

Abstract
Lymphocytes must promote protective immune responses while still maintaining self-tolerance. Stimulation through the T cell receptor (TCR1) can lead to distinct responses in naive and memory CD4 T cells. Whereas peptide antigen stimulates both naive and memory T cells, soluble anti-CD3 antibodies and bacterial superantigens stimulate only naive T cells to proliferate and secrete cytokines. Further, superantigens, like staphylococcal enterotoxin B (SEB), cause memory T cells to become anergic while soluble anti-CD3 does not. In the present report we show that signal transduction through the TCR is impaired in memory cells exposed to either anti-CD3 or SEB. A block in signaling leads to impaired activation of the kinase ZAP-70 so that downstream signals and cell proliferation do not occur. We further show that the signaling defect is unique to each agent. In anti-CD3-treated memory T cells, the src kinase Lck is only transiently activated and does not phosphorylate and activate ZAP-70. In SEB-treated memory T cells, ZAP-70 does not interact with the TCR/CD3 complex to become accessible to Lck. Finally, we provide evidence that alternative signaling pathways are initiated in SEB-treated memory cells. Altered signaling, indicated by an elevation in activity of the src kinase Fyn, may be responsible for memory cell anergy caused by SEB. Thus, differentiation of naive T cells into memory cells is accompanied by alterations in TCR-mediated signaling that can promote heightened recall immunity or specific tolerance.
Keywords: T Lymphocytes, Superantigens, Immunological memory, Signal transduction, Clonal anergy
Introduction
Adaptive immunity encompasses both vigorous responses to foreign pathogens and the imposition of immune tolerance to self-components. Much of protective immunity is due to the development of immunological memory, where memory B and T lymphocytes respond more quickly and more robustly to recall antigens (reviewed in Refs, [1–4]). In part, a more vigorous response by memory cells could be due to increased sensitivity to antigen through means such as a different or decreased requirement for accessory or costimulation signaling, increased adhesion molecules on the memory cell surface, or differential signaling through the antigen receptor [2]. Because memory cells are both more easily activated and more functionally potent than primary (naive) cells, it is especially important that improper responses, especially directed against self-antigens, be prevented. Hence, it is likely that additional or unique regulatory mechanisms are involved in memory cell activation.
Signaling through the TCR can be initiated by different stimuli, such as conventional peptide antigen, superantigen, or anti-TCR/CD3 antibodies. Each of these agents can lead to distinct functional outcomes in naive versus memory cells. For example, previous studies showed that in both humans and mice, exposure of naive and memory cells to immobilized antibodies against the TCR/CD3 complex preferentially activated memory cells [5–8]. In contrast, certain mitogens, soluble TCR/CD3-specific antibodies (presented by Fc receptor-bearing APCs), and superantigens induce vigorous proliferation by naive CD4 T cells but not by memory CD4 T cells [8–11]. Superantigens, including the Staphylococcal enterotoxins, such as SEB, are microbial products that can activate CD4+ and CD8+ T cells at high frequency [12]. Like conventional antigen, superantigens require presentation by a MHC Class II-bearing cell [12]. Exposure to superantigens in vivo leads to oligoclonal, TCRVβ-restricted T cell expansion followed by cell deletion or clonal anergy [13]. Because clonal anergy is an important mechanism for maintaining immune tolerance, the study of superantigens may reveal regulatory mechanisms utilized for self/non-self recognition.
As noted above, in vivo administration of superantigens induces both robust proliferation and anergy. In an earlier study, we observed that cell expansion and anergy were responses of two phenotypically distinct subpopulations of CD4 T cells [11]. Whereas proliferating T cells exhibited a naive phenotype, the recovered anergic cells expressed a phenotype consistent with those of “antigen-experienced” cells [11]. Further, we noted that resting memory cells were hyporesponsive to in vitro stimulation with SEB even if they had not been previously exposed to SEB in vivo [11;14;15]. Hence, as was also observed when CD4 cells were exposed to soluble anti-CD3 mAbs in vitro in normal and TCR transgenic mice [8;10;16], CD4 memory, but not naive, T cells fail to proliferate or secrete lymphokines when exposed to superantigens [11;16]. Further, we have recently shown that antigen-specific memory T cells fail to proliferate or secrete lymphokines when challenged with an agonist peptide antigen after prior exposure to superantigen [16]. In contrast, memory cells which had been exposed to anti-CD3 mAbs still retained the ability to respond to peptide antigen [16]. Hence, only the superantigen induced anergy in memory cells. Since, peptide antigen, superantigen, and anti-CD3 mAbs interact with the T cells through the same TCR/CD3 complex, yet, they lead to distinct biological outcomes, we wished to determine whether TCR-mediated signal transduction was different in memory cells exposed to these three stimuli. In the present report, we show that although peptide induces prototypical signaling paths (reviewed in Ref. [17]), also seen in naive cells responding to any of the stimuli, exposure to both superantigen (SEB) and anti-CD3 mAbs results in impaired signaling. Further, we report that the block in signaling differed depending on the stimulus, suggesting that superantigen-induced anergy results from the utilization of alternative signal transduction pathways.
Materials and Methods
Animals
The BALB/c ByJ and DO11.10 mice used in these experiments were bred and maintained at the Wadsworth Center Animal Core Facility under specific pathogen-free conditions. The majority of T cells in the DO11.10 mice are CD4+ cells which bear a TCR that recognizes a chicken ovalbumin-derived peptide, OVA(323–339) (hereafter referred to as OVA), presented by I-Ad [18]. This TCR is encoded by transgenes encoding Vβ8.2/Vα13.1 chains and can be identified by the anti-clonotypic mAb, KJ1-26 [19]. Unless otherwise indicated, the experiments were performed using 10–12 week old mice. Both male and female mice were used in different experiments with no discernible differences in the results. All mice used in these studies were bred and maintained in accordance with the guidelines of the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Resources, National Research Council (Washington, DC). All experiments were approved by the Wadsworth Center Institutional Animal Care and Use Committee.
Reagents and Antibodies
MAbs GK1.5, 2B6 (anti-CD4), 3.155 (anti-CD8), HO13.4 (anti-Thy1.2), KJ1-26 (anti-DO11.10 clonotype), C363.29B (anti-CD3ε), and 23G2 (anti-CD45RB) were prepared from the supernatants of hybridoma cell lines. Antibodies used for immunoprecipitation, immunoblotting and in vitro kinase assay included anti-CD3ζ, anti-Fyn, anti-Lck, anti-PLC-γ1, and anti-LAT (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti-phosphotyrosine (G410) (Upstate Biotechnology, Lake Placid, NY), and anti-ZAP-70, goat anti-mouse IgG-HRPO and goat anti-rabbit IgG-HRPO (Transduction Laboratories, San Diego, CA). An antibody reactive only with tyrosine-phosphorylated, activated ZAP-70 (anti-pY319 ZAP-70) was purchased from Cell Signaling Technology (Beverly, MA). Chicken OVA peptide (OVA323–339) was synthesized and supplied by the Wadsworth Center Peptide Synthesis Core Facility. Mitomycin C, PMA (Sigma Chemicals, St. Louis, MO), and SEB (Toxin Technology, Sarasota, FL) were purchased.
Preparation of cells
In all experiments, enriched populations of CD4+ T cells were prepared by negative selection procedures as previously described [20] and were 90–95% CD4+ and <3% sIg+ as determined by flow-cytometric analyses. Naive and memory cells were separated based upon CD45RB expression using mAb 23G2 supernatant and MACS (Miltenyi Biotec, Auburn, CA) sorting to separate the CD45RBhi (naive) and CD45RBlo (memory) populations [16]. Following separation, the sorting mAb was removed using a low pH buffer as described [20]. For most experiments OVA and SEB were presented to T cells using RT11-mB7 fibroblast cells [16]. This adherent cell line was originally obtained from Dr. C.T. Weaver (The University of Alabama at Birmingham, Birmingham, AL) and had previously been transfected to express both I-Ad and B7-1 molecules [21]. Cells bearing these molecules were positively selected in culture through G418 resistance. Alternatively, APCs were prepared by T cell depletion of splenocytes using anti-Thy1.2 and complement followed by anti-CD4 (mAb 2B6) and anti-CD8 plus complement [15]. Unless otherwise indicated, APCs were treated with mitomycin C (25 μg/ml) for 20 min at 37°C. For stimulation of T cells with anti-CD3, the cells were pre-coated with mAb C363.29B (rat anti-mouse CD3ε) [22] and Affinipure F(ab’)2 fragment mouse-anti-rat IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) was used to cross-link the mAb to initiate activation.
