig. 1.
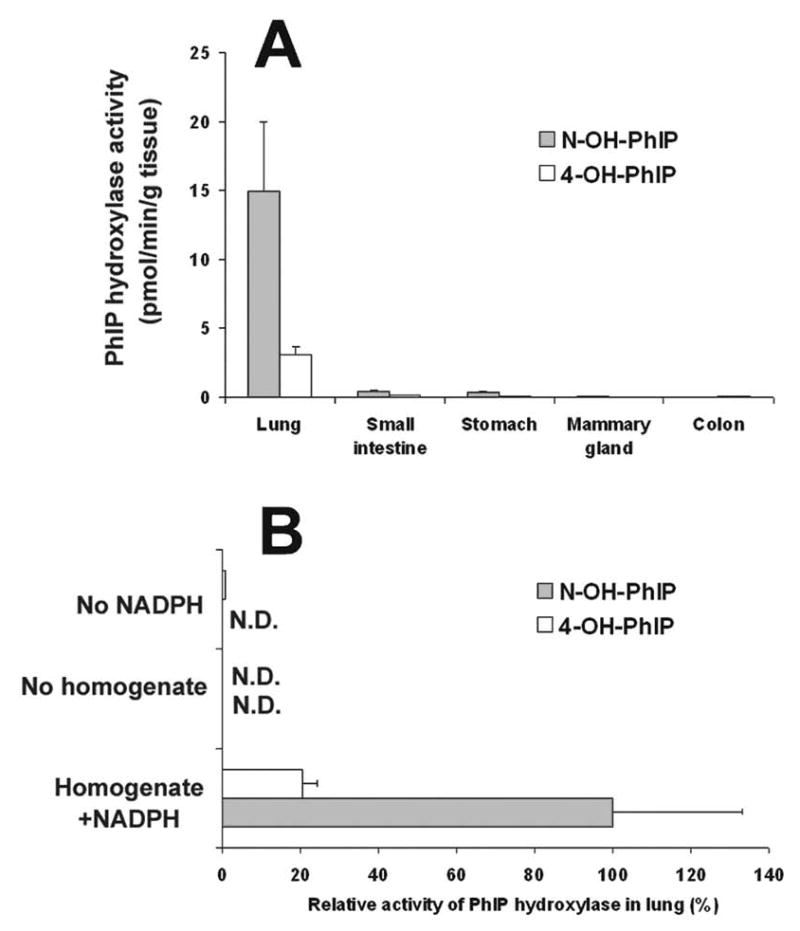
PhIP hydroxylase activity in extrahepatic organs of WT mice. (A) PhIP hydroxylase activity in homogenates of lung, small intestine, stomach, mammary gland and colon. (B) Relative PhIP hydroxylase activity in lung homogenates. The incubations were carried out with or without homogenates (20 mg tissue) and/or 1 mM NADPH. PhIP metabolites were detected by LC-MS/MS. Data are expressed as means ± SD (n=3).
PhIP metabolism in lung is CYP1A1-dependent
PhIP metabolism was assayed in lung homogenates prepared from Cyp1a1-null mice, Cyp1a2-null mice, and WT mice. There was a marked decrease in PhIP N2-hydroxylation and 4′-hydroxylation activities in Cyp1a1-null mouse lung homogenates (Figure 2B) compared to WT mice (Figure 2A). The ~10-fold lower PhIP N2-hydroxylase activity in Cyp1a1-null mouse lung homogenates contrasts with a ~50% higher PhIP N2-hydroxylase activity in Cyp1a2-null mouse lung homogenates (Figure 2C). The increase of PhIP N2-hydroxylase activity in Cyp1a2-null mouse lung homogenates can be significant inhibited (~90%) by monoclonal antibody (mAb) against CYP1A1. These data indicate that PhIP hydroxylation activity in lung is largely mediated by CYP1A1, rather than CYP1A2.
Fig. 2.
