

Abstract
Background
Smooth muscle cell (SMC) migration and proliferation are early and crucial events in the pathogenesis of intimal hyperplasia, the primary cause of restenosis following vascular intervention. We tested the hypothesis that Protein Kinase C–Delta (PKCδ ), a ubiquitously expressed intracellular protein kinase, regulates vascular SMC proliferation and migration.
Methods
Exogenous PKCδ was expressed in cultured SMCs via stable transfection or adenovirus-mediated gene transfer. Conversely, endogenous PKCδ was inhibited by means of targeted gene deletion (gene knock-out). Cell proliferation and migration were determined by 3H-thymidine incorporation and 24-well transwell assay, respectively.
Results
We isolated and examined three A10 SMC lines in which PKCδ were stably transfected. Compared to cells that were transfected with an empty vector, cells transfected with PKCδ exhibited reduced ability to proliferate. Moreover, PKCδ transfection inhibited SMC migration toward PDGF-BB. Similar inhibitory effects on proliferation and migration were also observed when PKCδ were introduced into primary aortic SMCs via an adenoviral vector. Interestingly, SMCs isolated from PKCδ knockout mice also displayed decreased chemotaxis and proliferation compared to PKCδ +/+ littermates, suggesting a complex yet critical role for PKCδ. We studied the MAP kinase ERK1/2 as a possible signaling pathway for PKCδ ’s inhibitory effect. PKCδ overexpression diminished ERK1/2 activity. Molecular restoration of ERK activation reversed the inhibitory effect of PKCδ on SMC proliferation and migration.
Conclusions
In summary, we demonstrate that while normal migration and proliferation is lessened in SMCs deficient in PKCδ, its prolonged activation also diminishes those behaviors. This suggests a dual, critical role for PKCδ in SMC proliferation and migration, and thus intimal hyperplasia and restenosis.
Clinical Relevance
Restenotic or hyperplastic lesions are typified by dedifferentiated vascular smooth muscle cells (VSMC) that demonstrate excessive proliferation and migration. We have previously demonstrated that PKCδ is a critical upstream factor leading to VSMC apoptosis. The present study show that PKCδ , when ectopically expressed in VSMCs, also inhibits proliferation and migration as well as ERK MAP kinase. Thus, gene transfer of PKCδ is a potential molecular therapeutic strategy to inhibit intimal hyperplasia.
INTRODUCTION
The intima of a normal human artery is almost entirely free of SMCs. Subsequent to the trauma and altered hemodynamics of vascular reconstruction, however, SMCs of the media are stimulated to migrate into the subintimal space. The proliferation of these cells within this layer contributes to the phenomenon of neointimal hyperplasia, the major obstacle to the long-term success of vascular interventions for treatment of atherosclerotic disease. The migration of SMCs into and their proliferation within the subintima is thus central to the pathogenesis of this costly, morbid and unsolved dilemma.
Members of the Protein Kinase C (PKC) family are activated by diverse stimuli as mediators of multiple processes such as cellular growth, differentiation, and apoptosis. In previous studies, we identified several PKC isotypes expressed by SMCs, including α , β I, β II, δ , ε , ζ , and λ .1 The novel PKC isoform Protein Kinase C - Delta (PKCδ ) is the only one of these that shown to be associated with the cytoskeleton,2 implying a possible role in migration. It has been demonstrated that PKCδ inhibits growth in many cell types including fibroblasts,3 capillary endothelial cells,4 and airway SMCs.5 The evidence from these investigations suggests that it PKCδ does so by halting the cell cycle at various points depending on the cell type. However, the detailed molecular mechanisms through which PKCδ influences cell cycle progression remain unclear. Similarly, little is known about how PKCδ regulates SMC migration.
The mitogen-activated protein kinases (MAPK) are mediators of nuclear and cytoplasmic responses to extracellular stimuli. They phosphorylate cytoplasmic substrates as well as activate specific genes by modulating a variety of transcription factors. The MAPK family consists of three major subfamilies with multiple members: the extracellular signal-regulated kinases (ERK), c-jun N-terminal kinases (JNK), and p38 MAP-kinases (p38). Each MAPK is activated in response to diverse extracellular stimuli by phosphorylation within a conserved Thr-X-Tyr motif in its activation loop. Ample in vitro studies using cultured vascular endothelial and SMCs as well as in vivo experiments with injured arteries link MAPK activation to cell proliferation, migration, and apoptosis.6–8 Specifically, the critical role of ERK1/2 in vascular SMC proliferation and intimal hyperplasia has been well established.9, 10
In this report, we examine the role of PKCδ in vascular SMC behavior by testing a hypothesis that high PKCδ activity inhibits both SMC proliferation and migration. We also elucidate the role of MAPK ERK1/2 as a downstream mediator following PKCδ activation. In total, these studies provide an explicit link between PKCδ and ERK1/2 in the process of SMC migration and proliferation. These findings have potential for improvement in approaches for the prevention and management of restenosis.
METHODS
General Materials
PDGF-BB was obtained from R & D Systems (Minneapolis, MN). Dimethyl sulfoxide (DMSO) along with other chemicals if not specified, was purchased from Sigma Chemical Co. Dulbecco’s Modified Eagles Medium (DMEM) and cell culture reagents were from Gibco BRL Life Technologies (Gaithesberg, MD). Mouse monoclonal anti-phospho-MBP was purchased from Upstate Biotechnologies Inc. (Lake Placid, NY). Rabbit polyclonal antibody to ERK1/2 and phospho-ERK1/2 were obtained from Cell Signal (Beverley, MA). Rabbit polyclonal anti-PKCδ was obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA).
SMC Culture
Rat aortic A10 SMCs, obtained from American Tissue Culture Collection, were grown as recommended, in DMEM modified to contain 4 mM L-glutamine, 4.5 g/L glucose, 1mM sodium pyruvate, and 1.5 g/L sodium bicarbonate supplemented with 10% FBS and antibiotics. The generation of PKCδ target deletion was described elsewhere.11 Both rat and mouse aortic SMCs were isolated from the thoracic aorta based on a protocol described by Clowes et al12 and maintained in DMEM containing 10% fetal FBS at 37°C with 5% CO2.
Immunoblotting
Protein extracts were resolved by electrophoresis as described previously.13 Equal amount of protein extracts were separated by SDS-PAGE and transferred to a polyvinylliden difluoride membrane and blotted with antibodies. Labeled proteins are visualized with an ECL system (Amersham Biosciences, Piscataway, NJ).
Proliferation Assay
Proliferation was assayed by evaluating DNA synthesis and counting cell numbers. In DNA synthesis assay, cells were seeded onto 24-well plates at the concentration of 10,000 cells/well in 10% FBS-DMEM and allowed to attach overnight. SMCs were then starved in 0.5 % FBS-DMEM for 48 h and then stimulated for 24 h with agonists as indicated. During the final 4 h of the assay, 2 μCi of [methyl 3H]-thymidine was added to each well and co-incubated. Protein was precipitated with 10 % trichloroacetic acid and radioactivity of incorporated 3H-thymidine was determined by using a liquid scintillation counter. For cell counting, cells were seeded onto 24 well plates at a density of 10,000 cells/well in 10 % FBS-DMEM (day 0) and medium was replaced every other day.1 Cell numbers were determined by hemacytometer on the indicated days.