Cell labeling and culture
DO11.10 naive and memory cells (1x105/well) were cultured in 96-well flat-bottom plates with irradiated (10,000R) RT11-mB7 cells (2x105/well) in 0.2 ml RPMI-1640 medium supplemented with 10% FBS, 50 mM 2-mercaptoethanol, 100 U/ml penicillin, 100 mg/ml streptomycin, and 2 mM glutamine. Where indicated, SEB (15 μg/ml) or OVA(323–339) (0.2 μg/ml) were added to the cultures with or without the addition of PMA (5 ng/ml). For anti-CD3ε stimulation, T cells were treated as described above. For proliferation analysis, the cells were cultured for 3 days followed by a 16-h pulse with [3H]-TdR (1 mCi/well). Cells were harvested using a 96-well automated harvester and radioactivity was measured using a BetaPlate (Wallac, Inc. Gaithersburg, MD).
Immunoprecipitation and phosphorylation analysis
Irradiated RT11-mB7 (5x106/well) cells were adhered to 24-well flat-bottom plates for 3-h at 37°C. Non-adherent cells were removed and remaining cells were pulsed with SEB (20 μg/ml) or OVA(323–339) (1.0 μg/ml) for 4-h. T cells (3x106/well) were added to the wells and plates were centrifuged (600xg) for 30 seconds to facilitate cell-cell contact. Plates were then incubated at 37°C for various times. For anti-CD3 activation, T cells (6x107/ml) were incubated in RPMI with mAb C363.29B (20 μg/ml) for 15 minutes on ice and then cross-linked with goat anti-rat F(ab’)2 (4 mg/3x106 cells) and incubated at 37°C. Signaling was quenched by placing the cells in ice-cold PBS followed by centrifugation at 4°C. The cells were solubilized in cell lysis buffer (20 mM Tris pH7.5, 150 mM NaCl, 1 mM EGTA, 5 mM EDTA, 1% Triton X-100, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 mM iodoacetamide, 5 mM benzamidine HCl, 1 mM AEBSF, 50 mM NaF, 10 mM Na4P2O7, 1 mM Na3VO4) for 30 min at 4°C. Lysates were precleared with Bio Mag Protein-G magnetic microbeads (Perseptive Biosystems, Framingham, MA) for 20-min at 4°C followed by removal of beads by adherence to magnets. Immunoprecipitations were carried out with 5 μg of the specified antibodies. Following lysate incubation with the precipitating antibody, Protein-G magnetic microbeads were incubated with the lysates for 30 min at 4°C followed by isolation using magnetic separation and repeated washing with lysis buffer containing 0.1% Triton X-100. For western blot analysis, SDS-sample buffer was added to the beads and boiled for 5 min followed by resolution of proteins on 8 or 12% SDS-PAGE gels. Proteins were transferred to PVDF membranes, blocked in 3.5% non-fat dry milk dissolved in TBST and probed with specific protein antibodies followed by appropriate secondary antibodies (goat anti-mouse IgG-HRPO or goat anti-rabbit IgG-HRPO). Immunoblots were developed with the ECL+ system and autoradiographed on ECL Hyperfilm (Amersham Pharmacia Biotech, Piscataway, NJ). The immunoblots were re-probed after removal of the bound antibodies with a stripping buffer (100 mM 2-ME, 2% SDS, 62.5 mM Tris-HCl pH 6.7) at 50°C for 30-min. The developed immunoblots were analyzed by scanning densitometry (CSPI Masterscan Interpretive Densitometer, Scanalytics, Billerica, MA).
In vitro tyrosine kinase assay
For analysis of kinase activity, immunoprecipitates were washed with kinase reaction buffer (25 mM Tris-HCl pH 7.2, 10 mM MgCl2, 10 mM MnCl2, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM AEBSF, 1 mM Na3VO4, 0.5 mM EGTA, 2 mM 2-ME) and assayed using a non-radioactive tyrosine kinase assay kit (Pierce Chemical, Rockford, IL). Briefly, a biotinylated substrate peptide was bound to avidin-coated wells and incubated with the immunoprecipitate and ATP/MgCl2 in reaction buffer. Detection was done using an HRP-conjugated anti-phosphotyrosine antibody followed by the supplied 1-STEP Turbo TMB ELISA substrate. Relative kinase activity was determined by conversion of absorbance (450 nm) values versus a standard curve derived from supplied standards (absorbance versus concentration of phosphopeptide standard (ng/mL).
Results
Both SEB and anti-CD3 mAbs promote partial TCR-mediated signal transduction in CD4 memory T cells
In an earlier study we showed that the addition of a phorbol ester, such as PMA, to cultures containing CD4 memory T cells, APCs and SEB permitted the memory cells to proliferate [14]. Further, PMA alone was insufficient to stimulate memory cells [14]. We speculated that SEB promoted partial signaling which did not lead to PKC activation. To determine whether anti-CD3 could, likewise, fully signal memory cells if PKC was activated, purified CD4+ T cells from DO11.10 mice were separated into naive and memory T cells based upon expression of CD45RB using mAb23G2 [23]. CD45RB is highly expressed on murine naive CD4 cells and weakly expressed on naive cells [20]. Each T cell population was then cultured with APCs plus OVA, SEB, or anti-CD3 in the presence or absence of PMA. As anticipated, naive cells proliferated in response to any of the three stimuli, regardless of whether PMA was added (Figure 1). In contrast, memory cells proliferated only in response to OVA and not to SEB or anti-CD3. However, proliferation occurred if PMA was also added with either SEB or anti-CD3. Since PMA by itself was ineffective, it was likely that both SEB and anti-CD3 initiated some signaling in memory cells but that signaling was not sufficient to promote the translocation of PKC to the plasma membrane and/or its subsequent activation. Further, it is likely that it is this failure to activate PKC which leads to the lack of proliferation of memory cells in response to anti-CD3 or SEB.
Figure 1. Addition of PMA restores the ability of memory CD4 T cells to proliferate in response to anti-CD3 and SEB.
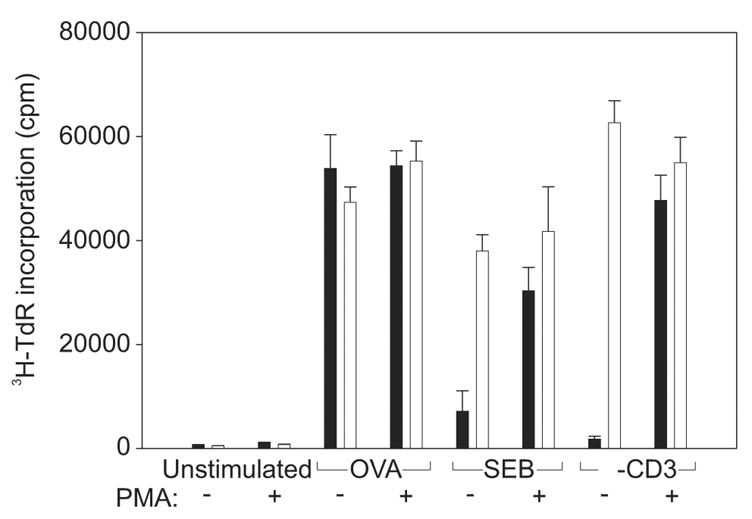
DO11.10 T cells were isolated and separated into naive (□ and memory (■) populations. The cells were left unstimulated or stimulated with OVA, SEB, or anti-CD3 with (+) or without (−) the addition of PMA. Proliferation was measured using a 16-h pulse with [3H]-TdR. Data shown is from one of two independent experiments.
TCR-mediated signal transduction is impaired in memory cells responding to either SEB or anti-CD3
Essential elements for PKC activation and translocation to the plasma membrane are the activation, via tyrosine phosphorylation, of the signaling proteins, LAT and PLC-γ1 [17;24]. Activated LAT interacts with PLC-γ1 and promotes translocation of PLC-γ1 to the plasma membrane where it can catalyze the hydrolysis of phosphatidyl (4,5) bisphosphate leading to diacylglycerol production and PKC activation [24;25]. We hypothesized that defective activation of LAT and/or PLC-γ1 would cause signaling to abort prior to PKC activation. Therefore, we examined the level of activation of these two molecules following TCR ligation. For these and subsequent experiments, naive and memory DO11.10 cells were exposed to either OVA or SEB by presentation using adherent RT11-mB7 cells that express both MHC class II (I-Ad) and B7-1 [21]. After stimulation, T cells could be recovered and used for subsequent analysis free from contaminating APCs [16]. Exposure to anti-CD3 was done by incubation with rat anti-mouse CD3ε followed by cross-linking with F(ab’)2 fragment of mouse anti-rat IgG [10]. Responses by T cells via these modes of activation were comparable to those seen when T-depleted splenocytes were used to present any of the three stimuli ([16] and data not shown). Following stimulation, the T cells were lysed and cell-associated LAT and PLC-γ1 were obtained by immunoprecipitation using specific Abs. Following SDS-PAGE, the immunoprecipitates and their corresponding activation states were identified by immunoblotting using specific mAbs or anti-phosphotyrosine mAb, respectively. Stimulation of memory cells with OVA and stimulation of naive cells with any of the three stimuli led to tyrosine phosphorylation of both LAT (Figure 2A) and PLC-γ1 (Figure 2B). In contrast, neither signaling molecule was activated in memory cells stimulated with anti-CD3 as compared to unstimulated cells, and activation of both molecules was greatly reduced in memory cells stimulated with SEB. Numbers of memory cells from DO11.10 mice are limiting and thus we do not present data for unstimulated memory cells, however, in additional experiments unstiimulated memory cells were assessed and found to be similar to unstimulated naive cells with regard to each examined protein (data not shown). Hence, neither SEB nor anti-CD3 initiate productive signaling via the TCR. Further, a signaling defect upstream of LAT activation prevents further activation of PLC-γ, the subsequent activation of PKC, and cell proliferation.