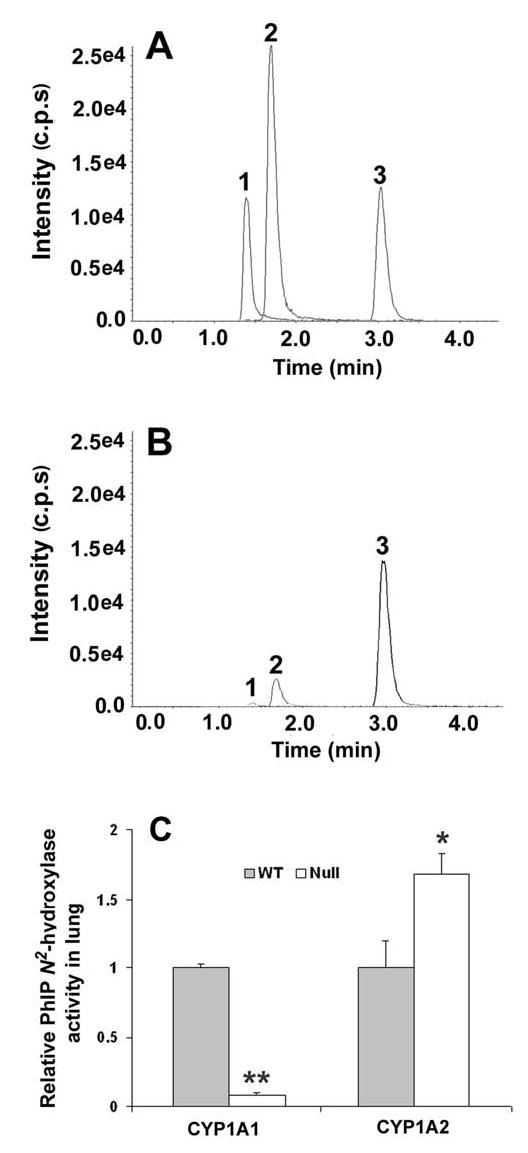
PhIP metabolism in lung homogenates. (A) Typical chromatography of PhIP metabolites in lung homogenates of WT mice. (B) Typical chromatography of PhIP metabolites in lung homogenates of Cyp1a1-null mice. PhIP metabolites were detected by LC-MS/MS, 241.2/226.2 for 4-OH-PhIP (1), 241.2/223.2 for N-OH-PhIP (2), and 267.0/208.4 for 6-Chloromelatonin (3, as an internal standard). (C) Relative quantification of PhIP N2-hydroxylase activity in lung homogenates of Cyp1a1-null mice and Cyp1a2-null mice. PhIP N2-hydroxylase activity in WT mouse lung homogenates was set as 1.0. Data are expressed as means ± SD (n=3). **P<0.01 and *P<0.05 compared with WT mice.
Effect of CYP1 inducer on PhIP hydroxylase activity in lung
In lung homogenates of WT mice pretreated with the CYP1 inducer TCDD, PhIP N2-hydroxylase activity was increased ~50-fold, to 636 ± 167 pmol/min/g tissue, compared to untreated WT controls (Figure 3A). Similar results were obtained for PhIP 4′-hydroxylation. PhIP N2-hydroxylation in lung homogenates of TCDD-treated Cyp1a1-null mice was ~80% lower than that of TCDD-treated WT mice (Figure 3B). Monoclonal antibody (mAb) against CYP1A1 inhibited PhIP N2-hydroxylation in WT lung by ~70%, but had no significant effect on that of Cyp1a1-null mice (Figure 3B). Consistent with the activity increase, lung Cyp1a1 mRNA was increased ~50-fold in WT mice after TCDD treatment. Moreover, Cyp1a1 mRNA was readily detectable in untreated WT mouse lung at ~ 2–2.5% the level of TCDD-treated WT mouse lung (Figure 4). While CYP1A1 protein was readily detected in TCDD-treated mouse lung, the protein was not detected in untreated mouse lung tissues, even though PhIP N2-hydroxylation activity was readily detected. This is due to the limited sensitivity of western blotting compared to the high sensitivity of the LC-MS for detection of the N2-hydroxy metabolite of PhIP.
