

Abstract
Two myelin oligodendrocyte glycoprotein (MOG92–106) monoclonal antibodies (mAbs) were produced from an A.SW mouse with progressive experimental autoimmune encephalomyelitis. Polyreactivity/specificity of the mAbs was demonstrated by ELISA. Functionality and a potential role in pathogenesis of systemic autoimmunity were demonstrated in vitro in a lymphocytotoxicity assay and in vivo upon injection into naïve mice. Injection of MOG mAb producing hybridomas into naïve mice resulted in immunoglobulin deposition in kidneys and liver. This model will be useful in determining whether transitional forms between CNS (organ)-specific and systemic autoimmune diseases exist, and whether progressive multiple sclerosis has features of a systemic autoimmune disease.
Keywords: Autoantibodies, Autoimmunity, CNS Demyelinating Autoimmune Diseases, Demyelinating Diseases, Histology
1. Introduction
Multiple sclerosis (MS) is an inflammatory demyelinating disease of the central nervous system (CNS) which encompasses a diverse spectrum of clinical disease (Willenborg and Staykova, 2003). The clinical course of MS can be categorized into four forms based on the presence or absence of relapses and remission and/or progression of neurological deficits: relapsing-remitting (RR), primary-progressive (PP), secondary progressive (SP) and progressive-relapsing (PR) (Lublin and Reingold, 1996). RR-MS is characterized by alternating disease relapses and periods of full recovery or sequelae. PP-MS is defined by its continuous disease progression from the onset. Patients with SP-MS initially present with RR disease which is then followed by disease progression. In contrast, PR-MS starts out progressive and is followed by definite acute relapses with or without full remission. Mechanisms of transition and differences between the various forms of MS are not well understood, due in part to the bias towards research on RR disease based on the availability of animal models (Thompson et al., 1997). PP-MS is clinically, epidemiologically, pathologically, and immunologically different from the other forms of MS (Pender et al., 2003; Sadatipour et al., 1998), which begs the question whether PP-MS and RR-MS are two distinct diseases (Larsen et al., 1985; McDonald, 1994; Olerup et al., 1989).
Myelin oligodendrocyte glycoprotein (MOG), a member of the immunoglobulin (Ig) superfamily (Gardinier et al., 1992), is encoded within the major histocompatibility complex (MHC) (Pham-Dinh et al., 1993) and can be detected on the surface of oligodendrocytes and myelin in the CNS (Linington et al., 1984). T and B cells reactive against MOG have been identified in demyelinating lesions and cerebrospinal fluid (CSF) of patients with MS (Genain et al., 1999; Reindl et al., 1999; Sun et al., 1991; Xiao et al., 1991), and MOG specific antibody has been found deposited within lesions (Genain et al., 1999). Both intact MOG protein and encephalitogenic peptides derived from MOG can induce experimental autoimmune encephalomyelitis (EAE) in mice (Amor et al., 1994; Mendel et al., 1995). In MOG induced EAE, CD4+ T helper (Th) cells appear to be essential for initiation of inflammation in the CNS, but MOG antibodies have been reported to be responsible for development of the large plaque-like demyelinating lesions. MOG antibody has been shown to increase demyelination in vivo when passively transferred (Berger and Reindl, 2000; Kojima et al., 1994; Lassmann et al., 1988; Linington et al., 1993), and to lyse oligodendrocytes in vitro (Linington et al., 1989).
We have previously demonstrated that the encephalitogenic peptide MOG92–106 can induce RR- and PP-EAE in two different strains of H-2s mice (Tsunoda et al., 2000). SJL/J mice immunized with MOG92–106 develop RR-EAE characterized by a mild demyelinating disease with T cell infiltration. In contrast, A.SW mice sensitized with MOG92–106 develop PP-EAE characterized by large areas of plaque-like demyelination and have been shown to have high titers of serum MOG specific antibody and Ig deposition in the CNS (Tsunoda et al., 2000). A correlation between anti-MOG IgG2a/IgG1 ratio and survival time has also been demonstrated in A.SW mice immunized with MOG92–106 (Tsunoda et al., 2000). These results indicate a role for MOG specific antibodies in disease progression.