Chemotaxis Assay
Chemotaxis was determined using a 24-well transwell (Poretics Corp, Livermore, CA) containing polycarbonate 8 μm-pore membrane filters. Cells, harvested using 0.05% trypsin/EDTA, were seed in upper wells in 0.5% FBS media at a density of 10,000 (A10) or 40,000 (rat and mouse aortic) cells/well. PDGF-BB, diluted in 0.5% FBS media to 5 ng/ml was applied into the lower wells. Cells were then incubated in the CO2 incubator 37°C for 4 (A10) or 6 h (rat and mouse aortic). On completion of the assay, membranes were removed, fixed in 70% ethanol at −20°C for 30 min, and stained with hematoxylin at room temperature for 30 min. The upper side of the membrane was scraped using a cotton swab to remove cells that had attached but not migrated, and the membrane was then mounted onto a microscope slide. Chemotaxis in each well was assessed by counting the number of cells in five independent high-power fields (200x magnification).
Construction of adenoviral vectors and infection
A recombinant adenoviral vector was constructed to express PKCδ . Briefly, a DNA fragment containing the desired sequence was generated by PCR using the human cDNA as a template. Following DNA sequencing, the PCR product was then cloned into an E1 and E3 deficient adenoviral vector (pEasy). Adenoviruses were propagated in HEK 293 cells and purified by CsCl density gradient centrifugation. SMCs were infected with adenoviruses as described previously.13
MAP kinase assay
The ERK activity was measured using a MAPK IP-kinase assay kit from Upstate Biotechnology. Cells were lysed with buffer containing 1% Nonidet P-40. Lysates were immunoprecipitated with an anti-ERK1/2 antibody immobilized with agarose. After four rounds of vigorous washing, the immunoprecipitated complexes were subjected to a phosphorylation assay using myelin basic protein as a substrate, which was analyzed by immunoblotting using an antibody specific for phosphorylated myelin basic protein.
Statistical analysis
Values were expressed as mean ± standard error. Unpaired Student’s t test was used to evaluate the statistical differences between control and treated groups. Values of p<0.05 were considered significant. All experiments were performed with n ≥ 3 and repeated at least three times.
RESULTS
PKCδ overexpressing vascular SMCs demonstrate decreased proliferation and migration
To evaluate the role of PKCδ in SMC proliferation and migration, two processes critical to the pathophysiology of intimal hyperplasia, we established PKCδ overexpressing SMC lines by stably transfecting A10 cells with a full-length human PKCδ cDNA inserted into the pTracerSV40 vector. The empty pTracerSV40 vector was used as a control. Transfected cells were selected for their resistance to the antibiotic Ziocine and the presence of pTracerSV40 vector sequences. Using such approach, we isolated three clones (PKCδ 3, 4 and 5) that were successfully transfected with PKCδ pTracerSV40 and two clones (PT3 and 4) transfected with the empty vector. The amount of PKCδ protein expressed in these clones was evaluated by immunoblotting with a PKCδ specific antibody. The level of PKCδ was significantly increased in PKCδ -transfected cells (PKCδ 3, 4, and 5) compared to wild-type A10 or empty vector PT3 and 4 cells (Figure 1A). Expression of other PKC isotypes was not affected by the PKCδ transfection (data not shown).
Figure 1. PKCδ overexpressing A10 SMC lines demonstrate decreased proliferation and chemotaxis.
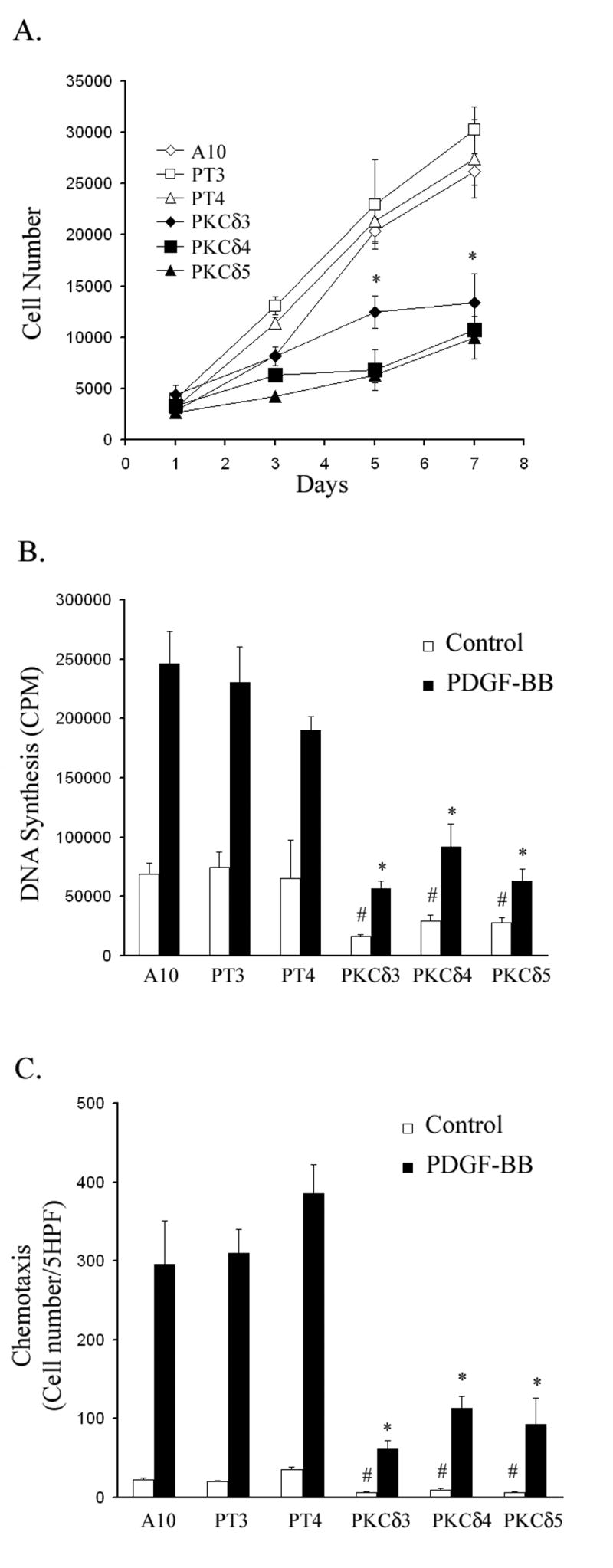
(A) A10, empty vector (PT3, PT4) or PKCδ (PKCδ 3, 4 & 5) cells were seeded into 24-well plates at a density of 10,000 cells per well on day 0 and maintained in 10% FBS. Cells were counted on days 1, 3, 5 and 7. (n=3, * p<0.05, compared to control A10 SMC line) (B) A10, empty vector or PKCδ cells were serum -starved for 48 h and then stimulated for 24 h with or without PDGF-BB (5 ng/ml). Incorporation of 3H-thymidine was measured. (C) A10, empty vector or PKCδ cells were made quiescent and then subjected to the chemotaxis assay as described in Experimental Procedures. (n=3, # p< 0.01, compared to A10 basal; * p< 0.01, compared to A10 treated with PDGF-BB).