Fig. 3.
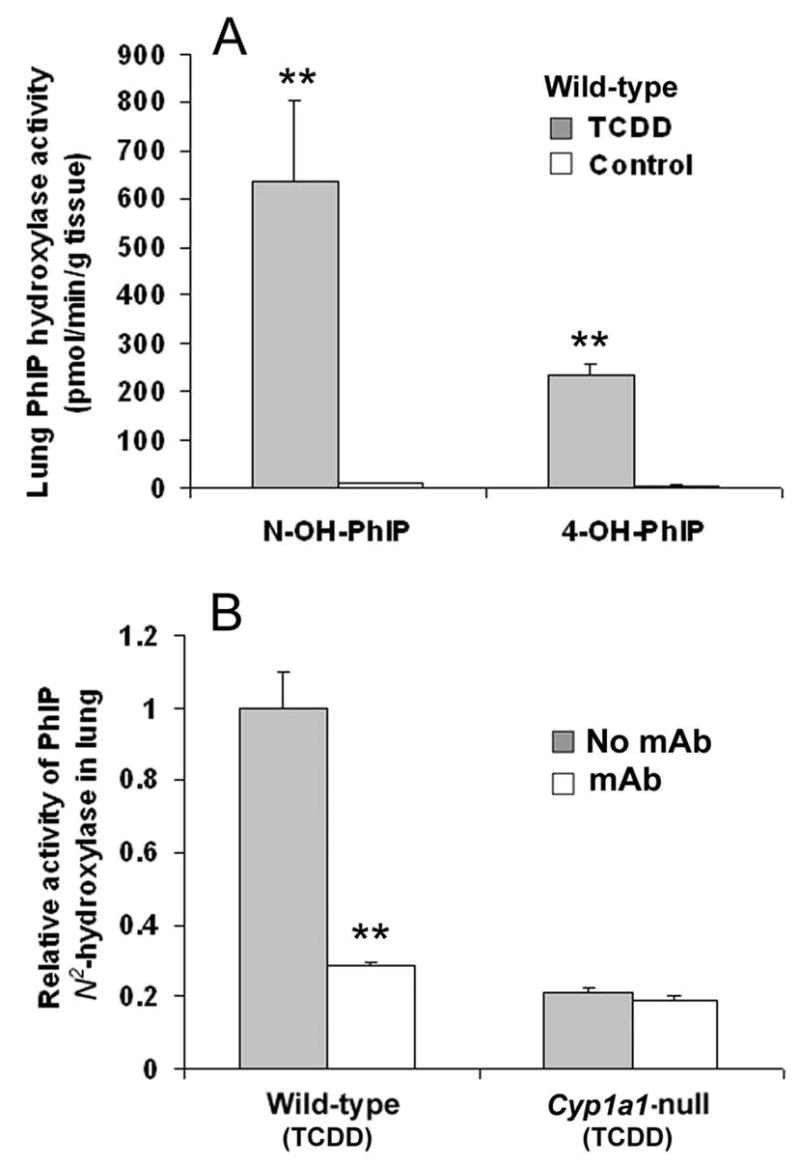
PhIP hydroxylase activity in lung homogenates of WT versus Cyp1a1-null mice, with or without TCDD pretreatment. (A) Comparison of PhIP hydroxylase activity in lung homogenates of untreated versus TCDD-treated WT mice. Data are expressed as means ± SD (n=3). **P<0.01 compared with that of untreated WT mice. (B) Comparison of PhIP N2-hydroxylase activity in lung homogenates of TCDD-treated WT versus Cyp1a1-null mice. PhIP N2-hydroxylase activity in WT lung homogenates without monoclonal antibodies (mAb) against CYP1A1 was set as 1.0. Data are expressed as means ± SD (n=3). **P<0.01 compared to that of WT without mAb.
Fig. 4.
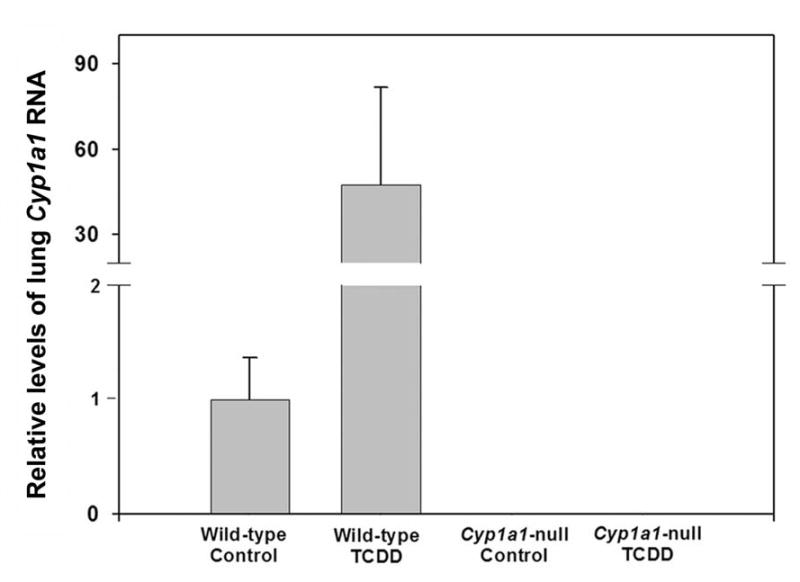
Relative levels of lung Cyp1a1 mRNA in untreated and TCDD-treated WT and Cyp1a1-null mice. Lung Cyp1a1 RNA was quantified by real time quantitative PCR (qPCR) using SYBR Green I chemistry. Data are graphed as relative values, normalized to the 18S rRNA content of each sample, and expressed as the means ± SE (n=6 for untreated, n=2 for TCDD-treated). Lung Cyp1a1 RNA in untreated WT group was set as 1.0.
PhIP distribution in mouse lung
30 min after oral PhIP treatment (40 mg/kg), mouse lung, liver, mammary gland and colon tissue were collected, and PhIP distribution was assayed by LC-MS/MS. Liver had the highest PhIP concentration at 46.4 nmol/g tissue (Figure 5). Interestingly, lung had similar PhIP level as liver (41.9 nmol/g tissue). PhIP concentration in mammary gland and colon was lower than that of liver and lung (22.1 and 29.4 nmol/g tissue, respectively). The high level of PhIP in lung raises the possibility of localized PhIP metabolic activation by lung CYP1A1. There was no significant difference in PhIP tissue distribution between WT, Cyp1a1-null, and Cyp1a2-null mice (data not shown).