In MS involvement of general organs and autoantibodies against non-myelin antigens is observed (Gardinier et al., 1992; Kabat and Freedman, 1950; Lowenthal et al., 1960; Tsunoda et al., 2000) and in systemic autoimmune disease, i.e., systemic lupus, CNS antibodies and inflammation are seen (Esiri, 1977; Woyciechowska and Brzosko, 1977).
To investigate the contribution of MOG92–106 antibodies in systemic autoimmunity in EAE, hybridoma cell lines were generated from the spleen of an A.SW mouse with progressive MOG92–106 induced EAE (P-EAE). Two hybridoma cell lines producing MOG92–106 antibodies of the IgM isotype were obtained and the monoclonal antibodies (mAbs) were purified. In EAE, antibodies are presumed to be involved in oligodendrocyte loss and myelin desolution, but these antibodies are mostly of the IgG isotype (Storch et al., 1998; Xiao et al., 1991). Recently, several autoantibodies of the IgM isotype, including a few oligodendrocyte-reactive antibodies, have been demonstrated to promote remyelination in the Theiler’s murine encephalomyelitis virus-induced model for MS (Asakura et al., 1998; Miller et al., 1994; Miller and Rodriguez, 1995). These antibodies promoting remyelination also have the characteristics of “natural autoantibodies” (Asakura et al., 1995; Asakura et al., 1998; Warrington et al., 2000). Natural autoantibodies are mostly of the IgM isotype, have polyreactivity toward multiple self and nonself antigens and are encoded by germline Ig genes with little or no mutations (Asakura et al., 1995; Kirschning et al., 1999; Miller and Rodriguez, 1995). Interestingly, MS patients have higher levels of natural autoantibodies in their CSF compared to patients with other neurological diseases and healthy controls (Matsiota et al., 1988).
We further characterized two MOG92–106 specific antibodies, A4ac and A4cd, that were previously shown to be of the IgM isotype and to be encoded by variable genes that are in the germline configuration (Libbey et al., 2006). We found that A4ac and A4cd both possess κ light chains. In this study, we first found Ig deposition in general organs of mice with P-EAE. Since A4ac and A4cd have characteristics of natural antibodies, this prompted us to investigate whether A4ac and A4cd have another characteristic of natural antibodies, polyreactivity, which could result in Ig deposition in multiple organs. A4ac and A4cd are polyreactive, and elicit complement dependent lymphocytotoxicity of splenic mononuclear cells (MNCs). A4ac and A4cd also bind myelin as well as antigens in various other tissues such as the kidney and liver in vivo. The detection of antibody deposition in the kidneys and lungs of A.SW mice with P-EAE and the isolation of antibodies from these mice that react with both MOG92–106 as well as DNA, histone H1a and proteins isolated from other mouse organs suggest that P-EAE has features of a systemic autoimmune disease. In addition, the demonstration that injection of the hybridomas into naïve mice resulted in antibody deposition in the liver and kidney supports the hypothesis that P-EAE is a systemic autoimmune disease. In MS involvement of general organs and autoantibodies against non-myelin antigens is observed (Gardinier et al., 1992; Kabat and Freedman, 1950; Lowenthal et al., 1960; Tsunoda et al., 2000), while in systemic autoimmune disease, systemic lupus erythematosus, antibodies against CNS antigens and inflammation in the CNS have been demonstrated (Esiri, 1977; Woyciechowska and Brzosko, 1977). This model will be useful in determining whether transitional forms between CNS (organ)-specific and systemic autoimmune diseases exist, and whether progressive MS has features of a systemic autoimmune disease.