Because of the important contribution of SMC proliferation to intimal hyperplasia, we evaluated the ability of PKCδ to influence proliferation of A10 SMCs. All cells were seeded on day 0 at the same density and were allowed to grow in a medium containing 10% FBS to days 1, 3, 5 and 7. As shown in Figure 1A, the growth rate of PKCδ overexpressing cells was markedly reduced compared to that of wild type or empty vector. At day 7, the number of PKCδ cells was less than 50% of the control cells. To confirm the inhibitory effect of PKCδ on SMC growth, we evaluated the effect of PKCδ on DNA synthesis using a 3H-thymidine incorporation assay. Wild-type A10 cells, empty vector clones (PT3 & 4) and PKCδ clones (PKCδ 3, 4 & 5) were made quiescent by incubation in 0.5% serum, then treated with PDGF-BB (5 ng/ml) or control solvent for 24 h. Compared to wild type A10 SMCs and empty vector controls, all cell lines overexpressing PKCδ demonstrated significantly reduced basal and PDGF-stimulated DNA synthesis (Figure 1B).
Since the migration of SMCs from the media to the intima is also essential for neotintima formation, we continued by evaluating SMC migration in our PKCδ overexpressing cell lines. Using a modified Boyden chamber assay, wild type A10 cells, empty vector clones (PT3 & 4) and PKCδ clones (PKCδ 3, 4 & 5) were seeded into the upper well of a microchemotaxis chamber (10,000 cells/well) and migration through an 8 μ m porous membrane was measured. PDGF-BB (5 ng/ml) was added to the lower well as the chemotactic stimulus. As shown in Figure 1C, all cell lines overexpressing PKCδ demonstrated significantly reduced basal and PDGF-stimulated chemotaxis when compared to empty vector controls (Figure 1C).
A10 SMCs are of embryonic origin and their behavior has been found to resemble SMCs derived from hyperplastic lesions rather than normal medial SMCs,14 therefore, we felt that it was necessary to confirm the effects of PKCδ on proliferation and migration in arterial SMCs isolated from adult animals. To this end, we employed the adenovirus-mediated gene transfer technique to overexpress PKCδ in SMCs isolated from the aorta of adult rats (RASMCs). We constructed a recombinant adenoviral vector to express the wild-type PKCδ (AdPKCδ ) from a replication-deficient adenovirus as described in the methods section. To confirm the expression of adenovirus-mediated expression of PKCδ , RASMCs were infected with AdPKCδ or an empty viral vector (AdNull) at a dosage of 30,000 particles per cell. Cell lysates were resolved by SDS-PAGE and immunoblotted with anti-PKCδ or anti-β -actin antibodies. As a result of transgene expression, higher levels of PKCδ were found in AdPKCδ -infected cells (Figure 2A).
Figure 2. PKCδ overexpression in RASMCs, via adenoviral transfection, inhibits proliferation and migration.
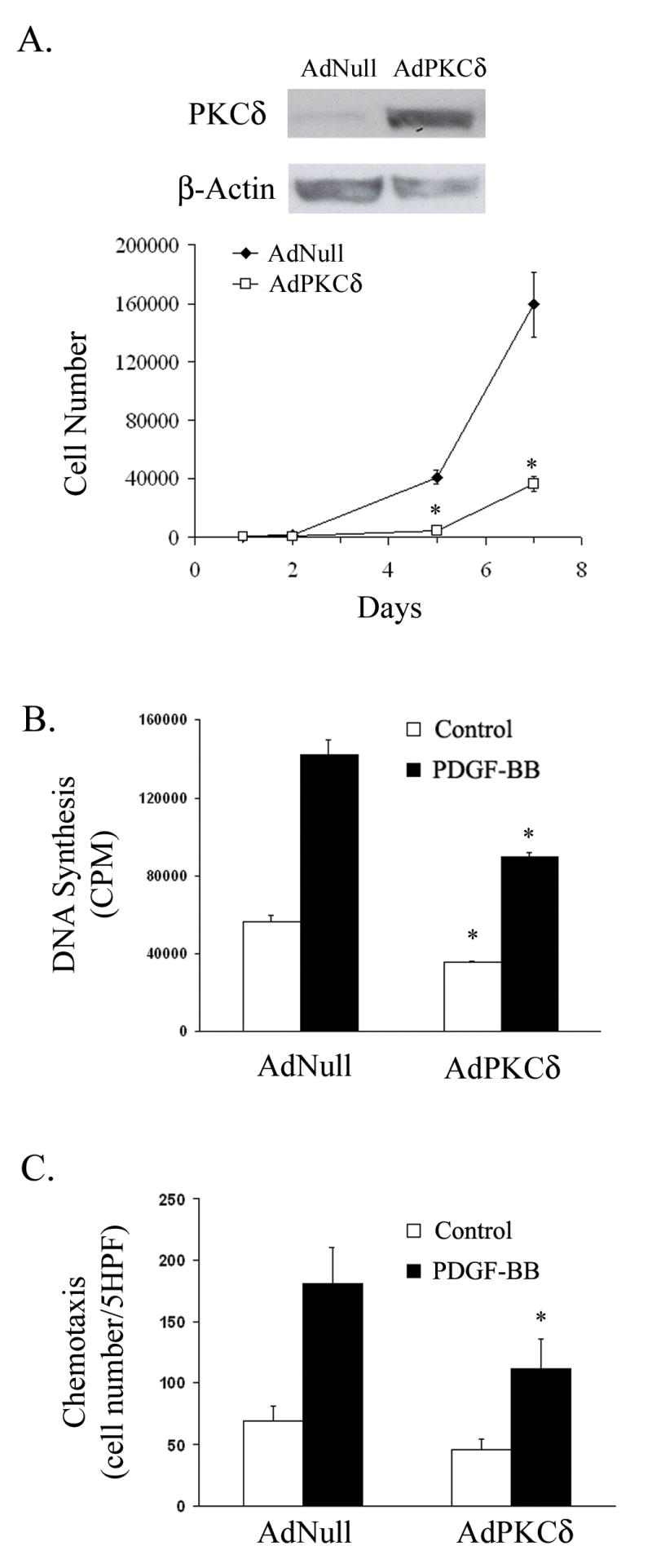
(A) RASMCs were infected with AdNull or AdPKCδ. Forty-eight hours after infection, cells were lysed and analyzed by immunoblotting. Additional infected cells were re-seeded into 24-well plates at a density of 10,000 cells per well on day 0 and maintained in 10% FBS. Cells were counted on days 1, 2, 5 or 7. (B) Following infection with AdNull or AdPKCδ, RASMCs were serum-starved for 48 h. Basal and PDGF-BB induced DNA synthesis was measured using the 3H-thymidine incorporation. (C) RASMCs were infected with AdNull or AdPKCδ. Forty-eight hours after infection, basal and PDGF-BB (5 ng/ml) induced chemotaxis was evaluated. (n=3, *p< 0.05, compared to AdNull control).
To assess cell proliferation, RASMCs were seeded at low density following infection with AdNull or AdPKCδ and allowed to grow in the presence of 10% serum. The number of cells was counted at day 1, 2, 5 and 7 after seeding. AdPKCδ significantly inhibited cell growth (Figure 2A). Similarly, basal and PDGF-stimulated DNA synthesis was also markedly suppressed by ectopic expression of PKCδ (Figure 2B). Taken together, these data indicate that PKCδ plays a similar inhibitory role in cell proliferation of adult arterial SMCs. Next, we examined the effect of adenoviral-mediated overexpression of PKCδ on migration of RASMCs. Forty-eight hours after viral infection, RASMCs were subjected to the chemotaxis assay in which the number of cells that migrated through a porous membrane toward PDGF-BB (5ng/ml) was measured. AdPKCδ significantly reduced the number of migrated RASMCs (Figure 2C). These data further supports PKCδ ’s inhibitory effect on vascular SMC migration.