Fig. 5.
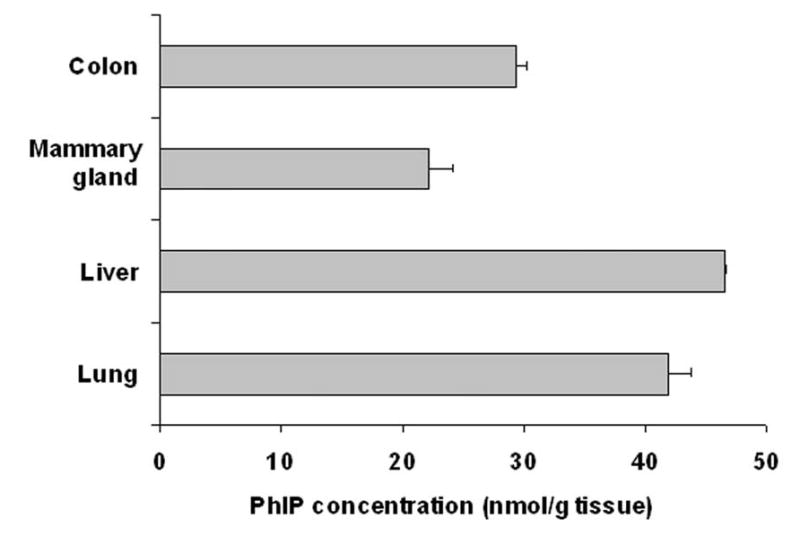
PhIP tissue distribution in WT mice, following oral administration of PhIP (40 mg/kg). 30 min after PhIP treatment, mouse lung, liver, mammary gland and colon were collected. PhIP concentration in various tissues was analyzed by LC-MS/MS. Data are expressed as means ± SD (n=3).
PhIP-DNA adducts in lung
PhIP-DNA adducts in lung of Cyp1a1-null mice, Cyp1a2-null mice, and WT mice were analyzed after a single dose of 100 μg/kg [14C]PhIP (6.3 μCi/kg). PhIP-DNA adducts were detected by accelerator mass spectrometry (AMS). PhIP-DNA adducts were significantly lower (46%) in Cyp1a1-null mouse lung than that in WT mouse lung (P=0.0028). In contrast, there was no significant change in PhIP-DNA adduct levels in Cyp1a2-null lung (P=0.177) (Figure 6).
Fig. 6.
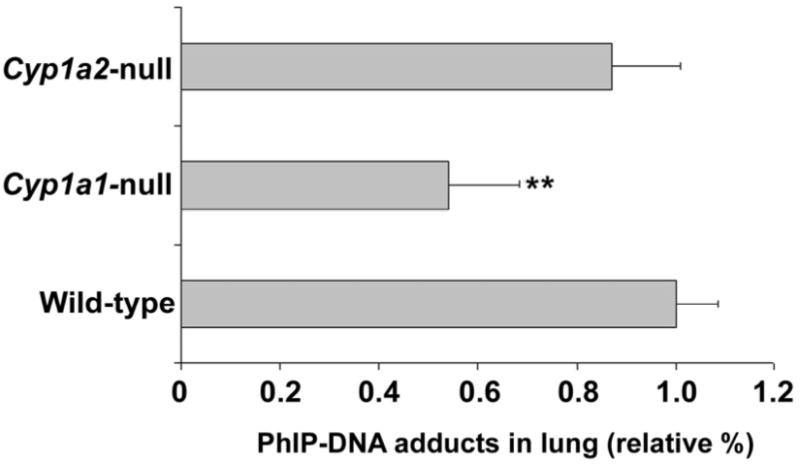
PhIP-DNA adducts in lung. Cyp1a1-null mice, Cyp1a2-null mice, and WT mice were treated orally with 100 μg/kg [14C]PhIP (6.3 μCi/kg). The mice were killed 18 h after administration, and lung DNA was isolated for PhIP-DNA adducts detection by accelerator mass spectrometry (AMS). PhIP-DNA adducts in WT lung were set as 1.0. Data are expressed as means ± SD (n=4). **P<0.01 compared with WT.