Figure 2. Decreased phosphorylation of LAT and PLC-γ1 in memory CD4 T cells stimulated with SEB and anti-CD3.
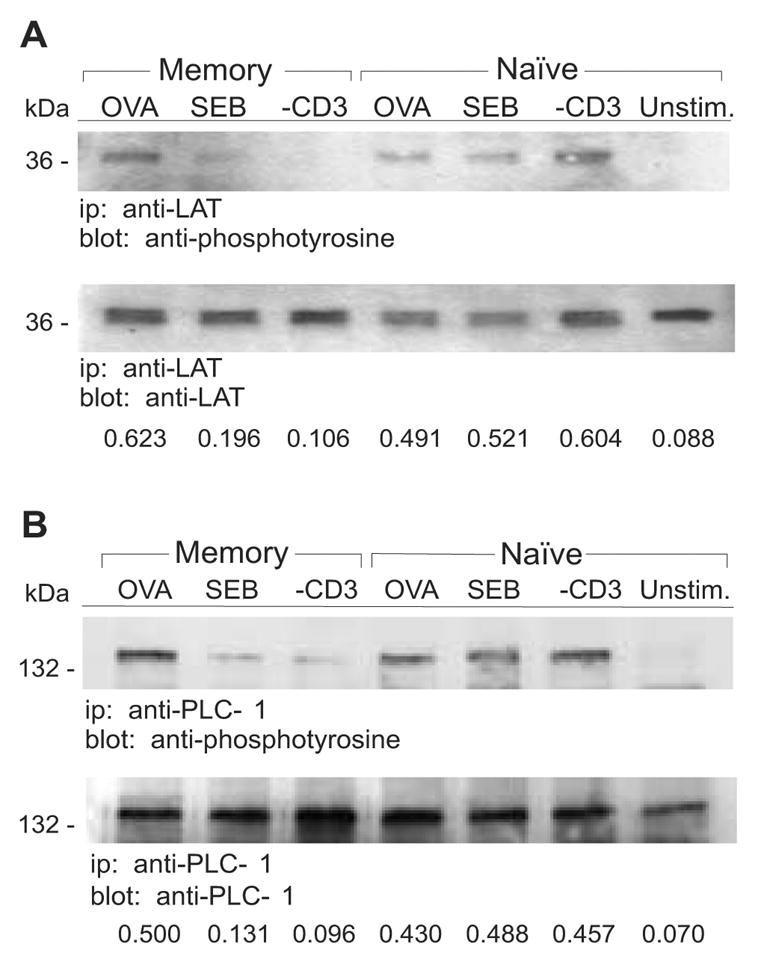
Naive and memory DO11.10 T cells were stimulated for 2 min with OVA, SEB, anti-CD3 or left unstimulated (Unstim.). A, LAT was immunoprecipitated (ip) from the T cell lysates and proteins were resolved on 12% SDS-PAGE gels followed by transfer to PVDF membranes. Immunoblotting (blot) was performed with anti-phosphotyrosine and anti-LAT Abs. B, PLC-γ1 was immunoprecipitated from the T cell lysates and proteins were resolved on 8% SDS-PAGE gels followed by transfer to PVDF membranes. Immunoblotting was performed with anti-phosphotyrosine and anti-PLC-γ1 Abs. Densitometry was performed on the immunoblots and the relative levels of phosphoprotein are expressed as a ratio relative to the total amount of precipitated protein. Data are representative of at least two independent experiments.
Signal transduction through the TCR leading to PLC-γ1 activation occurs through a cascade involving activation of several kinases. After peptide/MHC binding, ITAMS in the cytoplasmic domains of the TCR/CD3ζ-chains are phosphorylated by src kinases, such as Lck and Fyn, leading to the recruitment and activation of ZAP-70 [17]. ZAP-70 is subsequently phosphorylated on tyrosines by Lck and/or Fyn and then becomes itself capable of activating LAT and PLC-γ1 via tyrosine phosphorylation. We next evaluated the activation state of the TCR/CD3ζ-chains and ZAP-70 following stimulation of DO11.10 naive and memory CD4 T cells after engagement of their TCRs by OVA, SEB, or anti-CD3 mAb. Regardless of the cell type or stimulus, CD3ζ was phosphorylated in response to TCR ligation (Figure 3). Furthermore, signal transduction generally resulted in an increased level of p21 CD3ζ and an induction of p23 CD3ζ. We did note, however, that there was both a lower degree of p21 CD3ζ phosphorylation and an especially low amount of p23 found in anti-CD3-stimulated memory cells. In contrast to productive CD3ζ activation, ZAP-70 was only phosphorylated under conditions where the cells ultimately proliferated. For memory cells treated with anti-CD3 or SEB there was little to no tyrosine phosphorylation of ZAP-70 as compared to control, unstimulated cells (Figure 4A). In most experiments almost no phosphorylated ZAP-70 was observed when memory cells were stimulated with SEB. However, because in occasional experiments small amounts of phosphorylated ZAP-70 were observed in populations of SEB-treated memory cells (e.g. Figure 4A), we performed additional experiments to show that the decreased pZAP-70 in memory cells responding to SEB was not due to kinetic differences in signaling. When naive and memory cells were exposed to SEB for various times prior to examination of ZAP-70, we observed that the peak in activation occurred at similar times in both cell types (Figure 4B). However, the relative amount of phosphorylated ZAP-70 was consistently lower in memory cells. Hence, for both SEB and anti-CD3, insufficient ZAP-70 activation leads to impaired signal transduction and the resulting lack of proliferative response.
Figure 3. Tyrosine phosphorylation of TCR/CD3ζ-chain and ZAP-70 in naive and memory CD4 T cells.
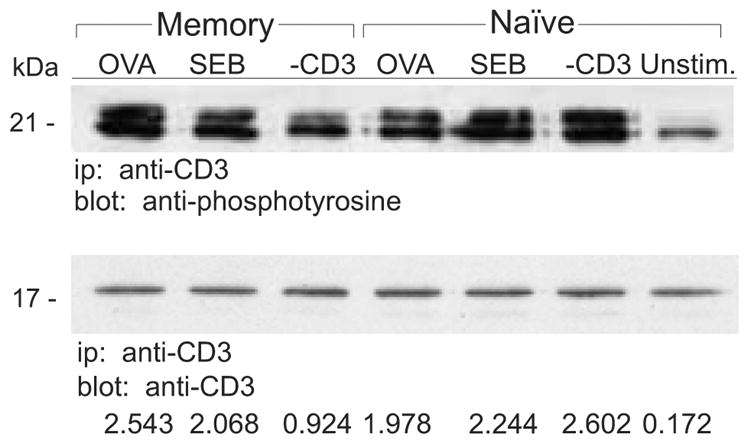
A, Naive and memory DO11.10 T cells were stimulated with for 2 min OVA, SEB, anti-CD3 or left unstimulated (Unstim.). The TCR/CD3ζ-chain complex was immunoprecipitated (ip) from the T cell lysates and proteins were resolved on 12% SDS-PAGE gels. Immunoblotting (blot) was performed with anti-phosphotyrosine and anti-CD3ζ Abs. Two forms of phosphorylated CD3ζ are detected, p21 and p23. When CD3ζ is fully activated, p23 is detected. To determine the amount of fully activated CD3ζ, densitometry was performed on the immunoblots and the relative levels of p23 phosphoprotein are expressed as a ratio relative to the total amount of precipitated protein. Data are representative of at least two independent experiments.
Figure 4. Decreased phosphorylation of ZAP-70 in memory cells stimulated with SEB and anti-CD3.