2. Materials and Methods
2.1 Animals
Female SJL/J and A.SW mice were purchased from the Jackson Laboratory (Bar Harbor, ME). SJL/J and A.SW mice were immunized subcutaneously in the base of the tail with 100 nmol of MOG92–106 peptide (DEGGYTCFFRDHSYQ) (Core Facility of the University of Utah, Salt Lake City, UT) (Amor et al., 1994; Papenfuss et al., 2004) in complete Freund’s adjuvant (CFA) containing 2 mg/ml Mycobacterium tuberculosis H37 Ra (Difco Laboratories, Detroit, MI) with or without 5 × 109 Bordetella pertussis cells (Michigan Department of Public Health, Lansing, MI). Mice were weighed and observed for clinical signs for four to six months. Classical EAE signs were assessed according to the following criteria: 0 = no clinical disease; 1 = loss of tail tonicity; 2 = mild hind leg paresis; 3 = moderate hind leg paralysis; 4 = complete paraplegia; and 5 = quadriplegia, moribund state or death (Tsunoda et al., 1998). A second clinical phenotype, ataxic form of EAE (Greer et al., 1996; Tsunoda et al., 2000), commenced with mice turning their heads or bodies to one side (scored 1 or 2 depending on the degree to which the head was turned) with or without a waddling gait. Score 3 = mice continuously rolled by twisting their bodies or rotated laterally in a circle; 4 = mice could not stand but would lie on their sides with or without rolling; and 5 = moribund state or death.
2.2 Establishment of MOG Specific B Cell Hybridomas
MOG specific B cell hybridomas were generated using Sp2/0-Ag14 (Shulman et al., 1978) [American Type Culture Collection (ATCC), Manassas, VA], a myeloma that does not synthesize or secrete any Ig chains, is resistant to 8-azaguanine and does not survive in hypoxanthine-aminopterin-thymidine (HAT) containing media (Köhler and Milstein, 1975; Ozaki, 1994). Splenocytes from mice with P-EAE were treated with NH4Cl solution for removal of red blood cells. Splenocytes and Sp2/0-Ag14 cells were incubated in 50% polyethylene glycol 1500 (Roche Diagnostics GmbH) at a 1:1 ratio. The cells were seeded at 1 × 105 cells/100 μl DMEM/well in 96-well flat-bottomed plates. On day 2, 5 × 106 thymocytes from normal SJL/J mice were added as feeder cells. Hybridoma cells were selected in HAT minimal medium and two weeks after fusion, supernatants were screened by enzyme-linked immunosorbent assay (ELISA) for the presence of anti-MOG antibodies. Wells that had a high MOG antibody titer, reflected by an optical density of greater than 1.0, were selected (number of positive wells: 25/238) and cells were cloned by limiting dilution onto 1 × 105 thymic feeder cells in DMEM-hypoxanthine-thymidine (HT) media supplemented with 20% fetal calf serum (FCS) in flat-bottom 96-well plates. Two hybridoma clones, A4ac and A4cd, out of 59 clones were selected for further work. Hybridoma cells were adapted to a BD Cell Mab Medium Serum-Free (BD Biosciences, San Jose, CA) in Erlenmeyer flasks (Corning Incorporated, Corning, NY) using an Orbit Environ-Shaker (LAB-LINE Instruments, Inc., Melrose Park, IL).
2.3 Purification of MOG92–106 Specific Antibodies
Cell culture supernatants were concentrated by Amicon ultrafiltration using YM-30 membranes (Millipore, Billerica, MA). Concentrated supernatants were then loaded onto S-200 gel filtration columns (Pharmacia, Uppsala, Sweden) and fractions were collected and analyzed by SDS-PAGE on a 5% gel under nonreducing conditions. Fractions containing antibody were pooled and concentrated again by Amicon ultrafiltration using YM-30 membranes. The concentrations of the proteins were determined by means of the BCA protein assay (Pierce, Rockford, IL), according to the manufacturer’s recommendations.
2.4 MOG Antibody Assay
MOG immunized mice were bled when sacrificed. We used an ELISA to measure the level of serum MOG antibody as described previously (Tsunoda et al., 2000). Briefly, 96-well flat-bottom Nunc-Immuno MaxiSorp™ plates (Nalge Nunc International, Rochester NY) were coated overnight with 10 μg/ml MOG92–106 peptide. After blocking with 10% FCS and 0.2% Tween 20, serial dilutions of supernatant fluids from hybridoma cell cultures or purified A4ac, A4cd or IgM κ isotype control (Purified Mouse IgM, κ Immunoglobulin Isotype Standard (anti-TNP), BD Biosciences) were added to the plates and incubated for 90 minutes at room temperature. After washing, a peroxidase-conjugated anti-mouse IgG (H + L) (Life Technologies, Gaithersburg, MD) or peroxidase-conjugated anti-mouse IgM (Stressgen Biotechnologies, Victoria, BC, Canada) was added for 90 minutes. Immunoreactive complexes were detected with o-phenylenediamine dihydrochloride (Sigma-Aldrich, St. Louis, MO) and were read at 492 nm in a Titertek Multiskan Plus MK II spectrophotometer (Flow Laboratories, McLean, VA).