Discussion
PhIP N2-hydroxylation is considered as the initiating step in PhIP carcinogenesis [1]. Previous studies showed PhIP metabolic activation by lung microsomes, using mutagenicity as an endpoint [21]. In the present study using Cyp1a1-null versus Cyp1a2-null mice, PhIP N2-hydroxylase activity in lung was shown to be CYP1A1-dependent. Expression of CYP1A1 in human lung has been well characterized [22,23]; a centrilobular expression of CYP1A1 mRNA and CYP1A protein was observed in peripheral lung [24]. Both constitutive and inducible lung CYP1A1 have been quantified, and the median levels of CYP1A1 were 15.5 pmol/mg microsomal protein in smokers, 6.0 pmol/mg microsomal protein in nonsmokers, and 19.0 pmol/mg microsomal protein in ex-smokers [25]. Lung CYP1A1 activity is increased ~100-fold in smokers as compared with that of nonsmokers [26,27]. In the current study, lung CYP1A1 expression and PhIP N2-hydroxylase activity were readily detectable in untreated WT mice. After TCDD treatment, lung CYP1A1 RNA and PhIP N2-hydroxylase activity were both markedly induced. These results raise the possibility of localized CYP1A1-mediated PhIP metabolic activation in lung that is of importance due to different routes of PhIP exposure to lung. The diet is considered the most common source of PhIP, and orally administered PhIP has been shown to localized in lung [28]. In the present study with oral PhIP treatment, lung had a PhIP concentration similar to that in liver. Besides oral exposure, inhalation is another important pathway for PhIP exposure to the lung. PhIP is detected in all brands of filter-tipped cigarettes, and the mean level of PhIP in cigarette mainstream smoke has been estimated at 16.4 ng/cigarette [29]. PhIP is also detected in airborne particles, diesel-exhaust particles, and incineration ash from garbage-burning plants [30]. PhIP-DNA adduct is considered a reliable biomarker for PhIP exposure, and it has been detected in lung in several previous studies. In monkeys after a single oral dose of PhIP, DNA adducts were highest in the liver followed by the lung [31]. PhIP-DNA adducts in mouse lung has also been reported, and these levels were significantly decreased by inhibiting the activity of CYP1A1 and CYP1A2 [32]. In the present study, lung PhIP-DNA adducts were significant lower in lung tissue of Cyp1a1-null mice, but not Cyp1a2-null mice, compared to WT mice, which highlights the importance of CYP1A1 in PhIP metabolic action in lung. These data also demonstrate the utility of comparing various knockout mouse lines instead of relying on CYP inhibitor studies [33]. Compared to the in vitro data supporting a role for lung CYP1A1 (~90%) in PhIP N2-hydroxylation, the apparent contribution of CYP1A1 (~50%) to PhIP-DNA adduct formation is not particularly dominant. Perhaps other factors or enzymes may also contribute to the formation of PhIP-DNA adducts, including N-acetyltransferases, prolyl-tRNA synthetases, phosphorylases, sulfotransferases, UDP-glucuronyltransferase, and enzymes involved in DNA repair [34,35].
Lung cancer is the leading cause of death among women worldwide, and cigarette smoking and second-hand smoking are regarded as the predominant risk factor [36–38]. However, lung cancer also strikes women absent from smoking [39]. For example, Chinese women have a high incidence of lung cancer despite a low smoking prevalence, and domestic exposure to cooking fumes is proposed as an important risk factor [40]. Associations between Chinese-style cooking and lung cancer has been confirmed in several epidemiological investigations [41–44]. Procarcinogens, including PhIP and 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline (MeIQx), were identified in smoke condensates from the frying meat [45]. In the present study, localized procarcinogen metabolic activation by CYP1A1 in mouse lung has been demonstrated which may be an important risk factor for occupationally relevant human lung cancer. Because of the high efficiency of CYP1A1 in PhIP N2-hydroxylation and high inducibility of human CYP1A1, especially in smokers, the role of lung CYP1A1 in PhIP carcinogenesis should be considered.
Acknowledgments
This research was supported by the NCI Intramural Research Program, the Superfund Basic Research Program at Boston University (NIH Grant 5 P42 ES07381 to D.J.W.), NIH R01 ES08147 (D.W.N.) and the NIH National Center for Research Resources, Biomedical Technology Program grant #P41 RR13461 and NCI Grant CA55861 (J.S.F). This work was partially performed under the auspices of the U.S. Department of Energy by the University of California, Lawrence Livermore National Laboratory at the Research Resource for Biomedical AMS under contract No. W-7405-Eng-48. J.R.I. is grateful to U.S. Smokeless Tobacco Company for a grant for collaborative research. We thank John R. Buckley for technical assistance.