Selective PKCδ gene deletion also decreases vascular SMC proliferation and migration
To further elucidate the effect of PKCδ on vascular SMC migration and proliferation, we employed PKCδ “knock-out” SMCs11 as an experimental approach to inhibit endogenous PKCδ activity. We first confirmed the absence of PKCδ in SMCs harvested from PKCδ −/− mice using Western blot analysis (Figure 3A). We next seeded SMCs, isolated from PKCδ null and wildtype littermates, at low density and allowed them to grow in the presence of 10% serum. The number of cells was counted at day 1, 2, 4 and 8 after seeding. PKCδ gene deletion significantly inhibited cell growth (Figure 3A). We then proceeded to evaluate the effect of PKCδ gene deletion on DNA synthesis using a 3H-thymidine incorporation assay. Vascular SMCs from both PKCδ +/+ and −/− mice were made quiescent by incubation in 0.5% serum, then treated with PDGF-BB (5 ng/ml) for 24 h. Compared to wild type SMCs, PKCδ null SMCs demonstrated both decreased basal and PDGF-stimulated DNA synthesis (Figure 3B).
Figure 3. PKCδ selective gene deletion downregulates SMC proliferation and migration.
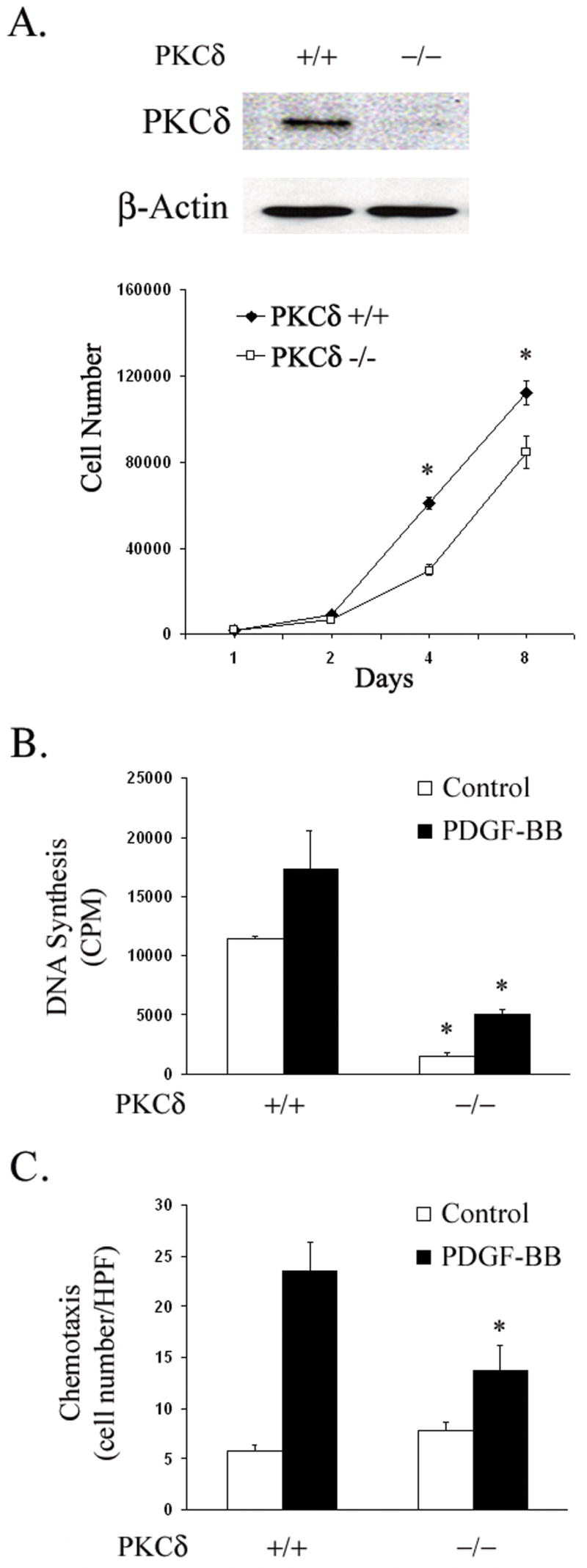
(A) Mouse SMCs were isolated from PKCδ −/− and +/+ littermates. SMCs from both groups were lysed and analyzed by immunoblotting. Additional cells were seeded into 24-well plates at a density of 10,000 cells/well on day 0 and maintained in 10% FBS. Cells were counted on days 1, 2, 4 and 8. (n=3, * p<0.05, compared to PKCδ +/+ control) (B) PKCδ −/− and +/+ SMCs were serum-starved for 48 h and then stimulated for 24 h with or without PDGF-BB (5 ng/ml). The incorporation of 3H-thymidine was measured. (B) PKCδ −/− and +/+ SMCs were serum-starved for 48h. Chemotaxis with and without PDGF-BB (5 ng/ml) was evaluated as described in Experimental Procedures. (n=3, *p< 0.05, compared to PKCδ +/+ control).
Next, we examined the effect of PKCδ gene deletion on migration of mouse aortic SMCs as in previous experiments. Without a chemotactic stimulus both PKCδ null and wild type cells demonstrated low rates of chemotaxis. However, in response to PDGF-BB (5 ng/ml) PKCδ −/− SMCs demonstrated significantly decreased chemotaxis (Figure 3C).
PKCδ overexpression inhibits ERK1/2 activity
To elucidate the mechanism by which ectopic expression of PKCδ inhibits SMC proliferation and migration, we utilized our stably transfected A10 SMCs and examined the MAPK ERK1/2, previously demonstrated to be essential for both SMC migration and proliferation.9, 15 Control A10 SMCs exhibited a detectable basal level of ERK1/2 activity as measured by phosphorylation of its substrate MBP. Stimulation with PDGF-BB (5 ng/ml for 15 min) greatly enhanced ERK1/2 activity as anticipated (Figure 4A). Both basal and PDGF-stimulated ERK1/2 activity was significantly diminished in PKCδ overexpressing cell lines cells with compatible amounts of ERK1/2 found in all clones (Figure 4A), suggesting that PKCδ had no effect on protein expression of ERK1/2 and the low activity was due to a downregulation of MAPK activity in PKCδ cells. We next tested whether manipulation of PKCδ in RASMCs would affect ERK1/2 activation. Forty-eight hours following adenoviral infection, cells were stimulated with PDGF-BB (5 ng/ml for 15 min) and ERK1/2 was immunoprecipitated from cell lysates. ERK1/2 activity in the immunocomplex was again measured by its ability to phosphorylate its substrate MBP. As in our prior experiment, PDGF-BB markedly increased ERK1/2 activity (Figure 4B). This PDGF-stimulated ERK1/2 activation was significantly diminished by AdPKCδ . These results are consistent with our previous findings in stably tranfected A10 cells. To confirm these findings, we next evaluated the phosphorylation of ERK1/2 as an additional assay of MAPK activity. We found that compared to RASMCs infected with the empty viral vector, those infected with AdPKCδ demonstrated significantly decreased amounts of PDGF-induced ERK1/2 phosphorylation (Figure 4C). Lastly, we evaluated basal and PDGF-BB stimulated ERK1/2 activity in PKCδ null SMCs. Compared to SMCs from their wild-type littermates, PKCδ gene deletion led to decreased PDGF-stimulated ERK1/2 activity (Figure 4D). These results mirror the data acquired from our previous migration and proliferation studies.
