

Abstract
Ovulation is a precisely timed process by which a mature oocyte is released from an ovarian follicle. This process is initiated by the pituitary surge of luteinizing hormone (LH), is temporally associated with transcriptional regulation of numerous genes, and is presumed to involve the synthesis and/or activation of specific proteases that degrade the follicle wall. The progesterone receptor (PR), a nuclear receptor transcription factor, is induced in granulosa cells of preovulatory follicles in response to the LH surge and has been shown to be essential for ovulation, because mice lacking PR fail to ovulate and are infertile. Using these mice as a model in which to elucidate PR-regulated genes in the ovulation process, we show that the matrix metalloproteinases MMP-2 and MMP-9 are not targets of PR during ovulation. In contrast, two other proteases, ADAMTS-1 (A disintegrin and metalloproteinase with thrombospondin-like motifs) and cathepsin L (a lysosomal cysteine protease), are transcriptional targets of PR action. ADAMTS-1 is induced after LH stimulation in granulosa cells of preovulatory follicles and depends on PR. Cathepsin L is induced in granulosa cells of growing follicles by follicle-stimulating hormone, but the highest levels of cathepsin L mRNA occur in preovulatory follicles in response to LH in a PR-dependent manner. The identification of two regulated proteases in the ovary, together with their abnormal expression in anovulatory PR knockout mice, suggests that each plays a critical role in follicular rupture and represents a major advance in our understanding of the proteolytic events that control ovulation.
Progesterone has long been regarded as the preeminent hormone regulating female fertility. Although usually associated with the maintenance of pregnancy, progesterone is also essential for ovulation (1). Ovulation in rats is inhibited by treatment with either anti-progesterone antiserum (2) or epostane (3), a compound that blocks the synthesis of progesterone. The progesterone receptor (PR) is a member of the nuclear receptor transcription factor superfamily (4). PR signaling is required for progesterone-mediated effects on ovulation, because the PR antagonist RU486 blocks ovulation in rats (5) and mice (6), and because the anti-progestogen Org-31710 blocks ovulation of in vitro cultured mouse follicles (7). In the rat ovary, PR mRNA is induced rapidly and only briefly by the surge of luteinizing hormone (LH); this induction occurs selectively in granulosa cells of preovulatory follicles destined to ovulate (8). A similar pattern of rapid yet transient induction of PR is observed in primary cultures of rat granulosa cells in response to LH (9, 10). Additionally, both isoforms of PR (PRA and PRB), differentially translated from a single transcript (11–14), are induced by LH, with PRA being the prevalent translated product (9). The significance of ovarian PR was demonstrated by the targeted disruption of the PR gene; PR knockout (PRKO) female mice lacking PR (PRA and PRB) fail to ovulate, even in response to exogenous hormones, and are completely infertile (15, 16).
The adult mammalian ovary contains follicles at all stages of development. However, only a few follicles mature to the preovulatory stage in which LH receptors (as well as other genes) are expressed in granulosa cells, thereby allowing them to respond to the LH surge and ultimately ovulate (17). Although much is now known about the functional characteristics of preovulatory follicles (17, 18), the biochemical mechanisms that control the complex process of ovulation are not well understood. Histological studies have shown that, in response to the ovulatory LH surge, tissue degradation and extracellular matrix remodeling occur at the apex of the preovulatory follicle, ultimately leading to follicular rupture on the outer edge of the ovary and release of the mature oocyte (1). These remodeling events implicate precisely controlled induction and activation of matrix-digesting proteases (1). Compounding the issue of identifying and analyzing the function of such proteases is that most proteases have overlapping as well as distinct substrate specificities and are synthesized as latent proenzymes requiring activation by additional proteases. The matrix metalloproteinases (MMPs) have received considerable attention as candidate proteases controlling ovulation (19). Of the 23 known MMPs (20), collagenases (MMP-13 and MMP-8), which degrade collagen type I, gelatinases (MMP-2 and MMP-9), which degrade collagen types IV/V, and stromelysin-like enzymes (MMP-19) have been shown to be present in the ovary (21–25). However, their relationship, if any, to each other or to ovulation has remained speculative. A membrane-bound MMP, MMP-14, which has been shown to activate MMP-2 (20), is expressed in granulosa cells of preovulatory follicles (23), but mice lacking MMP-14 have normal fertility (26); thus, this protease is not essential for ovarian function.
Analysis of the ovulatory defect of the PRKO mice affords a unique opportunity to elucidate the cell types and proteases critical for follicular rupture, because certain imperative ovulatory factors are lacking in these mice. The studies described herein demonstrate the precise temporal and cell-specific expression of PR in the mouse ovary as well as the specific defect in follicular rupture/ovulation in PRKO mice. In addition, the hormonal regulation of putative ovulatory proteinases MMP-2 and MMP-9 was analyzed. Importantly, the expression after ovulatory LH of two distinct proteases, ADAMTS-1 and cathepsin L, is shown to occur in granulosa cells of normal mice but not in anovulatory follicles of PRKO mice. As progesterone-responsive genes that are transcriptionally activated in granulosa cells at the time of ovulation, ADAMTS-1 and cathepsin L represent the first clues about which cell type and proteases impact the ovulation process.
Materials and Methods
Animals.