2.5 Determination of Ig Isotype
96-well plates were coated with 10 μg/ml cyto-anti-mouse IgG (H + L) (The Binding Site, Birmingham, UK) or MOG92–106 in phosphate buffered saline (PBS) and allowed to absorb overnight in a humidified box at 4°C. Nonspecific binding was blocked with 10% FCS and 0.2% Tween 20. Serial dilutions of A4ac and A4cd supernatant or purified antibody were added to the plates and incubated for 90 minutes at room temperature. After washing, peroxidase-conjugated goat anti-mouse IgM (Stressgen), IgG2a, IgG2b, κ or λ (Caltag Laboratories, Burlingame, CA) were added to the wells and incubated for 90 minutes at room temperature. Immunoreactive complexes were detected as described above.
2.6 ELISA Antigens
SJL/J mice were euthanized using halothane, perfused with PBS and tissues harvested and immediately frozen in liquid nitrogen. Protein was isolated from individual tissues using TRIzol® (Invitrogen, San Diego, CA) according to the manufacturer’s instructions. Protein concentration was determined using the Bio-Rad Protein Assay (Bio-Rad Laboratories, Inc., Hercules, CA) and a NanoDrop® ND-1000 spectrophotometer (NanoDrop Technologies, Rockland, DE). We used SJL/J mice for protein isolation, since autoantibody responses against proteins are generally independent of the strain of animal. MOG92–106 and myelin proteolipid protein (PLP)139–151 were synthesized by the Core Facility at the University of Utah (Salt Lake City, UT). Bovine axolemma enriched fraction (AEF) and bovine myelin enriched fraction (MEF) were isolated as described previously (Detskey et al., 1988). Monosialoganglioside GM1, monosialoganglioside GM3, disialoganglioside GD1b and calf thymus dsDNA were purchased from Sigma-Aldrich. Heat-denatured calf thymus DNA was used as ssDNA antigen. Vero cells (American Type Culture Collection (ATCC), Manassas, VA) were infected with mumps virus (Enders strain, ATCC) or mock infected as described previously (Fujinami and Oldstone, 1980). Infected cells were subjected to rapid freeze/thaw and sonication prior to partial purification by centrifugation. Protein concentration was determined as described above. Protein was isolated from baby hamster kidney (BHK)-21 cells (ATCC) using TRIzol® (Invitrogen) and concentration determined as described above.
2.7 Anti-Self and Nonself Protein ELISAs
Reactivity to the following antigens was determined by ELISA: MOG92–106, bovine AEF, bovine MEF, PLP139–151, dsDNA, ssDNA, Histone H1a, milk, mumps, Vero cell protein, BHK-21 cell protein and protein isolated from the blood, heart, kidney, liver, lung, muscle, pancreas, salivary gland and thymus of naïve SJL/J mice. Proteins were coated onto Nunc-Immuno MaxiSorp™ 96-well plates (Nalge Nunc International) at a concentration of 10 μg/ml in PBS and allowed to absorb overnight at 4°C in a humidified chamber. After blocking with 10% FCS and 0.2% Tween 20, serial dilutions of the primary antibodies were added to the plates and incubated for 90 minutes. After washing, peroxidase-conjugated anti-mouse IgM was added for 90 minutes. Immunoreactive complexes were detected as described above.
2.8 Ganglioside ELISAs
96-well plates were coated with monosialoganglioside GM1, monosialoganglioside GM3 and disialoganglioside GD1b (Sigma-Aldrich) at a concentration of 200 ng/ml in 100% ethanol and air dried prior to use. After blocking with 1% bovine serum albumin (BSA) in PBS, serial dilutions of the primary antibodies were added to the plates and incubated overnight at 4°C. The wells were then washed three times with PBS and incubated with peroxidase-conjugated anti-mouse IgM (Stressgen) for 90 minutes at room temperature. Immunoreactive complexes were detected as described above.