Figure 4. PKCδ overexpression inhibits ERK1/2 activity.
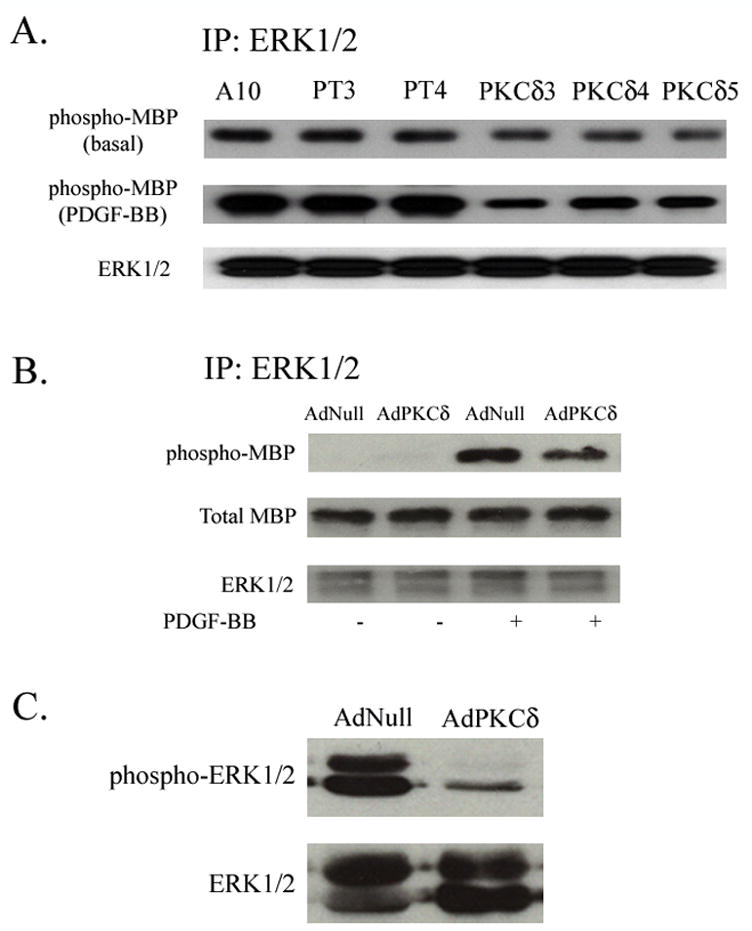
(A) A10, PKCδ or vector transfected cells were made quiescent by incubation in the low-serum media for 48 h. Cells were stimulated with PDGF-BB (5 ng/ml) for 15 min. Cell lysates were immuno-precipitated with an anti-ERK1/2 antibody. The activity of ERK in the IP complex was measured by its ability to phosphorylate MBP. (B) RASMCs were infected with AdNull or AdPKCδ. Following serum starvation for 48 h and PDGF-BB treatment (5 ng/ml for 15 min) were indicated, cell lysates were immunoprecipitated with an anti-ERK1/2 antibody and ERK activity in the IP complex was measured by its ability to phosphorylate MBP. (C) RASMCs were infected with AdNull or AdPKCδ. Following serum starvation and PDGF-BB treatment (5 ng/ml for 15 min), cell lysates underwent Western blot analysis with anti-ERK1/2 antibodies. (D) Mouse SMCs from PKCδ −/− and +/+ littermates underwent low-serum incubation and stimulation with PDGF-BB (5 ng/ml) for 15 min. Cell lysates were immunoprecipitated with an anti-ERK1/2 antibody and ERK activity in the IP complex was measured by MBP phosphorylation.
Overexpression of constitutively active MEK rescues PKCδ ’s inhibition of proliferation and migration
To provide further evidence that ectopic expression of PKCδ influences SMC behavior through its down regulation of ERK1/2, we restored ERK1/2 activity in SMCs overexpressing PKCδ by adenoviral transfection of a constitutively active MEK mutant (AdMEK), an approach we have previously used to selectively activate ERK1/2 in SMCs.13 A10 SMCs were infected with equal quantities of AdNull, AdPKCδ , or AdMEK (60,000 total viral particles per cell). Following low serum incubation, A10 cells, with or without PDGF-BB (5 ng/ml), underwent measurement of 3H-thymidine incorporation. As shown in Figure 5A, A10 SMCs receiving AdNull alone demonstrated low levels of DNA synthesis under basal conditions, but responded dramatically to PDGF stimulation. A10 cells receiving PKCδ in combination with AdNull responded with a blunted response to the mitogen, while PKCδ overexpressing cells that were transfected with AdMEK returned to normal levels of both basal and PDGF-stimulated 3H-thymidine incorporation (Figure 5A).
Figure 5. PKCδ’s inhibition of migration and proliferation can be overcome by restoring ERK1/2 activity.