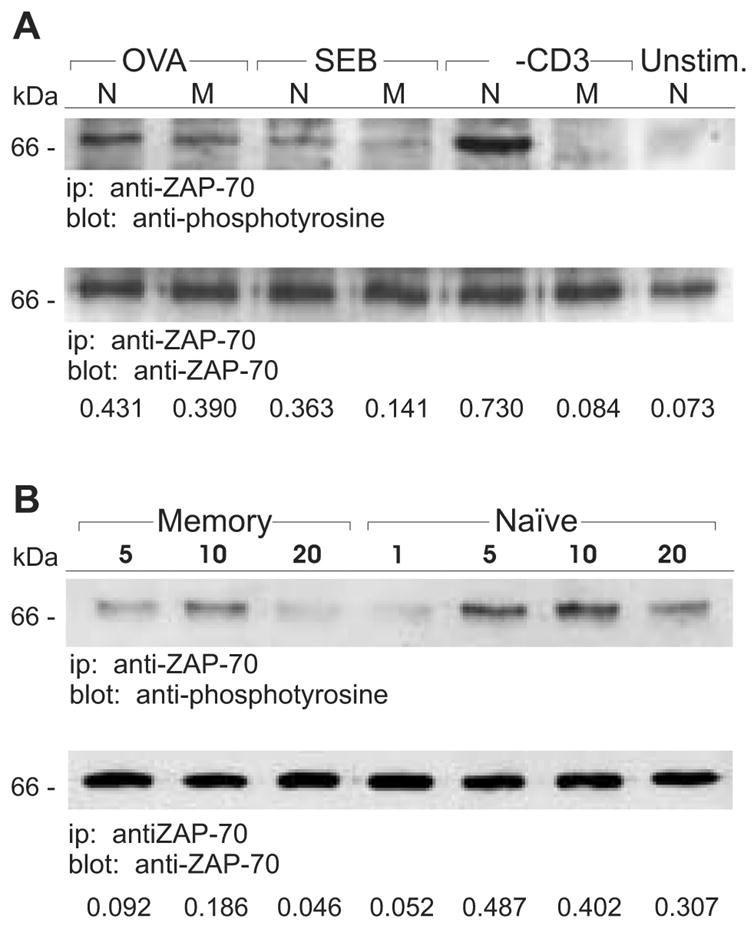
A, Naive (N) and memory (M) DO11.10 T cells were stimulated for 2 min with OVA, SEB, anti-CD3 or left unstimulated (Unstim.). ZAP-70 protein was immunoprecipitated from the T cell lysates and proteins were resolved on 8% SDS-PAGE gels. Immunoblotting was performed with anti-phosphotyrosine and anti-ZAP-70 Abs. B, Naive and memory DO11.10 T cells were stimulated with SEB for the indicated lengths of time (min). ZAP-70 protein was immunoprecipitated from the T cell lysates and resolved on 8% SDS-PAGE gels. Immunoblotting was performed with anti-phosphotyrosine and anti-ZAP-70 Abs. Densitometry was performed on the immunoblots and the relative levels of phosphoprotein are expressed as a ratio relative to the total amount of precipitated protein. Data are representative of at least two independent experiments.
Distinct signaling blockades in memory cells stimulated with SEB or anti-CD3
Both TCR/CD3ζ and ZAP-70 are substrates for the src kinases Lck and, to a lesser degree, Fyn. Since signal transduction in both SEB and anti-CD3 -treated memory T cells appeared to be blocked between phosphorylation of CD3ζ and ZAP-70 (SEB) or even prior to full CD3ζ phosphorylation (anti-CD3), we next examined src kinase activity in stimulated cells. Hence, DO11.10 naive and memory T cells were collected after exposure to OVA, SEB, or anti-CD3 mAb. The src kinases, Lck and Fyn were isolated from cell lysates by immunoprecipitation and their activation states were determined after immunoblotting with an anti-phosphotyrosine mAb. As indicated in Figure 5, active Fyn was found in lysates obtained from both naive and memory cells exposed to any of the three stimuli. Indeed, in memory cells exposed to SEB, slightly higher levels of activated Fyn were found when compared to the levels of phosphorylated Fyn associated with memory cells treated with OVA or anti-CD3 or from activated naive cells (Figure 5A). Although the observation of tyrosine phosphorylation of Fyn (and Lck) in our immunoblot analyses was dependent upon engagement of the TCRs on both naive and memory cells, it is noteworthy that inactive src kinases are also phosphorylated on an alternative (inhibitory) tyrosine residue. Hence, we also directly measured the level of src kinase activity using an in vitro assay. As we observed by immunoblot analysis, Fyn kinase activity was found associated with both naive and memory cells exposed to any of the three test stimuli (Figure 5B). Further, while Fyn activity was equivalent in naive cells and in OVA or anti-CD3-treated memory cells, SEB induced slightly higher levels of Fyn activity in memory cells. This increase in Fyn activation was not due to kinetic differences between naive and memory cells or with respect to the different stimuli as slightly higher Fyn activity levels were observed in memory cells exposed to SEB at all examined time intervals (data not shown). Hence, neither anti-CD3 nor SEB treatment impairs memory cells Fyn kinase activation; indeed, there is an elevation of memory cell Fyn activity in response to SEB.
Figure 5. Activation of Fyn is increased following stimulation of memory CD4 T cells with SEB.
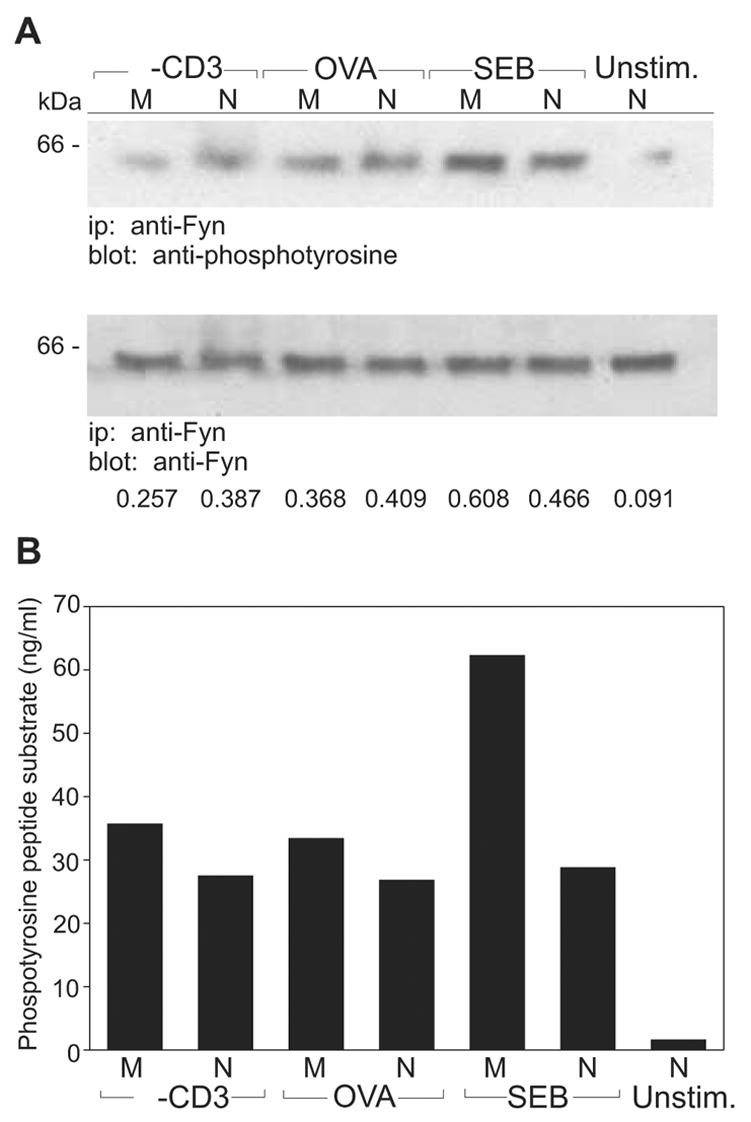
A, Naive (N) and memory (M) DO11.10 T cells were stimulated for 2 min with anti-CD3, OVA, SEB or left unstimulated (Unstim.). Fyn protein was immunoprecipitated (ip) from the T cell lysates and resolved on 8% SDS-PAGE gels. Immunoblotting (blot) was carried out with anti-phosphotyrosine and anti-Fyn Abs. Densitometry was performed on the immunoblots and the relative levels of phosphoprotein are expressed as a ratio relative to the total amount of precipitated protein. B, Naive and memory T cells were stimulated and Fyn protein was immunoprecipitated as in part A. Precipitated Fyn protein was used in ELISA-based tyrosine kinase assays to measure the activity of Fyn in phosphorylating peptide substrate. Phosphorylation values were determined by converting Absorbance measurements using a standard curve derived from supplied soluble phosphopeptide (ng/mL). Data are representative of three independent experiments.