PRKO mice were generated by insertion of a neomycin-resistance cassette into the first exon of the PR gene (15), resulting in a severely truncated noncoding message. Female mice homozygous for the mutation are completely infertile (15, 16). All mice were maintained in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Female mice at 22–24 days of age were injected intraperitoneally with 4 units of pregnant mare serum gonadotropin/gestyl (PMSG; Professional Compounding Center of America, Houston, TX), a follicle-stimulating hormone analog, to stimulate follicular growth and again 46 h later with 5 units of human CG/pregnyl (hCG; Organon), an LH analog, to trigger ovulation and luteinization.
Immunolocalization of PR.
Ovaries were fixed in 4% (vol/vol) paraformaldehyde, paraffin-embedded, and sectioned. Immunohistochemistry was performed as described (27) by using polyclonal anti-human PR antibody (Dako), which recognizes both PRA and PRB isoforms. Immunoreactivity was detected with peroxidase reagents (Vector Laboratories), and the sections were counterstained with the cytoplasmic stain eosin.
In-Gel Zymography.
Whole mouse ovaries were homogenized in cold zymography extraction buffer (50 mM Tris⋅HCl, pH 7.4/0.15 M NaCl/0.01 M CaCl2/0.25% Triton X-100/0.001% sodium azide) and centrifuged briefly; 5 μg of each supernatant and molecular mass markers were separated on a SDS/10% PAGE gel containing 1 mg/ml gelatin (Sigma). The gel was washed to remove SDS, incubated to stimulate protease activity, and stained with Coomassie blue (28). The gelatin substrate stains dark blue, whereas the activated gelatinase MMPs digest the substrate and appear as clear bands. Dried zymography gels were quantitated by scanning and densitometric analysis with imagequant software. Differences were expressed as fold increases above PMSG-treated control animals.
Reverse Transcription–PCR (RT-PCR).
Total ovarian RNA was prepared from whole mouse ovaries by using Trizol reagent (Life Technologies, Grand Island, NY). RT-PCR was performed as described (29). Total RNA [350 ng for cyclooxygenase-2 (COX-2) and 200 ng for cathepsin L and ADAMTS-1] was reverse transcribed by using oligo poly(dT) (Amersham Pharmacia) and AMV reverse transcriptase (Promega). Specific primers were used to amplify cDNAs: COX-2 sense, 5′-TGTACAAGCAGTGGCAAAGG-3′; COX-2 antisense, 5′-GCTGTGGATCTTGCACATTG-3′; cathepsin L sense, 5′-TGACACAGGGTTCGTGGATA-3′; cathepsin L antisense, 5′-ACCGCTACCCATCAATTCAC-3′; ADAMTS-1 sense, 5′-CAGTACCAGACCTTGTGCAGACCTT-3′; ADAMTS-1 antisense, 5′-CACACCTCACTGCTTACTGGTTTGA-3′. Primers for the ribosomal protein L19 (L19 sense, 5′-CTGAAGGTCAAAGGGAATGTG-3′; L19 antisense, 5′-GGACAGAGTCTTGATGATCTC-3′) were included in each experiment as an internal control (8). Dried gels were quantitated by using a Storm 860 PhosphorImager (Molecular Dynamics) and exposed to autoradiographic film. The mouse cathepsin L and ADAMTS-1 cDNA products were cloned into pCRII-TOPO vectors by using the TOPO TA Cloning kit (Invitrogen). Their authenticity was verified by sequence analyses.
In Situ Hybridization.
In situ hybridization was performed according to the detailed methods of Wilkensen (30) and as described (31). 35S-UTP-labeled antisense and sense probes for cathepsin L and ADAMTS-1 were synthesized from the cloned PCR cDNAs (above) by using the Riboprobe In Vitro Transcription Systems kit (Promega). Slides were dipped in photographic NTB-2 emulsion (Kodak) and exposed at 4°C for 1–3 days.
cDNA Array Differential Hybridization.
Mouse cDNA array membranes were purchased from CLONTECH. Poly(A)+ RNA was isolated from total ovarian RNA by Oligotex purification (Qiagen, Santa Clarita, CA), radiolabeled, and purified according to the CLONTECH protocol. Duplicate cDNA expression array membranes were hybridized overnight with equal counts of the labeled cDNAs derived either from PR+/− or from PRKO poly(A)+ RNA. Membranes were washed and exposed to Kodak X-Omat film, and signals from corresponding genes were compared.
Results
Ovarian Expression of PR.
The time course and cell-specific expression of PR protein in mouse ovary was analyzed by immunohistochemistry. Mice were treated with PMSG followed by hCG, which triggers ovulation approximately 14 h later. Immunoreactive PR was not detected in the ovaries of PMSG-treated mice (Fig. 1A). However, PR was present within 4 h and 8 h after hCG, specifically in granulosa cells of large preovulatory follicles (Fig. 1 B and D) and is localized to nuclei (Fig. 1E). Adjacent sections in which the primary antibody was omitted showed no detectable staining (Fig. 1F). Ovaries from PRKO littermates, similarly treated with PMSG and hCG, had large, antral follicles that did not have PR immunoreactivity (Fig. 1C). The expression of PR protein was down-regulated by 12 h after hCG treatment (not shown).
Figure 1.