References
- 1.Holme JA, Wallin H, Brunborg G, Soderlund EJ, Hongslo JK, Alexander J. Genotoxicity of the food mutagen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP): formation of 2-hydroxamino-PhIP, a directly acting genotoxic metabolite. Carcinogenesis. 1989;10:1389–96. doi: 10.1093/carcin/10.8.1389. [DOI] [PubMed] [Google Scholar]
- 2.Boobis AR, Lynch AM, Murray S, de la Torre R, Solans A, Farre M, Segura J, Gooderham NJ, Davies DS. CYP1A2-catalyzed conversion of dietary heterocyclic amines to their proximate carcinogens is their major route of metabolism in humans. Cancer Res. 1994;54:89–94. [PubMed] [Google Scholar]
- 3.Turesky RJ, Guengerich FP, Guillouzo A, Langouet S. Metabolism of heterocyclic aromatic amines by human hepatocytes and cytochrome P4501A2. Mutat Res. 2002;506–507:187–95. doi: 10.1016/s0027-5107(02)00165-3. [DOI] [PubMed] [Google Scholar]
- 4.Cheung C, Ma X, Krausz KW, Kimura S, Feigenbaum L, Dalton TP, Nebert DW, Idle JR, Gonzalez FJ. Differential metabolism of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in mice humanized for CYP1A1 and CYP1A2. Chem Res Toxicol. 2005;18:1471–8. doi: 10.1021/tx050136g. [DOI] [PubMed] [Google Scholar]
- 5.Stillwell WG, Sinha R, Tannenbaum SR. Excretion of the N(2)-glucuronide conjugate of 2-hydroxyamino-1-methyl-6-phenylimidazo[4,5-b]pyridine in urine and its relationship to CYP1A2 and NAT2 activity levels in humans. Carcinogenesis. 2002;23:831–8. doi: 10.1093/carcin/23.5.831. [DOI] [PubMed] [Google Scholar]
- 6.Kimura S, Kawabe M, Yu A, Morishima H, Fernandez-Salguero P, Hammons GJ, Ward JM, Kadlubar FF, Gonzalez FJ. Carcinogenesis of the food mutagen PhIP in mice is independent of CYP1A2. Carcinogenesis. 2003;24:583–7. doi: 10.1093/carcin/24.3.583. [DOI] [PubMed] [Google Scholar]
- 7.Snyderwine EG, Yu M, Schut HA, Knight-Jones L, Kimura S. Effect of CYP1A2 deficiency on heterocyclic amine DNA adduct levels in mice. Food Chem Toxicol. 2002;40:1529–33. doi: 10.1016/s0278-6915(02)00110-2. [DOI] [PubMed] [Google Scholar]
- 8.Murray BP, Edwards RJ, Murray S, Singleton AM, Davies DS, Boobis AR. Human hepatic CYP1A1 and CYP1A2 content, determined with specific anti-peptide antibodies, correlates with the mutagenic activation of PhIP. Carcinogenesis. 1993;14:585–92. doi: 10.1093/carcin/14.4.585. [DOI] [PubMed] [Google Scholar]
- 9.Edwards RJ, Murray BP, Murray S, Schulz T, Neubert D, Gant TW, Thorgeirsson SS, Boobis AR, Davies DS. Contribution of CYP1A1 and CYP1A2 to the activation of heterocyclic amines in monkeys and human. Carcinogenesis. 1994;15:829–36. doi: 10.1093/carcin/15.5.829. [DOI] [PubMed] [Google Scholar]
- 10.Hammons GJ, Milton D, Stepps K, Guengerich FP, Tukey RH, Kadlubar FF. Metabolism of carcinogenic heterocyclic and aromatic amines by recombinant human cytochrome P450 enzymes. Carcinogenesis. 1997;18:851–4. doi: 10.1093/carcin/18.4.851. [DOI] [PubMed] [Google Scholar]
- 11.Yamazaki Y, Fujita K, Nakayama K, Suzuki A, Nakamura K, Yamazaki H, Kamataki T. Establishment of ten strains of genetically engineered Salmonella typhimurium TA1538 each co-expressing a form of human cytochrome P450 with NADPH-cytochrome P450 reductase sensitive to various promutagens. Mutat Res. 2004;562:151–62. doi: 10.1016/j.mrgentox.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 12.Crofts FG, Sutter TR, Strickland PT. Metabolism of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine by human cytochrome P4501A1, P4501A2 and P4501B1. Carcinogenesis. 1998;19:1969–73. doi: 10.1093/carcin/19.11.1969. [DOI] [PubMed] [Google Scholar]
- 13.Crofts FG, Strickland PT, Hayes CL, Sutter TR. Metabolism of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) by human cytochrome P4501B1. Carcinogenesis. 1997;18:1793–8. doi: 10.1093/carcin/18.9.1793. [DOI] [PubMed] [Google Scholar]
- 14.Park SS, Fujino T, West D, Guengerich FP, Gelboin HV. Monoclonal antibodies that inhibit enzyme activity of 3-methylcholanthrene-induced cytochrome P-450. Cancer Res. 1982;42:1798–808. [PubMed] [Google Scholar]