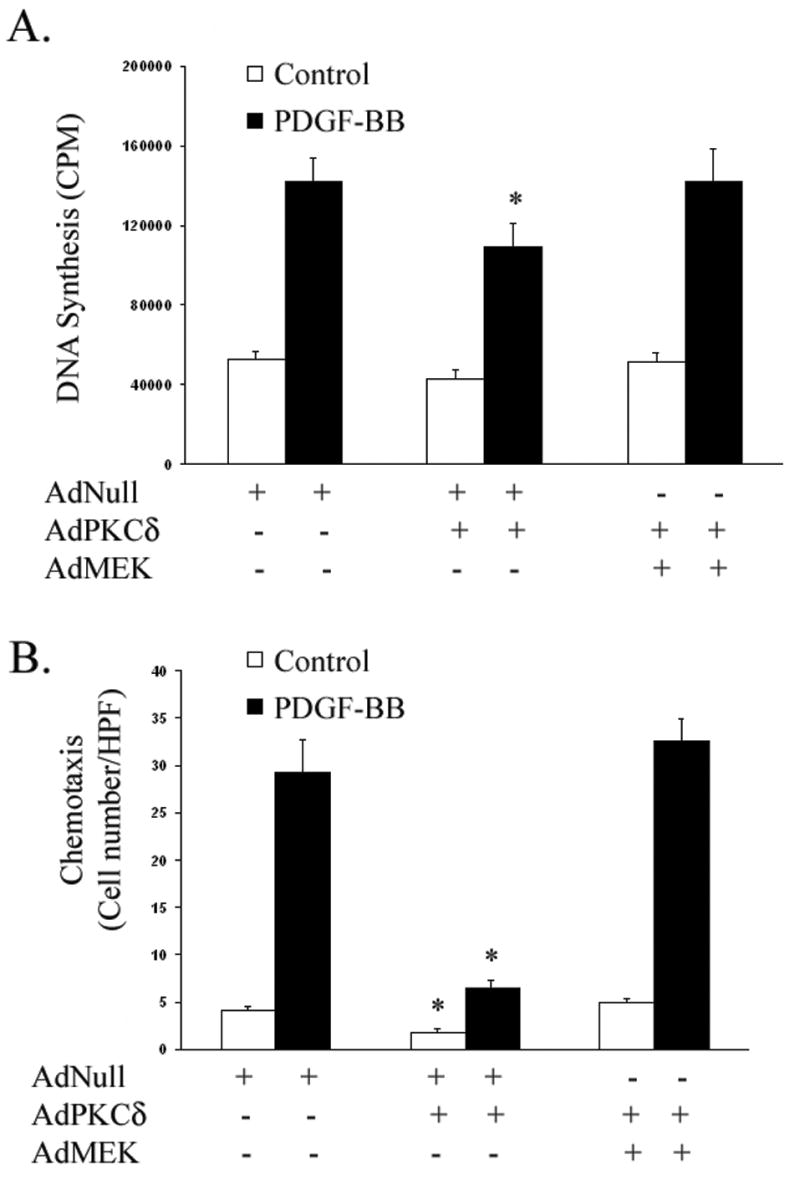
(A) Following low-serum incubation for 48 h, incorporation of 3H-thymidine was measured in A10 SMCs infected with equal quantities of AdNull, AdPKCδ, or AdMEK (60,000 total viral particles per cell) and stimulated for 24 h with or without PDGF-BB (5 ng/ml). (B) AdNull, AdPKCδ, or AdMEK infected A10 SMCs were serum-starved for 48 h. Chemotaxis with and without PDGF-BB (5 ng/ml) was evaluated. [n=3; *, p < 0.05 as compared to AdNull control]
Next, we evaluated A10 SMC migration under similar conditions. A10 cells were again infected with equal quantities of AdNull, AdPKCδ , or AdMEK (60,000 total viral particles per cell). Following low serum incubation, A10 cells underwent a transwell chemotaxis assay with and without PDGF-BB stimulation. As shown in Figure 6B, SMCs overexpressing PKCδ alone demonstrated significantly reduced basal and PDGF-stimulated chemotaxis, while A10 SMCs infected with both AdPKCδ and AdMEK exhibited migration similar to those infected with empty vector controls (Figure 5B).
DISCUSSION
We have demonstrated, using both genetic and molecular manipulation, that PKCδ is critical to proliferation and migration in vascular SMCs. PKCδ overexpression in both A10 and rat aortic SMCs, accomplished via stable transfection and adenoviral gene transfer, respectively, resulted in a suppression of proliferation and chemotaxis. We also provide evidence that PKCδ overexpression selectively suppresses activation of the ERK1/2 subclass of MAP kinases. This suppression may underlie the effect of PKCδ overexpression on proliferation and migration.
The role of PKCδ in cell proliferation has been studied in several cell types, including vascular SMCs. With the exception of certain transformed and cancerous cells, PKCδ was found to play a generally inhibitory role in proliferation.16 This ability of PKCδ to negatively or positively regulate proliferation may depend on its phosphorylation. As shown in NIH 3T3 cells, a PKCδ tyrosine mutant (Tyr155 → Phe) stimulates cell proliferation, while the wild-type PKCδ inhibits it.17 There are 19 tyrosine residues within the PKCδ molecule and at least 9 have been implicated as sites of phosphorylation. It is not clear, however, how PKCδ phosphorylation is related to its inhibition of vascular SMC proliferation. Additional studies using site-specific mutations are needed to clarify the effect of PKCδ phosphorylation on its regulation of SMC proliferation.
The observed reduction in cell number and DNA synthesis (~40%) could also be influenced by changes in cell apoptosis or necrosis. Indeed, we have previously found inhibition of PKCδ caused SMCs to become resistant to apoptotic stimuli.18 Conversely, overexpression of the wild-type PKCδ induced a small increase (~25%) in cell death in the absence of exogenous stimuli, such as PMA.18 Thus, both PKCδ -mediated cell apoptosis and growth inhibition could contribute to the reduction in cell number and DNA synthesis of PKCδ overexpression cells.
The importance of PKCδ to normal cell migration is only recently receiving increased attention, despite the PKC isotype having been shown over ten years ago to be intimately related to the cytoskeleton.2 In previous experiments, we found that phorbol ester-induced general activation of PKC in human saphenous vein SMCs resulted in a significant reduction in PDGF-stimulated chemotaxis. PKCδ is one of the four PKC isotypes subsequently identified as potential mediators of this effect.1
In this study, we showed that overexpression of exogenous PKCδ in vascular SMCs led to inhibition of PDGF-stimulated chemotaxis. This result, along with a recent report by Grahm et al19 in which prolonged PKCδ activation inhibits endothelial chemotaxis, suggests that the role of PKCδ in cell migration is inhibitory.
The decreased migration and proliferation seen in SMCs from PKCδ -deficient mice, however, suggests that a brief increase in PKCδ activation may be necessary for both processes. Similarly, Li et al reported diminished migration of similar SMCs when stimulated with mechanical stress.20 This dual role of PKCδ emphasizes the complexity of SMC chemotaxis and proliferation.
To elucidate how PKCδ overexpression affects proliferation and migration, two distinct cellular processes, we turned attention to the MAPK ERK1/2 pathway, an established mediator of both proliferation and migration in vascular SMCs. We showed in both A10 and adult RASMCs that ERK1/2 activation by PDGF-BB is greatly impaired by exogenous PKCδ expression. Given the critical role of ERK1/2 in proliferation and migration, it is plausible that prolonged activation of PKCδ inhibits SMC proliferation and migration by suppressing the ERK1/2 cascade. Protein Kinase C is a recognized stimulant of ERK1/2; specifically, several isotypes of PKC (α , β 1 and ε ) have been found to both activate ERK1/2 as well as stimulate proliferation in vascular SMCs.21, 22, 23Thus, our findings with PKCδ are novel and suggest an additional role of PKCδ , divergent from that of the other PKC isotypes. Moreover, we previously reported PKCδ is physically associated with another MAPK, p38, in SMCs.18 Protein sequence analysis, however, failed to disclose any consensus motifs within ERK1/2 that could serve as phosphorylation sites for PKCδ. This suggests that PKCδ may affect ERK1/2 indirectly.
This study illuminates one of several possible signaling pathways through which PKCδ induces growth arrest. Indeed, it is likely that PKCδ halts cell growth through multiple routes. We have previously shown that PKCδ upregulates p53 in vascular SMCs,18 suggesting another mechanism by which PKCδ could regulate the cell cycle and inhibit proliferation. This course has been studied in human breast cancer24 and lung adenocarcinoma25 cells. These studies show that PKCδ induces p21, a downstream target of p53, resulting in G1 phase arrest. p21 upregulation, however, occurs independently of p53 in many cell types;26, 27 additionally, conflicting reports exist regarding PKCδ ’s ability to upregulate p21 in vascular SMCs.28, 29
Similar to its dual effects on cell proliferation and migration, PKCδ may be both inhibitory and stimulatory to the ERK1/2 pathway. Recently, Ginnan and Singer showed, also in SMCS, that inhibition of PKCδ via a dominant negative mutant blocked activation of ERK1/2 in response to PDGF-BB, suggesting that PKCδ is necessary for the activation of this MAP kinase.30 Whether the diminished ERK1/2 activity underlies the reduction in proliferation and migration that we observed in PKCδ null cells remains to be investigated.