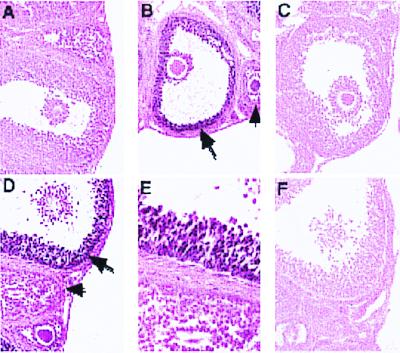
Expression and localization of ovarian PR. Immunohistochemistry with an anti-PR antibody and diaminobenzidine detection (dark nuclear staining) was performed on sections from wild-type (A and D–F), heterozygous (B), or PRKO (C) mice treated with PMSG only (A), PMSG and hCG for 4 h (B and C), or PMSG and hCG for 8 h (D–F). E is a higher magnification of D, and F is a serial section incubated without primary antibody. PR is expressed specifically in large, preovulatory follicles (large arrows) in response to hCG but not in neighboring small follicles (small arrows). Original images were taken at ×20 (A–C) and ×50 (D–F).
Ovarian Phenotype of PRKO Mice.
Preovulatory follicles of PRKO mice seem histologically indistinguishable from those of their wild-type and heterozygous littermates (Figs. 1 B and C and 2A Upper). The histological appearance of corpora lutea formed in response to PMSG and hCG also looked similar in the heterozygous and PRKO mice with the exception of entrapped oocytes in the PRKO ovary (ref. 15; Fig. 2A Lower).
Figure 2.

Ovarian phenotype of PRKO mice. (A Upper) PR+/− and PRKO littermates have large preovulatory follicles in response to PMSG treatment. (A Lower) In normal mice (PR+/−), follicles ruptured and expelled the oocyte and formed hypertrophic corpora lutea 48 h after ovulatory hCG. In similarly treated PRKO mice, the follicles do not ovulate, and the oocyte remains within the ovary, identified by periodic acid Schiff's reagent staining (dark pink color) of its zona pellucida layer. (B) RT-PCR analysis of the ovulatory marker COX-2. COX-2 was induced normally in both PR+/− and PRKO mice treated with PMSG and hCG (P, hCG 4 h). The ribosomal protein L19 was used as internal control. (C) In-gel zymography of ovarian extracts from hormonally treated PR+/− and PRKO mice showed active MMP-2 (clear bands at 66–72 kDa) throughout follicular development. MMP-9 (clear bands at 92 kDa) was induced and active 4 h after hCG treatment in both PR+/− and PRKO ovaries.
To analyze functional characteristics of the PRKO preovulatory follicles that might account for this ovulation defect, several molecular markers for specific stages of granulosa cell differentiation were analyzed by RT-PCR and in situ hybridization. Cytochrome P450 aromatase (CYP19), which catalyzes the production of estrogen, is a marker of steroidogenic function in granulosa cells of preovulatory follicles (32, 33). In PRKO ovaries, aromatase mRNA was induced by PMSG treatment and down-regulated by hCG in a pattern and magnitude indistinguishable from that of wild-type mice (not shown) and rats (32, 33). Similarly, follicle-stimulating hormone and PMSG-dependent induction of LH receptor mRNA in granulosa cells of preovulatory follicles and its expression in corpora lutea (34, 35) occurred normally in PRKO ovaries (not shown). Thus, granulosa cells of preovulatory follicles in PRKO mice have a normal ability to respond to gonadotropins.
To test LH responsiveness specifically, a gene associated with LH-induced ovulation, COX-2, was analyzed (17, 36, 37). COX-2 catalyzes the production of prostaglandins known to be necessary for ovulation (38); mice null for COX-2 have severely impaired ovulation (39, 40). COX-2 mRNA was undetectable by RT-PCR analysis in ovaries of PMSG-treated PR+/− and PRKO mice but was induced rapidly by hCG in ovaries of both genotypes (Fig. 2B). Thus, the anovulatory phenotype of PRKO mice is not due to the absence or aberrant temporal expression of COX-2 message. Despite the formation of corpora lutea (Fig. 2A Lower), which express normal genetic markers, such as cholesterol side-chain cleavage cytochrome P450 (CYP21A; ref. 41), the preovulatory follicles of the PRKO mice do not rupture; rather, the oocyte remains trapped within the luteinized structure. Although these oocytes are not ovulated, they are viable, can be fertilized in vitro, and grow to normal pups when implanted in foster mothers (41).
These data establish that the ovarian follicles of the PRKO mice develop normally to the preovulatory stage and differentiate in response to the LH surge but are defective in a critical step or steps essential in the ovulation process. Based on these results, we hypothesized that the LH-mediated induction of PR is essential for transcription of specific target genes, one or more of which is likely to be a protease (or protease activator) required to degrade the matrix proteins of the follicle wall and mediate rupture.
Degradation of two basement membranes (one surrounding the granulosa cells and one underlying the ovarian surface epithelium) is required for successful ovulation. Therefore, the activity of the gelatinases, MMP-2 (gelatinase A) and MMP-9 (gelatinase B), which digest the major component of basement membranes, collagen IV, was analyzed by in-gel zymography (Fig. 2C). In extracts of untreated mouse ovary, MMP-2 (migrating at 66/72 kDa) was present and active. In response to PMSG, MMP-2 remained present but migrated at a higher molecular mass (72 kDa), most likely as a nonprocessed pro(latent)-form of the enzyme. Treatment with hCG did not alter MMP-2 content or activity in ovarian extracts. In contrast, MMP-9 (migrating at 92 kDa) was induced rapidly (4 h) but transiently (<8 h) by ovulatory hCG, a pattern confirmed by Western blot analysis (not shown). Importantly, extracts from PRKO ovaries showed the same pattern of gelatinase expression and activity as their normal littermates (Fig. 2C). Analysis of additional samples showed that the induction of MMP-9 activity in PMSG, hCG 4-h ovaries was increased 1.90 ± 0.60-fold in PR+/− ovaries (n = 3) and 2.07 ± 0.39-fold in PRKO ovaries (n = 4) compared with PMSG-treated samples. Thus, neither MMP-2 nor MMP-9 is a target of PR action.