2.9 Competitive ELISAs
Competitive ELISAs were performed as described previously (Wang and Fujinami, 1997). Briefly, our standard ELISA was performed as described above with the following modifications. A4ac and A4cd were diluted to a concentration of 10 μg/ml in PBS and incubated with various concentrations of MOG92–106 or PLP139–151 peptide for 24 hours at 4°C. The mixtures were centrifuged at 10,000 rpm (8832 × g) for 5 minutes, and serial dilutions of the supernates reacted with dsDNA, liver protein or MEF coated wells.
2.10 Lymphocytotoxicity Assay
Spleens and lymph nodes were collected from mice immunized with MOG92–106, pressed through a 70-μm cell strainer (BD Biosciences) and cells pelleted by centrifugation at 1000 rpm (210 × g) for 5 minutes. The cells were resuspended in RPMI 1640, overlaid on Histopaque®-1083 (Sigma-Aldrich) and centrifuged at 1500 rpm (480 × g) for 30 minutes with no brake. MNCs were isolated at the interface and resuspended at a concentration of 1 × 107 cells/ml in RPMI 1640 supplemented with 3% BSA alone or 10 μg/ml A4ac, 10 μg/ml A4cd or 10 μg/ml IgM κ isotype control diluted in RPMI 1640 supplemented with 3% BSA and incubated on ice for 30 minutes. The cells were washed, centrifuged for 5 minutes at 1000 rpm (210 × g), resuspended in LOW-TOX-M Rabbit Complement (Cedarlane, Hornby, Ontario, Canada) and incubated at 4°C, 15°C, 25°C or 37°C for 3 hours and 30 minutes (Winfield et al., 1975). The cells were washed three times to remove the complement, and cell number and viability were determined by trypan blue exclusion.
2.11 Inoculation of MOG IgM Producing Hybridomas In Vivo
To determine whether MOG antibody induces demyelination in vivo (Barres et al., 1992; Rosenbluth et al., 1997; Rosenbluth et al., 1999; Schnell and Schwab, 1990; Zhou et al., 1998), we injected 1 × 106 MOG antibody producing hybridoma cells, A4ac or A4cd, or 1 × 106 isotype control antibody producing hybridoma cells, XMMEN-OE5 (kindly provided by Dr. Moses Rodriguez), in 20 μl of BD Cell Mab Medium into the right cerebral hemisphere or cerebellum of 22 naïve mice (Tsunoda et al., 1996). Mice were killed one week after inoculation. To see whether MOG92–106 antibodies bind other tissues in vivo we injected 1 × 107 A4ac, A4cd or 8.18C5 hybridoma cells (kindly provided by Dr. Harmut Wekerle) in 200 μl of PBS intraperitoneally into four naïve mice per hybridoma. Mice were killed one month after injection.
2.12 Histology
Mice were euthanized using halothane and perfused with PBS, followed by a buffered 4% paraformaldehyde solution and tissues were embedded in paraffin. We divided the brains into five coronal slabs and spinal cords into 10 to 12 transverse slabs. Four-micrometer thick tissue sections were stained with Luxol fast blue for myelin visualization, hematoxylin and eosin or Periodic acid Schiff.
2.13 Immunohistochemistry
Ig deposition was visualized using the avidin-biotin peroxidase complex (ABC) technique, with a biotin-conjugated mouse IgG (H + L) antibody (1:200 dilution, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) (Tsunoda et al., 2000). Surface IgM was visualized by immunofluorescence using Alexa Fluor® 568 goat anti-mouse IgM (μ chain) (1:500 dilution, Molecular Probes, Inc., Eugene, OR). Fluorescent images were collected and analyzed by laser-scanning confocal microscopy (Cell Image Facility, University of Utah, Salt Lake City, UT).