Although our current knowledge of PKCδ does not provide us with an explanation of how a single protein kinase can be both stimulatory and inhibitory to similar cellular functions, PKCδ would not be alone in possessing this property. The Rho family of regulatory GTPases also exhibit dual effects on SMC behavior. Activation of the Rho/Rho kinase pathway is necessary for stimulated proliferation and migration.31 If that activation is prolonged, however, migration is inhibited while proliferation remains increased.32 Interestingly, evidence supporting a relationship between PKCδ and Rho exists.20, 33
Our in vivo findings suggest that diminished PKCδ may in part contribute to the accumulation of SMCs in the neointima following arterial injury. Indeed, PKCδ -null mice, despite their normal and fertile appearance, showed increased B cell proliferation and autoimmunity and exacerbated lesion formation in a vein-graft vascular injury model.11, 34 In the vein graft study, intimal hyperplasia was found to be 50% greater in grafts derived from PKCδ -null mice than in wild-type grafts. At 8 weeks post grafting, there was an ~80% reduction in apoptosis within the PKCδ -null graft; surprisingly, no significant change in proliferation was observed. Whether PKCδ deletion affects SMC proliferation after vascular injury remains unclear, as proliferation peaks at an earlier time point than those evaluated.
In summary, we have demonstrated, through both genetic and molecular manipulation that prolonged PKCδ can be both inhibitory and stimulatory in its regulation of proliferation and migration in vascular SMCs. Furthermore, activation of the MAP kinase ERK1/2 is downregulated by overexpression of PKCδ , which could, at least in part, mediate PKCδ ’s inhibitory effect. Since it is known that PKCδ is activated by mitogenic stimuli such as PDGF-BB, PKCδ activation could serve as a negative feedback mechanism to keep SMC proliferation and migration under control. A diminution of this feedback pathway, such as that observed in injured arteries, could contribute to undesired SMC proliferation and migration, which in turn leads to the formation of intimal hyperplasia.
Acknowledgments
We thank Chunjie Wang and Sophia Chu for their technical assistance. We also thank Dr. N. Heckatte at The Gene Therapy Core Facility, Weill Cornell Medical College for assistance with adenovirus preparation.
Footnotes
This work was supported in part by a NHLBI, National Institutes of Health Grant HL-68673 (to K. C. K. and B. L.), American Heart Association Heritage Foundation Grant-in-aid 0455859T (to B. L.), and National Institutes of Health Training Grant T32 CA68971-07 (to E. J. R.).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Itoh H, Yamamura S, Ware JA, Zhuang S, Mii S, Liu B, et al. Differential effects of protein kinase C on human vascular smooth muscle cell proliferation and migration. Am J Physiol Heart Circ Physiol. 2001;281:H359–370. doi: 10.1152/ajpheart.2001.281.1.H359. [DOI] [PubMed] [Google Scholar]
- 2.Haller H, Lindschau C, Quass P, Distler A, Luft FC. Differentiation of Vascular Smooth Muscle Cells and the Regulation of Protein Kinase C-{alpha} Circ Res. 1995;76:21–29. doi: 10.1161/01.res.76.1.21. [DOI] [PubMed] [Google Scholar]
- 3.Acs P, Wang QJ, Bogi K, Marquez AM, Lorenzo PS, Biro T, et al. Both the Catalytic and Regulatory Domains of Protein Kinase C Chimeras Modulate the Proliferative Properties of NIH 3T3 Cells. J Biol Chem. 1997;272:28793–28799. doi: 10.1074/jbc.272.45.28793. [DOI] [PubMed] [Google Scholar]
- 4.Harrington EO, Loffler J, Nelson PR, Kent KC, Simons M, Ware JA. Enhancement of Migration by Protein Kinase Calpha and Inhibition of Proliferation and Cell Cycle Progression by Protein Kinase Cdelta in Capillary Endothelial Cells. J Biol Chem. 1997;272:7390–7397. doi: 10.1074/jbc.272.11.7390. [DOI] [PubMed] [Google Scholar]
- 5.Page K, Li J, Zhou L, Iasvoyskaia S, Corbit KC, Soh J-W, et al. Regulation of Airway Epithelial Cell NF-{kappa}B-Dependent Gene Expression by Protein Kinase C{delta} J Immunol. 2003;170:5681–5689. doi: 10.4049/jimmunol.170.11.5681. [DOI] [PubMed] [Google Scholar]
- 6.Bornfeldt KE, Campbell JS, Koyama H, Argast GM, Leslie CC, Raines EW, et al. The Mitogen-activated Protein Kinase Pathway Can Mediate Growth Inhibition and Proliferation in Smooth Muscle Cells. Dependence on the Availability of Downstream Targets. J Clin Invest. 1997;100:875–885. doi: 10.1172/JCI119603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pintucci G, Moscatelli D, Saponara F, Biernacki PR, Baumann FG, Bizekis C, et al. Lack of ERK activation and cell migration in FGF-2-deficient endothelial cells. FASEB J. 2002;16:598–600. doi: 10.1096/fj.01-0815fje. [DOI] [PubMed] [Google Scholar]
- 8.Gennaro G, Menard C, Michaud S-E, Deblois D, Rivard A. Inhibition of Vascular Smooth Muscle Cell Proliferation and Neointimal Formation in Injured Arteries by a Novel, Oral Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase Inhibitor. Circulation. 2004;110:3367–3371. doi: 10.1161/01.CIR.0000147773.86866.CD. [DOI] [PubMed] [Google Scholar]
- 9.Mii S, Khalil RA, Morgan KG, Ware JA, Kent KC. Mitogen-activated protein kinase and proliferation of human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 1996;270:H142–150. doi: 10.1152/ajpheart.1996.270.1.H142. [DOI] [PubMed] [Google Scholar]
- 10.Hu Y, Cheng L, Hochleitner B-W, Xu Q. Activation of Mitogen-Activated Protein Kinases (ERK/JNK) and AP-1 Transcription Factor in Rat Carotid Arteries After Balloon Injury. Arterioscler Thromb Vasc Biol. 1997;17:2808–2816. doi: 10.1161/01.atv.17.11.2808. [DOI] [PubMed] [Google Scholar]
- 11.Miyamoto A, Nakayama K, Imaki H, Hirose S, Jiang Y, Abe M, et al. Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cdelta. Nature. 2002;416:865–869. doi: 10.1038/416865a. [DOI] [PubMed] [Google Scholar]