We next examined whether the major T cell-associated src kinase, Lck, was fully activated in memory cells responding to OVA, SEB or anti-CD3 mAb. Again, we assessed Lck activation via both immunoblotting (Figure 6A) and direct measurement of kinase activity (Figure 6B). In naive cells and in memory cells responding to either OVA or SEB, the degree of tyrosine phosphorylation and the levels of kinase activity were comparable. In our initial experiments, we observed that memory cells exposed to anti-CD3 failed to activate Lck (Figure 6A and 6B). However, a more extended analysis of Lck activation revealed that Lck was transiently phosphorylated in anti-CD3-stimulated memory cells and that it became rapidly dephosphorylated (Figure 6C). Consistent with these results, in vitro kinase analysis of memory cells stimulated with anti-CD3 revealed that Lck activity was present early (at 2-min), but it rapidly decreased thereafter (Figure 6D). These data suggest that either Lck activation is impaired or that tyrosine phosphatase activity is enhanced in anti-CD3 treated memory cells. Thus, the failure to activate ZAP-70 and proceed with downstream signaling in memory cells exposed to anti-CD3 mAbs, is likely due to a failure of sustained Lck kinase activity.
Figure 6. Activation of Lck is compromised in anti-CD3 treated memory CD4 T cells.
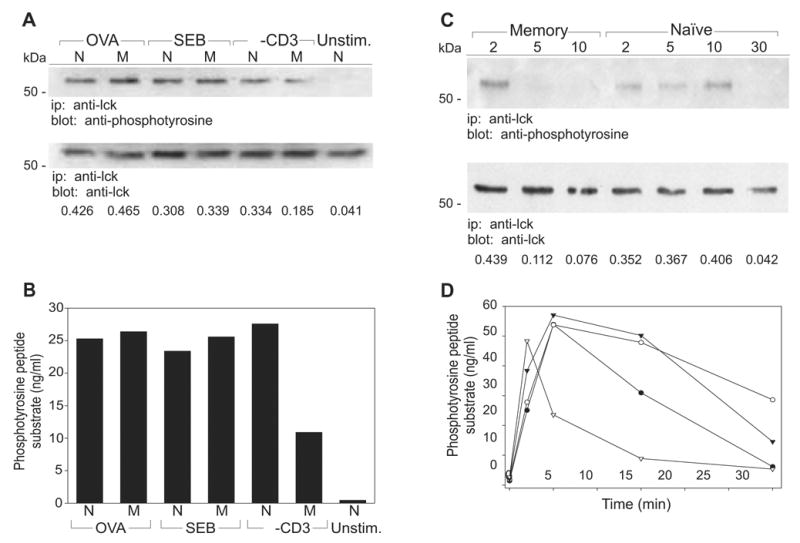
A–B, Naive (N) and memory (M) DO11.10 T cells were stimulated 2 min with OVA, SEB, anti-CD3 or left unstimulated (Unstim.). Lck protein was immunoprecipitated (ip) from the T cell lysates and (A) resolved on 8% SDS-PAGE gels and immunoblotted (blot) with anti-phosphotyrosine and anti-Lck Abs, or (B) used in tyrosine kinase assays. Phosphorylation values were determined by converting Absorbance measurements using a standard curve derived from supplied soluble phosphopeptide (ng/mL). C–D, Naive (○, ●) and memory (▽, ▼) DO11.10 T cells were stimulated with anti-CD3 (open) or SEB (filled) for the indicated lengths of time (min). Lck protein was immunoprecipitated from the T cell lysates and (C) resolved on 8% SDS-PAGE gels and immunoblotted as above, or (D) used in tyrosine kinase assays as above. Densitometry was performed on the immunoblots and the relative levels of phosphoprotein are expressed as a ratio relative to the total amount of precipitated protein. Data are representative of at least two independent experiments.
The previous experiments demonstrated that ZAP-70 was not activated in CD4 memory cells treated with either SEB or anti-CD3. Further, Lck activation appeared defective in anti-CD3 but not SEB-treated memory cells. Because an essential component step in the activation of ZAP-70 is its recruitment to the TCR/CD3ζ-chain complex prior to its phosphorylation by Lck and/or Fyn [26], we wanted to determine whether ZAP-70 and TCR/CD3ζ became associated during memory cell stimulation. Following stimulation, naive and memory DO11.10 CD4 cells were detergent solubilized and CD3ζ was isolated by immunoprecipitation using a specific anti-ZAP-70 mAb. Immunoblot analysis followed SDS-PAGE of the immunoprecipitates so that total ZAP-70 protein was identified. We also examined the blots for the presence of active ZAP-70, as determined by tyrosine phosphorylation, and we also determined whether the TCR/CD3ζ-chain protein was co-immunoprecipitated with ZAP-70. As indicated in Figure 7, ZAP-70 was found associated with CD3ζ in activated but not resting naive cells. Further, ZAP-70 and TCR/CD3ζ-chain were co-isolated in memory cells that were exposed to either OVA or anti-CD3 antibodies. In contrast, very little ZAP-70 was found associated with TCR/CD3ζ when memory cells were treated with SEB. Hence, in memory cells exposed to SEB, impaired activation of ZAP-70 is likely due to its failure to associate with the TCR/CD3 complex.
Figure 7. Decreased association between ZAP-70 and TCR/CD3ζ-chain in memory CD4 T cells stimulated with SEB.
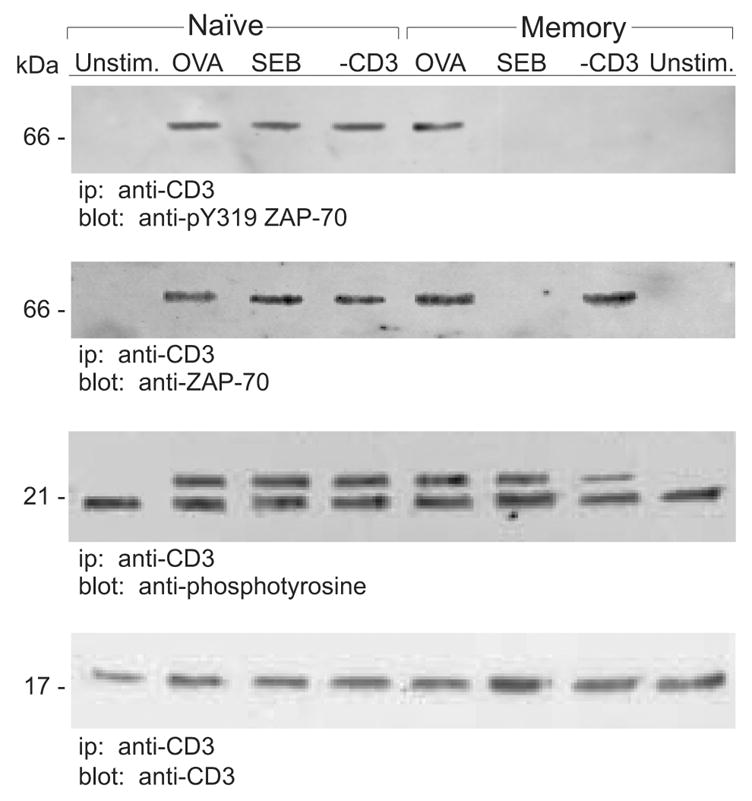
Naive and memory DO11.10 T cells were stimulated 2 min with OVA, SEB, anti-CD3 or left unstimulated (Unstim.) and CD3ζ-chain protein was immunoprecipitated. Immunoblotting was performed as indicated, using anti-phosphotyrosine, anti-CD3ζ Abs, anti-ZAP-70, or anti-pY319 ZAP-70 (activated ZAP-70). Data are representative of three independent experiments.
Discussion
Several studies have shown that naive and memory T cells can make different biological responses upon exposure to the same TCR-ligating agent. How this might control the disparate immune functions of these two cell types is unclear, but it is likely that the varied responses are linked to faster recall immunity and/or distinct regulatory mechanisms which prevent untoward autoimmune activation. We have previously reported that stimulation of memory CD4 T cells with peptide antigen, superantigen, and anti-CD3 mAbs leads to three distinct biological outcomes, namely, activation/proliferation, anergy, and “ignorance” [16]. Thus, the exposure of memory cells to peptide leads to classical cell activation that results in extensive cell division and lymphokine secretion. In contrast, the exposure of memory cells to superantigen does not promote activation, but instead leads to a prolonged non-responsive state where memory cells lose the ability to respond positively to antigen. Finally, like superantigens, anti-CD3 mAbs do not promote memory cell activation, however, unlike the case with superantigens, memory cells still retain the ability to respond to peptide [10;16]. In the present study, we have examined signal transduction through the TCR in naive and memory CD4 T cells exposed to each of these stimuli. Canonical TCR-mediated signal transduction was observed in naive cells responding to any of the stimuli and naive cells were activated by each stimulus. While canonical signaling was indeed observed in memory cells responding to peptide, both superantigen (SEB) and anti-CD3 caused aberrant signaling. These observations strongly suggest that anergy and ignorance are active processes which result from impaired or altered TCR-mediated signal transduction.