- 15.Yang TJ, Sai Y, Krausz KW, Gonzalez FJ, Gelboin HV. Inhibitory monoclonal antibodies to human cytochrome P450 1A2: analysis of phenacetin O-deethylation in human liver. Pharmacogenetics. 1998;8:375–82. doi: 10.1097/00008571-199810000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Dalton TP, Dieter MZ, Matlib RS, Childs NL, Shertzer HG, Genter MB, Nebert DW. Targeted knockout of Cyp1a1 gene does not alter hepatic constitutive expression of other genes in the mouse [Ah] battery. Biochem Biophys Res Commun. 2000;267:184–9. doi: 10.1006/bbrc.1999.1913. [DOI] [PubMed] [Google Scholar]
- 17.Liang HC, Li H, McKinnon RA, Duffy JJ, Potter SS, Puga A, Nebert DW. Cyp1a2(−/−) null mutant mice develop normally but show deficient drug metabolism. Proc Natl Acad Sci U S A. 1996;93:1671–6. doi: 10.1073/pnas.93.4.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beebe L, Park SS, Anderson LM. Differential enzyme induction of mouse liver and lung following a single low or high dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) J Biochem Toxicol. 1990;5:211–9. doi: 10.1002/jbt.2570050403. [DOI] [PubMed] [Google Scholar]
- 19.Wiwi CA, Gupte M, Waxman DJ. Sexually dimorphic P450 gene expression in liver-specific hepatocyte nuclear factor 4alpha-deficient mice. Mol Endocrinol. 2004;18:1975–87. doi: 10.1210/me.2004-0129. [DOI] [PubMed] [Google Scholar]
- 20.Dingley KH, Ubick EA, Vogel JS, Haack KW. DNA isolation and sample preparation for quantification of adduct levels by accelerator mass spectrometry. Methods Mol Biol. 2005;291:21–7. doi: 10.1385/1-59259-840-4:021. [DOI] [PubMed] [Google Scholar]
- 21.Hellmold H, Overvik E, Stromstedt M, Gustafsson JA. Cytochrome P450 forms in the rodent lung involved in the metabolic activation of food-derived heterocyclic amines. Carcinogenesis. 1993;14:1751–7. doi: 10.1093/carcin/14.9.1751. [DOI] [PubMed] [Google Scholar]
- 22.Hukkanen J, Pelkonen O, Hakkola J, Raunio H. Expression and regulation of xenobiotic-metabolizing cytochrome P450 (CYP) enzymes in human lung. Crit Rev Toxicol. 2002;32:391–411. doi: 10.1080/20024091064273. [DOI] [PubMed] [Google Scholar]
- 23.Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol. 2003;43:149–73. doi: 10.1146/annurev.pharmtox.43.100901.140251. [DOI] [PubMed] [Google Scholar]
- 24.Saarikoski ST, Husgafvel-Pursiainen K, Hirvonen A, Vainio H, Gonzalez FJ, Anttila S. Localization of CYP1A1 mRNA in human lung by in situ hybridization: comparison with immunohistochemical findings. Int J Cancer. 1998;77:33–9. doi: 10.1002/(sici)1097-0215(19980703)77:1<33::aid-ijc7>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 25.Kim JH, Sherman ME, Curriero FC, Guengerich FP, Strickland PT, Sutter TR. Expression of cytochromes P450 1A1 and 1B1 in human lung from smokers, non-smokers, and ex-smokers. Toxicol Appl Pharmacol. 2004;199:210–9. doi: 10.1016/j.taap.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 26.McLemore TL, Adelberg S, Liu MC, McMahon NA, Yu SJ, Hubbard WC, Czerwinski M, Wood TG, Storeng R, Lubet RA, et al. Expression of CYP1A1 gene in patients with lung cancer: evidence for cigarette smoke-induced gene expression in normal lung tissue and for altered gene regulation in primary pulmonary carcinomas. J Natl Cancer Inst. 1990;82:1333–9. doi: 10.1093/jnci/82.16.1333. [DOI] [PubMed] [Google Scholar]
- 27.Pasquini R, Sforzolini GS, Cavaliere A, Savino A, Monarca S, Puccetti P, Fatigoni C, Antonini G. Enzymatic activities of human lung tissue: relationship with smoking habits. Carcinogenesis. 1988;9:1411–6. doi: 10.1093/carcin/9.8.1411. [DOI] [PubMed] [Google Scholar]
- 28.Turteltaub KW, Vogel JS, Frantz CE, Shen N. Fate and distribution of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine in mice at a human dietary equivalent dose. Cancer Res. 1992;52:4682–7. [PubMed] [Google Scholar]