Regulation of ADAMTS-1.
ADAMTS-1, a member of the A disintegrin and metalloproteinase family of proteases, was shown recently to be induced by the LH surge in the rat ovary by a mechanism sensitive to the progesterone synthesis inhibitor epostane (42). Therefore, the expression of ADAMTS-1 mRNA was analyzed in both normal and PRKO mouse ovaries by RT-PCR. ADAMTS-1 message was not detected in ovaries from wild-type, untreated mice but was present at low levels in PMSG-stimulated ovaries (Fig. 3). In response to hCG for 7 h or 12 h, ADAMTS-1 mRNA was dramatically up-regulated in PR+/− ovaries but not in ovaries of PRKO mice. At 12 h after hCG treatment, near the time of ovulation, ADAMTS-1 expression was 12-fold lower in PRKO ovaries compared with that in their normal littermates.
Figure 3.

Ovarian expression of ADAMTS-1. As determined by RT-PCR analysis of total RNA from whole ovaries of untreated (unt), PMSG-treated, and PMSG/hCG-treated (P, hCG) PR+/− (+/−) and PRKO (KO) mice, ADAMTS-1 was dramatically up-regulated in response to hCG. The induction of ADAMTS-1 did not occur in PRKO ovaries. ADAMTS-1 expression was normalized to expression of ribosomal protein L19.
Tissue localization of ADAMTS-1 mRNA in PR+/− and PRKO ovaries was analyzed by in situ hybridization by using the cloned cDNA. Results confirmed the temporal regulation and PR-dependence demonstrated by RT-PCR analyses. ADAMTS-1 transcripts were not detected above background in ovaries of mice treated with PMSG alone (Fig. 4). After a 12-h hCG treatment, ADAMTS-1 mRNA was highly expressed in PR+/− ovaries, specifically in granulosa cells of the large ovulatory follicles, and this expression had declined by 16 h of hCG in the ruptured, luteinizing follicles. In PRKO ovaries, ADAMTS-1 mRNA induction by hCG did not occur and remained undetectable above background. These results indicate that LH induction of PR is essential for the induction and expression of ADAMTS-1 in follicles destined to ovulate.
Figure 4.

Regulated expression of ADAMTS-1 in preovulatory follicles. In situ hybridization with a radiolabeled antisense probe showed undetectable ADAMTS-1 expression in ovaries of PMSG-treated PR+/− and PRKO mice. In response to hCG, ADAMTS-1 expression was induced in granulosa cells of the large, ovulating follicles of PR+/− mice (black arrows) but not in similar large follicles of PRKO mice (red arrows). ADAMTS-1 expression in ovulating follicles was transient and had decreased in PR+/− granulosa cells by 16 h after hCG.
Regulation of Cathepsin L.
To identify additional PR-regulated genes, immobilized cDNA arrays were used to compare mRNA populations present in ovaries of normal PR+/− and PRKO mice. Mice were primed with PMSG and hCG for 8 h; poly(A)+ RNA was prepared; and matching arrays were probed with the radiolabeled cDNAs. Expression of one protease was present in the RNA prepared from PR+/− ovaries but was reduced markedly in the RNA from the PRKO mice (arrays not shown). This protease was identified as cathepsin L, a lysosomal cysteine proteinase.
Analysis of the expression of cathepsin L mRNA by RT-PCR detected cathepsin L mRNA in ovaries from untreated wild-type mice (Fig. 5); cathepsin L mRNA was up-regulated by PMSG in both PR+/− and PRKO mice. Further up-regulation by hCG (7 h and 12 h) in PR+/− mice did not occur in PRKO mice. Immediately before ovulation (12 h), cathepsin L mRNA was 4-fold lower in PRKO ovaries than in PR+/− ovaries.
Figure 5.

Ovarian expression of cathepsin L. As determined by RT-PCR analysis of total RNA from whole ovaries of untreated (unt), PMSG-treated, and PMSG/hCG-treated (P, hCG) PR+/− (+/−) and PRKO (KO) mice, cathepsin L was induced by PMSG and to highest levels by hCG. The induction of cathepsin L in response to hCG did not occur in PRKO ovaries. Cathepsin L expression was normalized to expression of ribosomal protein L19.
In situ hybridization experiments showed cathepsin L expressed in the small follicles as well as in the largest, preovulatory follicles of PMSG-treated mice (Fig. 6). This expression pattern was identical in PR+/− and PRKO ovaries. In mice treated with PMSG and hCG for 12 h, cathepsin L mRNA was specifically detected in the large, ovulating follicles. Similar large follicles of the PRKO ovaries did not express detectable levels of cathepsin L mRNA, although it was expressed in adjacent smaller follicles. The presence of cathepsin L transcripts in small follicles of PRKO ovaries explains the only partial reduction of cathepsin L message observed in whole ovary RNA analysis by RT-PCR. Similarly, 16 h after hCG, cathepsin L mRNA was highly expressed in ruptured follicles of normal ovaries but was not detected in the large nonovulated follicles of PRKO ovaries. Therefore, cathepsin L expression is localized to the granulosa cells of the ovary. Interestingly, PR is required for the enhanced cathepsin L expression in ovulating follicles, whereas the lower expression levels in small follicles are independent of PR.