Signal transduction in memory T cells is, in general, not well understood. To a large extent, this results from the lack of sufficient numbers of specific memory cells with which to study responses to peptide antigens. Even in most TCR-transgenic mouse strains there is a paucity of antigen-specific memory cells [27]. The DO11.10 mouse model is useful because of the relatively abundant numbers of OVA-specific memory CD4 cells. These memory cells can be obtained from mice that had not been previously immunized with OVA. They are generated in response to environmental antigens which trigger a dual-expressed TCR composed of the transgenic TCR Vβ chain paired with an endogenous TCR Vα chain [15]. As we have previously described [16], these cells are typical memory CD4 cells. Given the lack of memory cell source material in prior studies, many of the assumptions about signal transduction in memory cells are based upon data obtained using naive T cells or cloned T-cell lines. Insight has also come from studies, which employed polyclonal stimuli, such as anti-CD3 mAbs, but it is not clear whether this also reflects pathways triggered by peptide stimulation, given that the polyclonal stimuli can sometimes lead to different biological outcomes [5;8]. Still, very detailed studies, primarily coming from the Farber laboratory, indicate that signaling in memory cells is different than in naive cells [28–31]. Those studies have primarily used the anti-CD3 mAbs (under activating conditions) to induce memory cell signaling. In the current study, we have examined signaling in naive and memory cells responding to peptide antigens and we have identified no significant differences in a straightforward path from TCR-ligation down to PKC activation. However, we should note that we have recently reported differences in peptide-induced signaling in naive and memory cells with respect to the formation of signaling complexes at the T-cell: APC synapse [32].
It is generally accepted that an increased intracellular calcium content along with activation of PKC is necessary and sufficient to induce lymphocyte proliferation. Prior studies have indicated that SEB and OVA induce comparable increases in intracellular calcium levels in DO11.10 memory T cells [33]. Further examination showed that several of the biological consequences of SEB (and the anti-CD3) exposure (e.g. induction of cell surface markers) were solely due to calcium-mediated signaling [33]. Since the addition of PMA along with either SEB or anti-CD3 led to memory cell proliferation, we hypothesized that signal transduction was blocked prior to the stage where PKC was activated. The lack of PLC-γ1 activation supports this hypothesis. Since PLC-γ1 catalyzes the hydrolysis of phosphatidyl (4,5) bisphosphate leading to diacylglycerol production and PKC activation [24;34], as well as promoting the release of intracellular calcium stores through inositol-1,4,5-trisphosphate, we speculate that the increase in intracellular calcium concentrations in response to anti-CD3 [33;35;36] and SEB ([33] and unpublished observations) are largely due to an influx of extracellular calcium. Further, because PMA directly activates PKC and because both the SEB- and the anti-CD3-treated memory cells proliferated upon addition of PMA, we suggest that signal transduction downstream of PKC is normal and that the failure to proliferate is due to the upstream defects that we have identified in this study. At present, it is unclear as to why, in the absence of ZAP-70 and PLC-γ1 activation, there is sufficient intracellular calcium to promote an influx of extracellular calcium or to promote proliferation in the presence of PMA. The levels of intracellular calcium are generally lower in memory cells even when responding to an activating stimulus [35]. It may be that small amounts of calcium can enter the cell in the absence of changes in intracellular calcium stores and, since memory cells do not require as much calcium, the amount entering is sufficient to promote activation. We do note that increases in marker expression in SEB- or anti-CD3-exposed memory cells can be prevented by chelating extracellular calcium or by using putative calcium channel blockers ([33] and A.W., unpublished observations).
In memory CD4 cells stimulated with either anti-CD3 or SEB, signal transduction terminates because ZAP-70 does not become activated. As noted above, in a small number of experiments we have observed a low level of p-ZAP-70 in preparations of memory cells exposed to SEB (e.g. Figure 4). However, in most experiments we cannot detect any p-ZAP-70 (e.g. Figure 7). At present we believe that the occasional activated ZAP-70 that we observe may result from small numbers of non-memory cells (e.g. contaminating naive cells or CD45RBlo effector CD4 cells) within our cell preparations. In anti-CD3 or SEB-treated memory cells, because ZAP-70 is not activated signaling events downstream do not occur as indicated by a lack of tyrosine phosphorylation of LAT and PLC-γ1 [37]. However, the mechanism underlying the failure to phosphorylate ZAP-70 differs depending on the mode of stimulation. In memory cells stimulated by anti-CD3, Lck kinase activation is not sustained and Lck is not able to phosphorylate ZAP-70. Previous studies likewise showed defective ZAP-70 phosphorylation in anti-CD3-treated memory cells [28;31]. In the study by Farber et al [10] the failure to activate anti-CD3-treated memory cells was attributed to an inhibitory signal transduced through CD4 via MHC Class II molecules. Further studies showed that such negative signaling was dependent upon unique molecular interactions between CD3, CD4, and CD45 which were regulated by the memory cell-specific isoform of CD45 [38;39]. Given the data presented in the current study, we speculate that negative signaling via CD4 occurs through activation of CD45 or some other tyrosine phosphatase associated with CD4. This putative phosphatase would permit initial Lck activation which would in turn lead to CD3ζ phosphorylation and docking of ZAP-70. However, shortly thereafter dephosphorylation of Lck would lead to abrupt termination of Lck activity before ZAP-70 phosphorylation and activation. Because of the importance of CD45 in TCR-mediated signaling, we considered that our use of an antibody to CD45RB (mAb 23G2) to initially separate naive and memory cells might have influenced our findings. However, we note that little to no anti-CD45RB binds to memory cells and it is these cells that respond differently to the various stimuli used in this study. In contrast, naive cells which express large amounts of CD45RB respond similarly to the various stimuli. Further, independent isolation of memory cells based on high expression of CD44, instead of low expression of CD45RB, resulted in similar functional characteristics in all experiments (WTL, unpublished observations).
Unlike with anti-CD3, presentation of SEB to CD4 T cells requires MHC Class II molecules [40]. Consequently, TCR-independent interactions between MHC Class II molecules and CD4, and inhibitory signal transduction are less likely to occur. Further, unlike with anti-CD3 [41], SEB does not activate memory cells which lack CD4 (WTL, unpublished observations). Finally, both the means by which ZAP-70 fails to become activated and the ultimate biological outcome (ignorance versus anergy) differ between anti-CD3 and SEB stimulation. In SEB-treated memory CD4 cells Lck activation appears to be normal and Lck is able to phosphorylate CD3ζ. Phosphorylation of ZAP-70 is impaired, however, because ZAP-70 does not become accessible to Lck. The reason why ZAP-70 is not recruited to the CD3ζ-Lck complex is unclear at present. Since CD3ζ seems to be phosphorylated to a similar level in both OVA and SEB-treated memory cells, we would expect that the ability of ZAP-70 to bind to the phosphorylated ITAMs of CD3ζ would not be impaired. Rather, we hypothesize that ZAP-70 does not translocate to the appropriate membrane microdomain that would enable physical interactions with the CD3 complex. We are currently testing this hypothesis. We also speculate that Fyn kinase is important to the ZAP-70 recruitment process. Based upon the slightly higher levels of Fyn activity we observed only in memory cells that were exposed to SEB, we have examined cells in which Fyn activity was absent or suppressed. Our preliminary findings show that CD3ζ-ZAP-70 association and cell activation are restored in these cells (WTL and AROW, manuscript in preparation).
The mechanisms by which proximal signaling is impaired in anti-CD3 and SEB-treated memory cells differ, but with either agent a failure to phosphorylate and activate ZAP-70 leads to a lack of initial cell proliferation. Since memory cells exposed to anti-CD3, but not SEB, remain responsive to subsequent stimulation by OVA, it is unlikely that a block in initial proliferation per se leads to anergy. Hence, it is likely that additional mechanisms promote SEB-induced anergy in memory cells. In previous studies, anergy was induced in naive primary CD4 cells and in CD4 T cell lines using either TCR stimulation in the absence of costimulation or altered peptide ligands (reviewed in Refs. [42;43]). Similar to the present study, several prior studies showed aberrant proximal signaling that resulted in a failure to phosphorylate ZAP-70 [44–46]. In part, the block in ZAP-70 activation was attributable to altered CD3ζ chain ITAM phosphorylation. While differential CD3ζ phosphorylation may have led to activation of alternative, inhibitory signaling pathways, decreased CD3ζ phosphorylation may also have prevented ZAP-70 coupling and availability to Lck [45]. As noted above, CD3ζ chain phosphorylation does not appear to be impaired in SEB-treated memory cells which suggests that the underlying mechanism for inducing anergy is unique in these cells. At present, we are examining the role of Fyn in memory cell anergy. In the current study we found Fyn activity to be elevated in memory cells exposed to SEB. Previous reports have suggested that Fyn plays an important role in T cell activation [47]. While some studies have shown that over-expression of Fyn can lead to hyperactivity of antigen-specific T cells [48], other reports have shown that in anergic T cells there is an elevation of Fyn activity [49–53]. It is interesting to note that one Fyn substrate, c-cbl [51;54;54–57], can serve as a negative regulator of ZAP-70 [54;58]. Thus it is tempting to speculate that in memory cells SEB induces strong Fyn activation that leads to c-cbl-mediated inhibition of ZAP-70 activation. We are currently testing this hypothesis.