- 29.Manabe S, Tohyama K, Wada O, Aramaki T. Detection of a carcinogen, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), in cigarette smoke condensate. Carcinogenesis. 1991;12:1945–7. doi: 10.1093/carcin/12.10.1945. [DOI] [PubMed] [Google Scholar]
- 30.Manabe S, Kurihara N, Wada O, Izumikawa S, Asakuno K, Morita M. Detection of a carcinogen, 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine, in airborne particles and diesel-exhaust particles. Environ Pollut. 1993;80:281–6. doi: 10.1016/0269-7491(93)90049-t. [DOI] [PubMed] [Google Scholar]
- 31.Snyderwine EG, Schut HA, Sugimura T, Nagao M, Adamson RH. DNA adduct levels of 2-amino-1-methyl-6-phenylimidazo-[4,5-b]pyridine (PhIP) in tissues of cynomolgus monkeys after single or multiple dosing. Carcinogenesis. 1994;15:2757–61. doi: 10.1093/carcin/15.12.2757. [DOI] [PubMed] [Google Scholar]
- 32.Arimoto-Kobayashi S, Ishida R, Nakai Y, Idei C, Takata J, Takahashi E, Okamoto K, Negishi T, Konuma T. Inhibitory effects of beer on mutation in the Ames test and DNA adduct formation in mouse organs induced by 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) Biol Pharm Bull. 2006;29:67–70. doi: 10.1248/bpb.29.67. [DOI] [PubMed] [Google Scholar]
- 33.Gonzalez FJ. The use of gene knockout mice to unravel the mechanisms of toxicity and chemical carcinogenesis. Toxicol Lett. 2001;120:199–208. doi: 10.1016/s0378-4274(01)00296-x. [DOI] [PubMed] [Google Scholar]
- 34.Schut HA, Snyderwine EG. DNA adducts of heterocyclic amine food mutagens: implications for mutagenesis and carcinogenesis. Carcinogenesis. 1999;20:353–68. doi: 10.1093/carcin/20.3.353. [DOI] [PubMed] [Google Scholar]
- 35.Stevnsner T, Frandsen H, Autrup H. Repair of DNA lesions induced by ultraviolet irradiation and aromatic amines in normal and repair-deficient human lymphoblastoid cell lines. Carcinogenesis. 1995;16:2855–8. doi: 10.1093/carcin/16.11.2855. [DOI] [PubMed] [Google Scholar]
- 36.Alberg AJ, Brock MV, Samet JM. Epidemiology of lung cancer: looking to the future. J Clin Oncol. 2005;23:3175–85. doi: 10.1200/JCO.2005.10.462. [DOI] [PubMed] [Google Scholar]
- 37.Lesmes GR, Donofrio KH. Passive smoking: the medical and economic issues. Am J Med. 1992;93:38S–42S. doi: 10.1016/0002-9343(92)90626-m. [DOI] [PubMed] [Google Scholar]
- 38.Baldini EH, Strauss GM. Women and lung cancer: waiting to exhale. Chest. 1997;112:229S–234S. doi: 10.1378/chest.112.4_supplement.229s. [DOI] [PubMed] [Google Scholar]
- 39.Brownson RC, Alavanja MC, Caporaso N, Simoes EJ, Chang JC. Epidemiology and prevention of lung cancer in nonsmokers. Epidemiol Rev. 1998;20:218–36. doi: 10.1093/oxfordjournals.epirev.a017982. [DOI] [PubMed] [Google Scholar]
- 40.Gao YT, Blot WJ, Zheng W, Ershow AG, Hsu CW, Levin LI, Zhang R, Fraumeni JF., Jr Lung cancer among Chinese women. Int J Cancer. 1987;40:604–9. doi: 10.1002/ijc.2910400505. [DOI] [PubMed] [Google Scholar]
- 41.Metayer C, Wang Z, Kleinerman RA, Wang L, Brenner AV, Cui H, Cao J, Lubin JH. Cooking oil fumes and risk of lung cancer in women in rural Gansu, China. Lung Cancer. 2002;35:111–7. doi: 10.1016/s0169-5002(01)00412-3. [DOI] [PubMed] [Google Scholar]
- 42.Ko YC, Cheng LS, Lee CH, Huang JJ, Huang MS, Kao EL, Wang HZ, Lin HJ. Chinese food cooking and lung cancer in women nonsmokers. Am J Epidemiol. 2000;151:140–7. doi: 10.1093/oxfordjournals.aje.a010181. [DOI] [PubMed] [Google Scholar]
- 43.Wang TJ, Zhou BS, Shi JP. Lung cancer in nonsmoking Chinese women: a case-control study. Lung Cancer, 14 Suppl. 1996;1:S93–8. doi: 10.1016/s0169-5002(96)90214-7. [DOI] [PubMed] [Google Scholar]
- 44.Zhong L, Goldberg MS, Gao YT, Jin F. Lung cancer and indoor air pollution arising from Chinese-style cooking among nonsmoking women living in Shanghai, China. Epidemiology. 1999;10:488–94. [PubMed] [Google Scholar]
- 45.Thiebaud HP, Knize MG, Kuzmicky PA, Hsieh DP, Felton JS. Airborne mutagens produced by frying beef, pork and a soy-based food. Food Chem Toxicol. 1995;33:821–8. doi: 10.1016/0278-6915(95)00057-9. [DOI] [PubMed] [Google Scholar]