Figure 6.

Regulated expression of cathepsin L in preovulatory follicles. In situ hybridization with a radiolabeled antisense probe showed cathepsin L expression in small follicles of PMSG-treated PR+/− and PRKO mice. In response to hCG, cathepsin L was induced in the large, ovulating follicles of PR+/− mice (black arrows) but was not induced in similar large follicles of PRKO ovaries (red arrows).
Discussion
The biochemical events that control the process of ovulation are not yet known. However, the profound changes in follicle structure and the extensive tissue remodeling that occur during the ovulatory process almost certainly require the action of specific and tightly regulated proteases. The data presented herein indicate that the LH-induced transcription factor PR is required for the production of two such regulated proteases, ADAMTS-1 and cathepsin L. In normal mice, these proteases are dramatically up-regulated in granulosa cells of LH-stimulated ovulatory follicles, but in anovulatory PRKO mice, these proteases remain at preovulatory levels. The spatial/temporal expression of these two proteases and their specific dependency on the actions of LH and PR make them highly attractive candidates for mediating specific steps in the ovulatory process.
ADAMTS-1 is a modular protein (43), consisting of a disintegrin domain and three thrombospondin type I motifs, in addition to a metalloproteinase domain (10). Unlike other ADAM family members, ADAMTS-1 does not have a transmembrane region but is secreted from cells in a catalytically active form; however, an in vivo substrate or substrates have yet to be identified (44, 45). Disintegrin domains, which generally act to inhibit integrin function and intracellular signaling cascades by blocking their binding to adhesive ligands (46), may play a role in the disruption of cellular interactions with the collagenous matrix or the inhibition of granulosa cell interactions mediated by integrins (47). ADAMTS-1 has been shown to interact with extracellular matrix via the thrombospondin type I motifs (44), a likely mechanism by which it is targeted in tissue to specific sites of action. Thrombospondin type I has been shown to inhibit endothelial cell proliferation, migration, and angiogenesis (48) and is also present in the ovary (41). Interestingly, the human ortholog of ADAMTS-1 (METH-1) as well as its homolog METH-2 have angioinhibitory activity (49)—even greater than that of thrombospondin itself. After ovulation, the ruptured follicles rapidly reorganize to form corpora lutea, a highly vascularized tissue. Therefore, any role for ADAMTS-1 at the time of ovulation as an angiostatic factor would need to be of limited duration, consistent with the transient expression pattern of ADAMTS-1 in ovulating follicles (Fig. 5; ref. 42). The distinct matrix-interacting capability and multiple domain structure of ADAMTS-1 most likely enable it to localize and exert multiple biological effects simultaneously. The generation of site-specific mutations within each domain as well as the generation of mice null for ADAMTS-1 will be required to determine the contribution of each domain to its biological activity and to elucidate roles for ADAMTS-1 in specific tissues. In the ovary, perhaps the ADAMTS-1 protease domain mediates aspects of cellular degradation and dissolution of the follicle wall, whereas the thrombospondin and disintegrin domains control its localization and aspects of inflammation, differentiation, and/or neovascularization, thereby coordinating many facets of follicle rupture and corpus luteum formation. Recently, other members of the ADAMTS family have been identified (50, 51). However, the dramatic ovarian expression pattern of ADAMTS-1 and the absence of expression in the anovulatory PRKO mice suggest a particular significance in ovulation.
Cathepsin L may also possess complex functions. Although commonly known as a lysosomal protease, cathepsin L is also secreted from cells (52), and its substrates include collagen (I and IV), elastin, and fibronectin. Cathepsin L therefore has the potential to act on extracellular matrix proteins, such as those that maintain the integrity of the follicle wall. Although secreted as a proenzyme, cathepsin L can be activated when complexed with extracellular glycosaminoglycans, which are known to be present in follicular fluid and extracellular matrices (53). Both the proenzyme and the cleaved form of cathepsin L can degrade matrix proteins but with different specificities (54). Cathepsin L has also been observed to stimulate DNA synthesis (55) and steroidogenesis (56), perhaps by activating or inactivating locally secreted cytokines. Also of interest, cathepsin L was identified as a progesterone-induced secretory protein in the cat uterus (57).
The transcriptional mechanisms by which PR regulates the promoters of ADAMTS-1 and cathepsin L remain to be resolved. The mouse cathepsin L gene does not have a consensus PR binding site in the 240 bp of known upstream sequence (GenBank accession no. L06427). Nor does the murine ADAMTS-1 gene have a consensus PR binding element within the 440 bp of its known 5′ sequence (GenBank accession no. AB001735). Based on these rather limited data, one cannot resolve whether the effects of PR are direct or indirect. Both genes have multiple Sp1 binding sites, and for cathepsin L, this site has been shown to be important for expression in the 3Y1 cell line (58). Sp1 is present at high levels in the ovary (59) and directly regulates the expression of serum and glucocorticoid-inducible kinase (sgk) in granulosa cells. In addition, PR regulates expression of the p21Cip1 gene in a DNA-independent mechanism by interacting with Sp1 bound to the promoter (60). Thus, if a consensus PR regulatory site is not identified in additional upstream or intronic sequences, PR may regulate the expression of cathepsin L and/or ADAMTS-1 by similar, indirect mechanisms.