3.0 Results
3.1 Presence of Antibody Deposition in Organs Outside the CNS in Mice with P-EAE
A.SW mice sensitized with MOG92–106 develop PP-EAE characterized by large areas of plaque-like demyelination, high titers of serum MOG specific antibody and Ig deposition in the white matter of the CNS (Tsunoda et al., 2000). We then investigated whether Ig deposition could also be detected in general organs in mice with PP-EAE. Interestingly, we found that 14 of 15 mice (93%) with PP-EAE had antibody deposition in general organs outside the CNS, including the lung and glomeruli in the kidney (Figure 1b–c). In contrast, prior to the onset of PP-EAE, none of 15 mice sensitized with MOG had antibody deposition in the CNS and only four of 15 mice (27%) had very mild antibody deposition in the lung or kidney (Figure 1a). This suggests that autoantibody responses against general organs could be present in mice with PP-EAE mice and that the autoantibody responses were associated with MOG-specific antibody deposition in the CNS.
Figure 1.
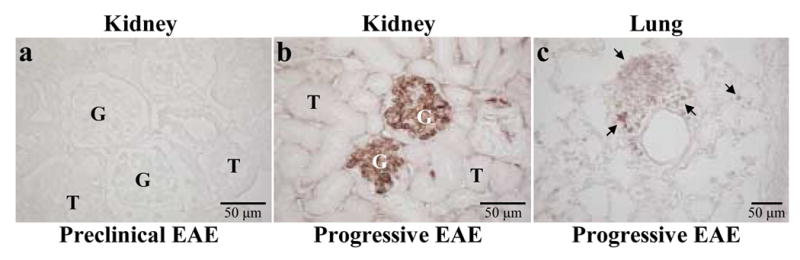
Immunoglobulin (Ig) deposition in the kidney and lung of mice with progressive experimental autoimmune encephalomyelitis (EAE). Kidney and lung sections were immunostained for Ig deposition. (a) No Ig deposition was seen in glomeruli (G) or tubules (T) in the kidneys of MOG92–106 sensitized mice during the preclinical period. (b) Ig deposition was localized to the glomeruli of the kidney in mice with progressive EAE. (c) Ig positive inflammatory cells (arrows) were detected in the lung of mice with progressive EAE.
3.2 MOG Reactive IgM Antibodies from Hybridoma Cell Lines
The results described above prompted us to characterize MOG92–106-specific antibody responses in mice with PP-EAE. For this purpose, we first decided to establish hybridoma cell lines that produce MOG92–106 antibody from EAE mice. From the hybridomas, we selected two clones, A4ac and A4cd, which have significant reactivity with MOG92–106 for further analysis. Using ELISA, we found that A4ac and A4cd were of the IgM isotype and have κ light chains (Figure 2a–b).
Figure 2.
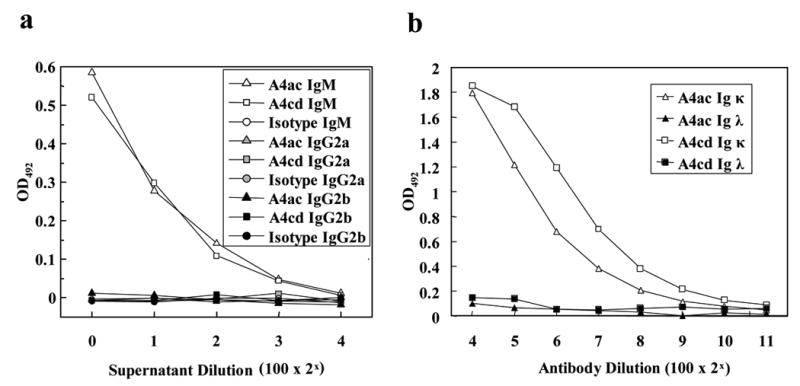
ELISA analyses of the MOG antibodies, A4ac and A4cd. (a) Plates were coated with MOG92–106, and antibody isotype was determined using peroxidase-conjugated anti-mouse IgM, IgG2a or IgG2b antibody. (b) Plates were coated with capture anti-mouse Ig, and the isotype of the light chain was determined by titration of purified antibody using peroxidase-conjugated anti-mouse Ig κ or λ antibody.
Establishment of IgM (not IgG) producing hybridomas was intriguing, since the hybridomas were derived from a mouse during the late chronic stage of EAE. In addition, our previous independent experiments demonstrated that A4ac and A4cd are encoded by variable genes that are in the germline configuration (Libbey et al., 2006). The IgM isotype and germline configulation are two characteristics of natural antibody. The third and most important characteristic of natural antibody is reactivity against multiple antigens (polyreactivity). Thus, we hypothesized that MOG92–106 antibodies, A4ac and A4cd, could be polyreactive and that the autoantibody deposition in general organs could be due to the presence of polyreactive MOG natural antibodies similar to A4ac and A4cd. If this hypothesis is correct, this would lead to future experiments to determine whether similar natural autoantibody responses against MOG can contribute to involvement of general organs in EAE.