- 12.Clowes M, Lynch C, Miller A, Miller D, Osborne W, Clowes A. Long-term biological response of injured rat carotid artery seeded with smooth muscle cells expressing retrobirally introduced human genes. J Clin Invest. 1994;93:644–651. doi: 10.1172/JCI117016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakakibara K, Kubota K, Worku B, Ryer EJ, Miller JP, Koff A, et al. PDGF-BB Regulates p27 Expression through ERK-dependent RNA Turn-over in Vascular Smooth Muscle Cells. J Biol Chem. 2005;280:25470–25477. doi: 10.1074/jbc.M502320200. [DOI] [PubMed] [Google Scholar]
- 14.Rao R, Miano J, Olson E, Seidel C. The A10 cll line: a model for neonatal, neointimal, or differentiated vascular smooth muscle cells? Cardiovasc Res. 1997;36:118–126. doi: 10.1016/s0008-6363(97)00156-9. [DOI] [PubMed] [Google Scholar]
- 15.Nelson P, Yamamura S, Mureebe L, Itoh H, Kent K. Smooth muscle cell migration and proliferation are mediated by distic phases of activation of the intracellular messeger mitogen-activated protein kinase. J Vasc Surg. 1998;27:117–125. doi: 10.1016/s0741-5214(98)70298-8. [DOI] [PubMed] [Google Scholar]
- 16.Jackson DN, Foster DA. The enigmatic protein kinase C{delta}: complex roles in cell proliferation and survival. FASEB J. 2004;18:627–636. doi: 10.1096/fj.03-0979rev. [DOI] [PubMed] [Google Scholar]
- 17.Kronfeld I, Kazimirsky G, Lorenzo PS, Garfield SH, Blumberg PM, Brodie C. Phosphorylation of Protein Kinase Cdelta on Distinct Tyrosine Residues Regulates Specific Cellular Functions. J Biol Chem. 2000;275:35491–35498. doi: 10.1074/jbc.M005991200. [DOI] [PubMed] [Google Scholar]
- 18.Ryer EJ, Sakakibara K, Wang C, Sarkar D, Fisher PB, Faries PL, et al. Protein kinase C delta induces apoptosis of vascular smooth muscle cells through induction of the tumor suppressor p53 by both p38 dependent and independent mechanisms. J Biol Chem. 2005 doi: 10.1074/jbc.M507187200. [DOI] [PubMed] [Google Scholar]
- 19.Chaudhuri P, Colles SM, Fox PL, Graham LM. Protein Kinase C{delta}-Dependent Phosphorylation of Syndecan-4 Regulates Cell Migration. Circ Res. 2005;97:674–681. doi: 10.1161/01.RES.0000184667.82354.b1. [DOI] [PubMed] [Google Scholar]
- 20.Li J, O'Connor KL, Greeley GH, Jr, Blackshear PJ, Townsend CM, Jr, Evers BM. Myristoylated Alanine-rich C Kinase Substrate-mediated Neurotensin Release via Protein Kinase C-{delta} Downstream of the Rho/ROK Pathway. J Biol Chem. 2005;280:8351–8357. doi: 10.1074/jbc.M409431200. [DOI] [PubMed] [Google Scholar]
- 21.Aoyama E, Yoshihara R, Tai A, Yamamoto I, Gohda E. PKC- and PI3K-dependent but ERK-independent proliferation of murine splenic B cells stimulated by chondroitin sulfate B. Immunology Letters. 2005;99:80–84. doi: 10.1016/j.imlet.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 22.Campbell M, Trimble ER. Modification of PI3K- and MAPK-Dependent Chemotaxis in Aortic Vascular Smooth Muscle Cells by Protein Kinase C{beta}II. Circ Res. 2005;96:197–206. doi: 10.1161/01.RES.0000152966.88353.9d. [DOI] [PubMed] [Google Scholar]
- 23.Heidkamp MC, Bayer AL, Martin JL, Samarel AM. Differential Activation of Mitogen-Activated Protein Kinase Cascades and Apoptosis by Protein Kinase C {epsilon} and {delta} in Neonatal Rat Ventricular Myocytes. Circ Res. 2001;89:882–890. doi: 10.1161/hh2201.099434. [DOI] [PubMed] [Google Scholar]
- 24.Shanmugam M, Krett NL, Maizels ET, Murad FM, Rosen ST, Hunzicker-Dunn M. A role for protein kinase C [delta] in the differential sensitivity of MCF-7 and MDA-MB 231 human breast cancer cells to phorbol ester-induced growth arrest and p21WAFI/CIP1 induction. Cancer Letters. 2001;172:43–53. doi: 10.1016/s0304-3835(01)00602-4. [DOI] [PubMed] [Google Scholar]
- 25.Nakagawa M, Oliva JL, Kothapalli D, Fournier A, Assoian RK, Kazanietz MG. Phorbol Ester-induced G1 Phase Arrest Selectively Mediated by Protein Kinase C{delta}-dependent Induction of p21. J Biol Chem. 2005;280:33926–33934. doi: 10.1074/jbc.M505748200. [DOI] [PubMed] [Google Scholar]
- 26.Akashi M, Osawa Y, Koeffler H, Hachiya M. p21WAF1 expression by an activator of protein kinase Cis regulated mainly at the post-trancriptional level in cells lacking p53: important role of RNA stabilization. Biochem J. 1999;337:607–616. [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng Y, el-Deiry W. Regulation of p21WAF1/CIP1 expression by p53-independent pathways. Oncogene. 1996;12:1557–1564. [PubMed] [Google Scholar]
- 28.Fukumoto S, Nishizawa Y, Hosoi M, Koyama H, Yamakawa K, Ohno S, et al. Protein Kinase C delta †Inhibits the Proliferation of Vascular Smooth Muscle Cells by Suppressing G1 Cyclin Expression. J Biol Chem. 1997;272:13816–13822. doi: 10.1074/jbc.272.21.13816. [DOI] [PubMed] [Google Scholar]
- 29.Wakino S, Kintscher U, Liu Z, Kim S, Yin F, Ohba M, et al. Peroxisome Proliferator-activated Receptor gamma Ligands Inhibit Mitogenic Induction of p21Cip1 by Modulating the Protein Kinase Cdelta Pathway in Vascular Smooth Muscle Cells. J Biol Chem. 2001;276:47650–47657. doi: 10.1074/jbc.M108719200. [DOI] [PubMed] [Google Scholar]
- 30.Ginnan R, Singer HA. PKC-{delta}-dependent pathways contribute to PDGF-stimulated ERK1/2 activation in vascular smooth muscle. Am J Physiol Cell Physiol. 2005;288:C1193–1201. doi: 10.1152/ajpcell.00499.2004. [DOI] [PubMed] [Google Scholar]
- 31.Seasholtz TM, Majumdar M, Kaplan DD, Brown JH. Rho and Rho Kinase Mediate Thrombin-Stimulated Vascular Smooth Muscle Cell DNA Synthesis and Migration. Circ Res. 1999;84:1186–1193. doi: 10.1161/01.res.84.10.1186. [DOI] [PubMed] [Google Scholar]
- 32.Liu B, Itoh H, Louie O, Kubota K, Kent KC. The signaling protein Rho is necessary for vascular smooth muscle migration and survival but not for proliferation. Surgery. 2002;132:317–325. doi: 10.1067/msy.2002.125786. [DOI] [PubMed] [Google Scholar]
- 33.Obara K, Nishizawa S, Koide M, Nozawa K, Mitate A, Ishikawa T, et al. Interactive Role of Protein Kinase C-d with Rho-Kinase in the Development of Cerebral Vasospasm in a Canine Two-Hemorrhage Model. Journal of Vascular Research. 2005;42:67–76. doi: 10.1159/000083093. [DOI] [PubMed] [Google Scholar]
- 34.Leitges M, Mayr M, Braun U, Mayr U, Li C, Pfister G, et al. Exacerbated vein graft arteriosclerosis in protein kinase C{delta}-null mice. J Clin Invest. 2001;108:1505–1512. doi: 10.1172/JCI12902. [DOI] [PMC free article] [PubMed] [Google Scholar]