MMP-9 and MMP-13 have long been considered to be potential mediators in the ovulatory process. The induction of MMP-9 by hCG at the time of ovulation, reported herein and by others (23), suggests a role in follicle remodeling but not an essential role in ovulation per se, because mice lacking MMP-9 are fertile (61) and have completely normal ovarian histology (Z. Werb, personal communication). Of course other MMPs with overlapping substrate specificities, such as MMP-2, may account for the lack of a more dramatic ovarian phenotype in the MMP-9 null mice. Based on increased collagenase (against collagen I) activity in ovaries of ovulating rats (21, 22) and bovine follicles (62), it is highly likely that MMP-13 plays a role in ovulation. However, the temporal pattern of MMP-13 expression in the rodent ovary (24) and its presence in PRKO mice (J.S.R., unpublished data) indicate that it is not a PR-regulated gene. It is possible, however, that MMP-13, an additional collagenase such MMP-8 (25), or MMP-19 (23) acts in conjunction with the PR-regulated proteases (ADAMTS-1 and cathepsin L) to generate a proteolytic cascade that ultimately degrades the ovarian follicle wall and ovarian surface epithelium.
In summary, PRKO mice fail to ovulate and have an ovarian phenotype in which the expression and/or activation of key ovulatory proteases is impaired. We have identified two candidate proteases, ADAMTS-1 and cathepsin L, which are induced in the ovary at the time of ovulation and depend on PR for normal expression. Our demonstration of markedly reduced expression of ADAMTS-1 and cathepsin L in anovulatory PRKO follicles suggests an important role for each enzyme in the ovulatory process, providing new perspectives for future studies on the proteolytic events that impact the ovulation process.
Acknowledgments
This work was supported in part by National Institutes of Health Grants HD-16229 and HD-07495 (to J.S.R. and B.W.O.) and National Science Foundation Grant 9870793 (to L.L.E.).
Abbreviations
- LH
luteinizing hormone
- PR
progesterone receptor
- PRKO
PR knockout
- PMSG
pregnant mare serum gonadotropin/gestyl
- hCG
human CG/pregnyl
- RT-PCR
reverse transcription–PCR
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.080073497.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.080073497
References
- 1.Espey L L, Lipner H. In: The Physiology of Reproduction. Knobil E, Neill J D, editors. New York: Raven; 1994. pp. 725–780. [Google Scholar]
- 2.Mori T, Suzuki A, Nishimura T, Kambegawa A. J Endocrinol. 1977;73:185–186. doi: 10.1677/joe.0.0730185. [DOI] [PubMed] [Google Scholar]
- 3.Snyder B W, Beecham G D, Schane H P. Proc Soc Exp Biol Med. 1984;176:238–242. doi: 10.3181/00379727-176-41865. [DOI] [PubMed] [Google Scholar]
- 4.Schrader W T, O'Malley B W. J Biol Chem. 1972;247:51–59. [PubMed] [Google Scholar]
- 5.van der Schoot P, Bakker G H, Klijn J G. Endocrinology. 1987;121:1375–1382. doi: 10.1210/endo-121-4-1375. [DOI] [PubMed] [Google Scholar]
- 6.Loutradis D, Bletsa R, Aravantinos L, Kallianidis K, Michalas S, Psychoyos A. Hum Reprod. 1991;6:1238–1240. doi: 10.1093/oxfordjournals.humrep.a137519. [DOI] [PubMed] [Google Scholar]
- 7.Rose U M, Hanssen R G, Kloosterboer H J. Biol Reprod. 1999;61:503–511. doi: 10.1095/biolreprod61.2.503. [DOI] [PubMed] [Google Scholar]
- 8.Park O K, Mayo K E. Mol Endocrinol. 1991;5:967–978. doi: 10.1210/mend-5-7-967. [DOI] [PubMed] [Google Scholar]
- 9.Natraj U, Richards J S. Endocrinology. 1993;133:761–769. doi: 10.1210/endo.133.2.8344215. [DOI] [PubMed] [Google Scholar]
- 10.Clemens J W, Robker R L, Kraus W L, Katzenellenbogen B S, Richards J S. Mol Endocrinol. 1998;12:1201–1214. doi: 10.1210/mend.12.8.0157. [DOI] [PubMed] [Google Scholar]
- 11.Tung L, Mohamed M K, Hoeffler J P, Takimoto G S, Horwitz K B. Mol Endocrinol. 1993;7:1256–1265. doi: 10.1210/mend.7.10.8123133. [DOI] [PubMed] [Google Scholar]
- 12.Vegeto E, Shahbaz M M, Wen D X, Goldman M E, O'Malley B W, McDonnell D P. Mol Endocrinol. 1993;7:1244–1255. doi: 10.1210/mend.7.10.8264658. [DOI] [PubMed] [Google Scholar]