Acknowledgments
The authors would like to acknowledge Mr. G. Pasos, Mr. K. Moynehan, Ms. C. Love, and Ms. E. Lapkowicz for their expert technical assistance. We also acknowledge the Wadsworth Center Immunology Core, the Wadsworth Center Peptide Synthesis Core, and the Wadsworth Center Molecular Genetics Core. This work was supported by National Institutes of Health grant AI-35583.
Abbreviations used
- TCR
T cell receptor
- SEB
Staphylococcal enterotoxin B
- PVDF
polyvinylidene difluoride
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vitetta ES, Berton MT, Burger C, Kepron M, Lee WT, Yin XM. Memory T and B cells. AnnRevImmunol. 1991;9:193–217. doi: 10.1146/annurev.iy.09.040191.001205. [DOI] [PubMed] [Google Scholar]
- 2.Swain SL, Bradley LM, Croft M, Tonkonogy S, Atkins G, Weinberg A, Duncan DD, Hedrick SM, Dutton RW, Huston G. Helper T cell subsets: phenotype, function and the role of lymphokines in regulating their development. ImmunolRev. 1991;123:115–144. doi: 10.1111/j.1600-065x.1991.tb00608.x. [DOI] [PubMed] [Google Scholar]
- 3.Swain SL. CD4 T cell development and cytokine polarization: An overview. JLeukocyte Biol. 1995;57:795–798. doi: 10.1002/jlb.57.5.795. [DOI] [PubMed] [Google Scholar]
- 4.Swain SL. Generation and in vivo persistence of polarized Th1 and Th2 memory cells. Immunity. 1994;1:543–552. doi: 10.1016/1074-7613(94)90044-2. [DOI] [PubMed] [Google Scholar]
- 5.Byrne JA, Butler JL, Reinherz EL, Cooper MD. Virgin and memory T cells have different requirements for activation via the CD2 molecule. IntImmunol. 1989;1:29–35. doi: 10.1093/intimm/1.1.29. [DOI] [PubMed] [Google Scholar]
- 6.Byrne JA, Butler JL, Cooper MD. Differential activation requirements for virgin and memory T cells. JImmunol. 1988;141:3249–3257. [PubMed] [Google Scholar]
- 7.Sanders ME, Makgoba MW, June CH, Young HA, Shaw S. Enhanced responsiveness of human memory T cells to CD2 and CD3 receptor-mediated activation. EurJImmunol. 1989;19:803–807. doi: 10.1002/eji.1830190504. [DOI] [PubMed] [Google Scholar]
- 8.Luqman M, Bottomly K. Activation requirements for CD4+ t-cells differing in CD45r expression. JImmunol. 1992;149:2300–2306. [PubMed] [Google Scholar]
- 9.Lerner A, Yamada T, Miller RA. PgP-1hi T lymphocytes accumulate with age in mice and respond poorly to concanavalin A. EurJImmunol. 1989;19:977–982. doi: 10.1002/eji.1830190604. [DOI] [PubMed] [Google Scholar]
- 10.Farber DL, Luqman M, Acuto O, Bottomly K. Control of memory CD4 T cell activation: MHC class II molecules on APCs and CD4 ligation inhibit memory but not naive CD4 cells. Immunity. 1995;2:249–259. doi: 10.1016/1074-7613(95)90049-7. [DOI] [PubMed] [Google Scholar]
- 11.Lee WT, Vitetta ES. Memory T cells are anergic to the superantigen, staphylocoocal enterotoxin B. JExpMed. 1992;176:575–580. doi: 10.1084/jem.176.2.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janeway CA, Jr, Yagi J, Conrad PJ, Katz ME, Jones B, Vroegop S, Buxser S. T-cell responses to M1s and to bacterial proteins that mimic its behavior. ImmunolRev. 1989;107:61–88. doi: 10.1111/j.1600-065x.1989.tb00003.x. [DOI] [PubMed] [Google Scholar]
- 13.Kawabe Y, Ochi A. Selective anergy of Vbeta8+, CD4 + T cells in staphylococcus enterotoxin B-primed mice. JExpMed. 1990;172:1065–1070. doi: 10.1084/jem.172.4.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee WT, Thrush GR, Vitetta ES. Staphylococcal enterotoxin B induces the expression of activation markers on murine memory T cells in the absence of proliferation or lymphokine secretion. CellImmunol. 1995;162:26–32. doi: 10.1006/cimm.1995.1047. [DOI] [PubMed] [Google Scholar]
- 15.Lee WT, Cole-Calkins J, Street NE. Memory T cell development in the absence of specific antigen priming. JImmunol. 1996;157:5300–5307. [PubMed] [Google Scholar]
- 16.Watson AR, Mittler JN, Lee WT. Staphylococcal enterotoxin B induces anergy to conventional peptide in memory T cells. Cell Immunol. 2003;222:144–155. doi: 10.1016/s0008-8749(03)00117-5. [DOI] [PubMed] [Google Scholar]
- 17.Chan AC, Desai DM, Weiss A. The role of protein tyrosine kinases and protein tyrosine phosphatases in T cell antigen receptor signal transduction. AnnuRevImmunol. 1994;12:555–592. doi: 10.1146/annurev.iy.12.040194.003011. [DOI] [PubMed] [Google Scholar]
- 18.Murphy KM, Heimberger AB, Loh DY. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 1990;250:1720–1722. doi: 10.1126/science.2125367. [DOI] [PubMed] [Google Scholar]
- 19.Haskins K, Kubo R, White J, Pigeon M, Kappler J, Marrack P. The major histocompatibility complex-restricted antigen receptor on T cells. I. Isolation with a monoclonal antibody. JExpMed. 1983;157:1149–1169. doi: 10.1084/jem.157.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee WT, Yin XM, Vitetta ES. Functional and ontogenetic analysis of murine CD45Rhi and CD45Rlo CD4+ T cells. JImmunol. 1990;144:3288–3295. [PubMed] [Google Scholar]
- 21.Zhou T, Weaver C, Linsley PS, Mountz JD. T cells of staphylococcal enterotoxin B-tolerized autoimmune MRL-lpr/lpr mice require co-stimulation through the B7- CD28/CTLA-4 pathway for activation and can be reanergized in vivo by stimulation of the T cell receptor in the absence of this co-stimulatory signal. EurJImmunol. 1994;24:1019–1025. doi: 10.1002/eji.1830240502. [DOI] [PubMed] [Google Scholar]
- 22.Portoles P, Rojo J, Golby A, Bonneville M, Gromkowski S, Greenbaum L, Janeway CA, Jr, Murphy DB, Bottomly K. Monoclonal antibodies to murine CD3 epsilon define distinct epitopes, one of which may interact with CD4 during T cell activation. JImmunol. 1989;142:4169–4175. [PubMed] [Google Scholar]
- 23.Birkeland ML, Johnson P, Trowbridge IS, Pure E. Changes in CD45 isoform expression accompany a nt igen-induced murine T cell activation. ProcNatlAcadSci(USA) 1989;86:6734–6839. doi: 10.1073/pnas.86.17.6734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiss A, Koretzky G, Schatzman RC, Kadlecek T. Functional activation of the T-cell antigen receptor induces tyrosine phosphorylation of phospholipase C-gamma 1. ProcNatlAcadSciUSA. 1991;88:5484–5488. doi: 10.1073/pnas.88.13.5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilliland LK, Schieven GL, Norris NA, Kanner SB, Aruffo A, Ledbetter JA. Lymphocyte lineage-restricted tyrosine-phosphorylated proteins that bind PLC gamma 1 SH2 domains. JBiolChem. 1992;267:13610–13616. [PubMed] [Google Scholar]
- 26.Chan AC, Irving BA, Weiss A. New insights into T-cell antigen receptor structure and signal transduction. CurrOpinImmunol. 1992;4:246–251. doi: 10.1016/0952-7915(92)90072-m. [DOI] [PubMed] [Google Scholar]
- 27.Kearney ER, Pape KA, Loh DY, Jenkins MK. Visualization of peptide-specific immunity and peripheral tolerance induction in vivo. Immunity. 1994;1:327–339. doi: 10.1016/1074-7613(94)90084-1. [DOI] [PubMed] [Google Scholar]
- 28.Farber DL, Acuto O, Bottomly K. Differential T cell receptor-mediated signaling in naive and memory CD4 T cells. Eur J Immunol. 1997;27:2094–2101. doi: 10.1002/eji.1830270838. [DOI] [PubMed] [Google Scholar]
- 29.Ahmadzadeh M, Hussain SF, Farber DL. Heterogeneity of the memory CD4 T cell response: persisting effectors and resting memory T cells. JImmunol. 2001;166:926–935. doi: 10.4049/jimmunol.166.2.926. [DOI] [PubMed] [Google Scholar]