- 13.Lessey B A, Alexander P S, Horwitz K B. Endocrinology. 1983;112:1267–1274. doi: 10.1210/endo-112-4-1267. [DOI] [PubMed] [Google Scholar]
- 14.Kraus W L, Montano M M, Katzenellenbogen B S. Mol Endocrinol. 1993;7:1603–1616. doi: 10.1210/mend.7.12.8145766. [DOI] [PubMed] [Google Scholar]
- 15.Lydon J P, DeMayo F J, Funk C R, Mani S K, Hughes A R, Montgomery C A, Shyamala G, Conneely O M, O'Malley B W. Genes Dev. 1995;9:2266–2278. doi: 10.1101/gad.9.18.2266. [DOI] [PubMed] [Google Scholar]
- 16.Lydon J P, DeMayo F J, Conneely O M, O'Malley B W. J Steroid Biochem Mol Biol. 1996;56:67–77. doi: 10.1016/0960-0760(95)00254-5. [DOI] [PubMed] [Google Scholar]
- 17.Richards J S. Endocrinol Rev. 1994;15:725–751. doi: 10.1210/edrv-15-6-725. [DOI] [PubMed] [Google Scholar]
- 18.Richards J S, Fitzpatrick S L, Clemens J W, Morris J K, Alliston T, Sirois J. Recent Prog Horm Res. 1995;50:223–254. doi: 10.1016/b978-0-12-571150-0.50014-7. [DOI] [PubMed] [Google Scholar]
- 19.Richards J S, Russell D L, Robker R L, Dajee M, Alliston T N. Mol Cell Endocrinol. 1998;145:47–54. doi: 10.1016/s0303-7207(98)00168-3. [DOI] [PubMed] [Google Scholar]
- 20.Nagase H, Woessner J F., Jr J Biol Chem. 1999;274:21491–21494. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- 21.Curry T E, Jr, Dean D D, Woessner J F, Jr, LeMaire W J. Biol Reprod. 1985;33:981–991. doi: 10.1095/biolreprod33.4.981. [DOI] [PubMed] [Google Scholar]
- 22.Reich R, Tsafriri A, Mechanic G L. Endocrinology. 1985;116:522–527. doi: 10.1210/endo-116-2-522. [DOI] [PubMed] [Google Scholar]
- 23.Hagglund A C, Ny A, Leonardsson G, Ny T. Endocrinology. 1999;140:4351–4358. doi: 10.1210/endo.140.9.7002. [DOI] [PubMed] [Google Scholar]
- 24.Balbin M, Fueyo A, Lopez J M, Diez-Itza I, Velasco G, Lopez-Otin C. J Endocrinol. 1996;149:405–415. doi: 10.1677/joe.0.1490405. [DOI] [PubMed] [Google Scholar]
- 25.Balbin M, Fueyo A, Knauper V, Pendas A M, Lopez J M, Jimenez M G, Murphy G, Lopez-Otin C. J Biol Chem. 1998;273:23959–23968. doi: 10.1074/jbc.273.37.23959. [DOI] [PubMed] [Google Scholar]
- 26.Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov S A, Mankani M, Robey P G, Poole A R, Pidoux I, et al. Cell. 1999;99:81–92. doi: 10.1016/s0092-8674(00)80064-1. [DOI] [PubMed] [Google Scholar]
- 27.Tibbetts T A, Mendoza-Meneses M, O'Malley B W, Conneely O M. Biol Reprod. 1998;59:1143–1152. doi: 10.1095/biolreprod59.5.1143. [DOI] [PubMed] [Google Scholar]
- 28.Russell D L, Salamonsen L A, Findlay J K. Endocrinology. 1995;136:3657–3664. doi: 10.1210/endo.136.8.7628406. [DOI] [PubMed] [Google Scholar]
- 29.Orly J, Rei Z, Greenberg N M, Richards J S. Endocrinology. 1994;134:2336–2346. doi: 10.1210/endo.134.6.7514996. [DOI] [PubMed] [Google Scholar]
- 30.Wilkensen D G. In: Essential Developmental Biology, A Practical Approach. Stern C D, Holland P W H, editors. New York: Oxford Univ. Press; 1993. pp. 258–263. [Google Scholar]
- 31.Robker R L, Richards J S. Mol Endocrinol. 1998;12:924–940. doi: 10.1210/mend.12.7.0138. [DOI] [PubMed] [Google Scholar]
- 32.Hickey G J, Chen S A, Besman M J, Shively J E, Hall P F, Gaddy-Kurten D, Richards J S. Endocrinology. 1988;122:1426–1436. doi: 10.1210/endo-122-4-1426. [DOI] [PubMed] [Google Scholar]
- 33.Fitzpatrick S L, Richards J S. Endocrinology. 1991;129:1452–1462. doi: 10.1210/endo-129-3-1452. [DOI] [PubMed] [Google Scholar]
- 34.Camp T A, Mayo K E. In: Follicle Stimulating Hormone: Regulation of Secretion and Molecular Mechanisms of Action. Hunzicker-Dunn M, Schwartz N B, editors. New York: Springer; 1992. pp. 345–350. [Google Scholar]
- 35.Segaloff D L, Wang H Y, Richards J S. Mol Endocrinol. 1990;4:1856–1865. doi: 10.1210/mend-4-12-1856. [DOI] [PubMed] [Google Scholar]
- 36.Wong W Y, Richards J S. Endocrinology. 1992;130:3512–3521. doi: 10.1210/endo.130.6.1317786. [DOI] [PubMed] [Google Scholar]
- 37.Sirois J, Richards J S. J Biol Chem. 1992;267:6382–6388. [PubMed] [Google Scholar]