- 30.Krishnan S, V, Warke G, Nambiar MP, Wong HK, Tsokos GC, Farber DL. Generation and biochemical analysis of human effector CD4 T cells: alterations in tyrosine phosphorylation and loss of CD3zeta expression. Blood. 2001;97:3851–3859. doi: 10.1182/blood.v97.12.3851. [DOI] [PubMed] [Google Scholar]
- 31.Hussain SF, Anderson CF, Farber DL. Differential SLP-76 expression and TCR-mediated signaling in effector and memory CD4 T cells. JImmunol. 2002;168:1557–1565. doi: 10.4049/jimmunol.168.4.1557. [DOI] [PubMed] [Google Scholar]
- 32.Watson AR, Lee WT. Differences in signaling molecule organization between naive and memory CD4+ T lymphocytes. JImmunol. 2004;173:33–41. doi: 10.4049/jimmunol.173.1.33. [DOI] [PubMed] [Google Scholar]
- 33.Lee WT, Mittler JN, Watson ARO, Li X. Actiivation and anergy in memory CD4 T lymphocytes. RecResDevImmunol. 2002;4:303–329. [Google Scholar]
- 34.Majerus PW, Neufeld EJ, Wilson DB. Production of phosphoinositide-derived messengers. Cell. 1984;37:701–703. doi: 10.1016/0092-8674(84)90405-7. [DOI] [PubMed] [Google Scholar]
- 35.Miller RA, Flurkey K, Molloy M, Luby T, Stadecker MJ. Differential sensitivity of virgin and memory T lymphocytes to calcium ionophores suggests a buoyant density separation method and a model for memory cell hyporesponsiveness to Con A. JImmunol. 1991;147:3080–3086. [PubMed] [Google Scholar]
- 36.Leitenberg D, Constant S, Lu DD, Smith BR, Bottomly K. CD4 and CD45 regulate qualitatively distinct patterns of calcium mobilization in individual CD4+ T cells. EurJImmunol. 1995;25:2445–2451. doi: 10.1002/eji.1830250906. [DOI] [PubMed] [Google Scholar]
- 37.Gelfand EW, Weinberg K, Mazer BD, Kadlecek TA, Weiss A. Absence of ZAP-70 prevents signaling through the antigen receptor on peripheral blood T cells but not on thymocytes. JExpMed. 1995;182:1057–1065. doi: 10.1084/jem.182.4.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leitenberg D, Novak TJ, Farber D, Smith BR, Bottomly K. The extracellular domain of CD45 controls association with the CD4-T cell receptor complex and the response to antigen-specific stimulation. JExpMed. 1996;183:249–259. doi: 10.1084/jem.183.1.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leitenberg D, Boutin Y, Lu DD, Bottomly K. Biochemical association of CD45 with the T cell receptor complex: regulation by CD45 isoform and during T cell activation. Immunity. 1999;10:701–711. doi: 10.1016/s1074-7613(00)80069-2. [DOI] [PubMed] [Google Scholar]
- 40.Yagi J, Baron J, Buxser S, Janeway CA., Jr Bacterial proteins that mediate the association of a defined subset of T cell receptor:CD4 complexes with Class II MHC. JImmunol. 1990;144:892–901. [PubMed] [Google Scholar]
- 41.Metz DP, Bottomly K. Function and regulation of memory CD4 T cells. ImmunolRes. 1999;19:127–141. doi: 10.1007/BF02786482. [DOI] [PubMed] [Google Scholar]
- 42.Schwartz RH. T cell anergy. AnnuRevImmunol. 2003;21:305–334. doi: 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- 43.Sloan-Lancaster J, Allen PM. Altered peptide ligand-induced partial T cell activation: Molecular mechanisms and role in T cell biology. AnnuRevImmunol. 1996;14:1–27. doi: 10.1146/annurev.immunol.14.1.1. [DOI] [PubMed] [Google Scholar]
- 44.Migita K, Eguchi K, Kawabe Y, Tsukada T, Ichinose Y, Nagataki S. Defective TCR-mediated signaling in anergic T cells. JImmunol. 1995;155:5083–5087. [PubMed] [Google Scholar]
- 45.Sloan-Lancaster J, Shaw AS, Rothbard JB, Allen PM. Partial T cell signaling: Altered phospho-zeta and lack of zap70 recruitment in APL-induced T cell anergy. Cell. 1994;79:913–922. doi: 10.1016/0092-8674(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 46.Smith JA, Tso JY, Clark MR, Cole MS, Bluestone JA. Nonmitogenic anti-CD3 monoclonal antibodies deliver a partial T cell receptor signal and induce clonal anergy. J Exp Med. 1997;185:1413–1422. doi: 10.1084/jem.185.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Utting O, Teh SJ, Teh HS. T cells expressing receptors of different affinity for antigen ligands reveal a unique role for p59fyn in T cell development and optimal stimulation of T cells by antigen. JImmunol. 1998;160:5410–5419. [PubMed] [Google Scholar]
- 48.Tang Q, Subudhi SK, Henriksen KJ, Long CG, Vives F, Bluestone JA. The Src family kinase Fyn mediates signals induced by TCR antagonists. JImmunol. 2002;168:4480–4487. doi: 10.4049/jimmunol.168.9.4480. [DOI] [PubMed] [Google Scholar]
- 49.Appleman LJ, Tzachanis D, Grader-Beck T, van Puijenbroek AA, Boussiotis VA. Helper T cell anergy: from biochemistry to cancer pathophysiology and therapeutics. JMolMed. 2001;78:673–683. doi: 10.1007/s001090000180. [DOI] [PubMed] [Google Scholar]
- 50.Boussiotis VA, Barber DL, Lee BJ, Gribben JG, Freeman GJ, Nadler LM. Differential association of protein tyrosine kinases with the T cell receptor is linked to the induction of anergy and its prevention by B7 family-mediated costimulation. JExpMed. 1996;184:365–376. doi: 10.1084/jem.184.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boussiotis VA, Freeman GJ, Berezovskaya A, Barber DL, Nadler LM. Maintenance of human T cell anergy: blocking of IL-2 gene transcription by activated Rap1. Science. 1997;278:124–128. doi: 10.1126/science.278.5335.124. [DOI] [PubMed] [Google Scholar]
- 52.Utting O, Priatel JJ, Teh SJ, Teh HS. p59fyn (Fyn) promotes the survival of anergic CD4-CD8- alpha beta TCR+ cells but negatively regulates their proliferative response to antigen stimulation. JImmunol. 2001;166:1540–1546. doi: 10.4049/jimmunol.166.3.1540. [DOI] [PubMed] [Google Scholar]
- 53.Gajewski TF, Fields P, Fitch FW. Induction of the increased Fyn kinase activity in anergic T helper type 1 clones requires calcium and protein synthesis and is sensitive to cyclosporin A. EurJImmunol. 1995;25:1836–1842. doi: 10.1002/eji.1830250707. [DOI] [PubMed] [Google Scholar]
- 54.Thien CB, Langdon WY. c-Cbl: a regulator of T cell receptor-mediated signalling. ImmunolCell Biol. 1998;76:473–482. doi: 10.1046/j.1440-1711.1998.00768.x. [DOI] [PubMed] [Google Scholar]
- 55.Deckert M, Elly C, Altman A, Liu YC. Coordinated regulation of the tyrosine phosphorylation of Cbl by Fyn and Syk tyrosine kinases. JBiolChem. 1998;273:8867–8874. doi: 10.1074/jbc.273.15.8867. [DOI] [PubMed] [Google Scholar]
- 56.Hunter S, Burton EA, Wu SC, Anderson SM. Fyn associates with Cbl and phosphorylates tyrosine 731 in Cbl, a binding site for phosphatidylinositol 3-kinase. JBiolChem. 1999;274:2097–2106. doi: 10.1074/jbc.274.4.2097. [DOI] [PubMed] [Google Scholar]
- 57.Tezuka T, Umemori H, Fusaki N, Yagi T, Takata M, Kurosaki T, Yamamoto T. Physical and functional association of the cbl protooncogen product with an src-family protein tyrosine kinase, p53/56lyn, in the B cell antigen receptor-mediated signaling. JExpMed. 1996;183:675–680. doi: 10.1084/jem.183.2.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murphy MA, Schnall RG, Venter DJ, Barnett L, Bertoncello I, Thien CB, Langdon WY, Bowtell DD. Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. MolCell Biol. 1998;18:4872–4882. doi: 10.1128/mcb.18.8.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]