- 38.Mikuni M, Pall M, Peterson C M, Peterson C A, Hellberg P, Brannstrom M, Richards J S, Hedin L. Biol Reprod. 1998;59:1077–1083. doi: 10.1095/biolreprod59.5.1077. [DOI] [PubMed] [Google Scholar]
- 39.Morham S G, Langenbach R, Loftin C D, Tiano H F, Vouloumanos N, Jennette J C, Mahler J F, Kluckman K D, Ledford A, Lee C A, et al. Cell. 1995;83:473–482. doi: 10.1016/0092-8674(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 40.Dinchuk J E, Car B D, Focht R J, Johnston J J, Jaffee B D, Covington M B, Contel N R, Eng V M, Collins R J, Czerniak P M, et al. Nature (London) 1995;378:406–409. doi: 10.1038/378406a0. [DOI] [PubMed] [Google Scholar]
- 41.Robker R L, Richards J S. In: Ovulation: Evolving Scientific and Clinical Concepts. Adashi E, Stouffer R, editors. New York: Springer; 2000. , in press. [Google Scholar]
- 42.Espey L L, Yoshioka S, Russell D L, Fujii S, Richards J S. Biol Reprod. 2000;62:1090–1095. doi: 10.1095/biolreprod62.4.1090. [DOI] [PubMed] [Google Scholar]
- 43.Kuno K, Iizasa H, Ohno S, Matsushima K. Genomics. 1997;46:466–471. doi: 10.1006/geno.1997.5064. [DOI] [PubMed] [Google Scholar]
- 44.Kuno K, Matsushima K. J Biol Chem. 1998;273:13912–13917. doi: 10.1074/jbc.273.22.13912. [DOI] [PubMed] [Google Scholar]
- 45.Kuno K, Terashima Y, Matsushima K. J Biol Chem. 1999;274:18821–18826. doi: 10.1074/jbc.274.26.18821. [DOI] [PubMed] [Google Scholar]
- 46.Niewiarowski S, McLane M A, Kloczewiak M, Stewart G J. Semin Hematol. 1994;31:289–300. [PubMed] [Google Scholar]
- 47.Fujiwara H, Maeda M, Honda T, Yamada S, Ueda M, Kanzaki H, Suginami H, Mori T. Horm Res. 1996;46:24–30. doi: 10.1159/000185177. [DOI] [PubMed] [Google Scholar]
- 48.Adams J C. Int J Biochem Cell Biol. 1997;29:861–865. doi: 10.1016/s1357-2725(96)00171-9. [DOI] [PubMed] [Google Scholar]
- 49.Vazquez F, Hastings G, Ortega M A, Lane T F, Oikemus S, Lombardo M, Iruela-Arispe M L. J Biol Chem. 1999;274:23349–23357. doi: 10.1074/jbc.274.33.23349. [DOI] [PubMed] [Google Scholar]
- 50.Abbaszade I, Liu R Q, Yang F, Rosenfeld S A, Ross O H, Link J R, Ellis D M, Tortorella M D, Pratta M A, Hollis J M, et al. J Biol Chem. 1999;274:23443–23450. doi: 10.1074/jbc.274.33.23443. [DOI] [PubMed] [Google Scholar]
- 51.Hurskainen T L, Hirohata S, Seldin M F, Apte S S. J Biol Chem. 1999;274:25555–25563. doi: 10.1074/jbc.274.36.25555. [DOI] [PubMed] [Google Scholar]
- 52.Ishidoh K, Kominami E. Biol Chem. 1998;379:131–135. doi: 10.1515/bchm.1998.379.2.131. [DOI] [PubMed] [Google Scholar]
- 53.Salustri A, Camaioni A, Di Giacomo M, Fulop C, Hascall V C. Hum Reprod Update. 1999;5:293–301. doi: 10.1093/humupd/5.4.293. [DOI] [PubMed] [Google Scholar]
- 54.Ishidoh K, Kominami E. Biochem Biophys Res Commun. 1995;217:624–631. doi: 10.1006/bbrc.1995.2820. [DOI] [PubMed] [Google Scholar]
- 55.Kasai M, Shirasawa T, Kitamura M, Ishido K, Kominami E, Hirokawa K. Cell Immunol. 1993;150:124–136. doi: 10.1006/cimm.1993.1184. [DOI] [PubMed] [Google Scholar]
- 56.Boujrad N, Ogwuegbu S O, Garnier M, Lee C H, Martin B M, Papadopoulos V. Science. 1995;268:1609–1612. doi: 10.1126/science.7777858. [DOI] [PubMed] [Google Scholar]
- 57.Jaffe R C, Donnelly K M, Mavrogianis P A, Verhage H G. Mol Endocrinol. 1989;3:1807–1814. doi: 10.1210/mend-3-11-1807. [DOI] [PubMed] [Google Scholar]
- 58.Ishidoh K, Taniguchi S, Kominami E. Biochem Biophys Res Commun. 1997;238:665–669. doi: 10.1006/bbrc.1997.7349. [DOI] [PubMed] [Google Scholar]
- 59.Alliston T N, Maiyar A C, Buse P, Firestone G L, Richards J S. Mol Endocrinol. 1997;11:1934–1949. doi: 10.1210/mend.11.13.0033. [DOI] [PubMed] [Google Scholar]
- 60.Owen G I, Richer J K, Tung L, Takimoto G, Horwitz K B. J Biol Chem. 1998;273:10696–10701. doi: 10.1074/jbc.273.17.10696. [DOI] [PubMed] [Google Scholar]
- 61.Vu T H, Shipley J M, Bergers G, Berger J E, Helms J A, Hanahan D, Shapiro S D, Senior R M, Werb Z. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson M L, Murdoch J, Van Kirk E A, Kaltenbach J E, Murdoch W J. Biol Reprod. 1999;61:1581–1585. doi: 10.1095/biolreprod61.6.1581. [DOI] [PubMed] [Google Scholar]