3.3 Polyreactivity to Self- and Nonself-Antigens
Binding of the mAbs to a variety of autoantigens was determined by ELISA to assess the polyreactivity of A4ac and A4cd. In order to characterize reactivity with self-antigens, proteins were isolated from heart, lung, muscle, kidney, liver, blood, salivary glands, pancreas and thymus of naïve mice. A4ac and A4cd both exhibited binding with proteins extracted from various tissues albeit to different extents. A4ac had significant binding to pancreas, liver, salivary gland and thymus as compared to the IgM κ control antibody. Similarly, A4cd exhibited significant binding to muscle, kidney, blood, salivary glands, pancreas, thymus and lung as compared to the IgM κ control antibody (Figure 3a). Reactivity of A4ac and A4cd with myelin peptides (MOG92–106 and PLP139–151) and proteins (AEF and MEF) was also examined by ELISA. A4cd bound all of the myelin peptides and proteins tested to a greater extent than the IgM κ control antibody. However, A4ac bound all but the AEF and MEF to a greater extent than the IgM κ control antibody (Figure 3a). A4cd bound monosialylated gangliosides, GM1 and GM3, and a disialylated ganglioside, GD1b, to a greater extent than the IgM κ control antibody (Figure 3b). A4ac reacted with gangliosides at a level similar to that of the IgM κ control antibody (Figure 3b). Both A4ac and A4cd reacted with dsDNA, ssDNA and histone H1a to a much greater extent than the IgM κ control antibody (Figure 3b). A4ac and A4cd were both found to react with several nonself antigens including milk and protein isolated from Vero and BHK-21 cell lines, but not with mumps antigen, FCS or PBS (Figure 3b). Therefore, A4ac and A4cd both appeared to be polyreactive.
Figure 3.
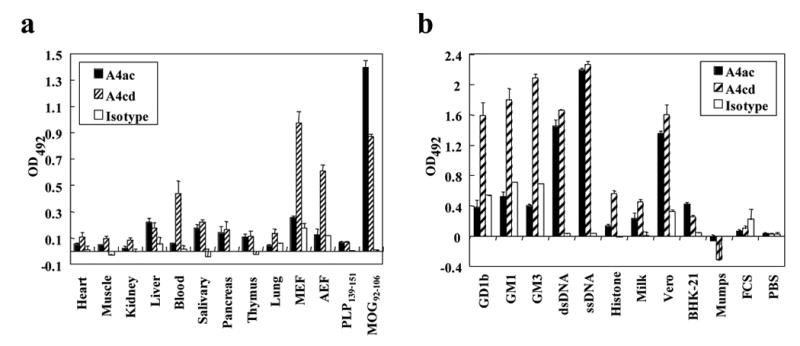
Polyreactivity of MOG antibodies. (a) ELISA analyses showed that MOG antibodies reacted with proteins from a variety of mouse organs, myelin enriched fraction (MEF), and axolemma enriched fraction (AEF), as compared with an IgM κ isotype control (1:400 dilution). (b) ELISA analyses demonstrated that MOG IgM antibodies reacted with various self- and nonself-antigens, as compared to an isotype control (1:400 dilution). High reactivitities were detected against gangliosides (GD1b, GM1, GM3), Vero cells and nuclear antigens (histone H1a, dsDNA, ssDNA). All experiments were performed in triplicate. Data shown are mean ± SD.
A4ac and A4cd were incubated with various concentrations of MOG92–106 or PLP139–151 peptides, and ELISAs were performed to confirm their specificity for MOG. Reactivity of A4ac and A4cd with dsDNA, protein isolated from the liver or MEF could be blocked by incubating the antibodies with MOG92–106, but not with PLP139–151, prior to performing the ELISAs (Figure 4a–c). This data supports the concept that these antibodies are polyreactive, not polyclonal. The results of the MEF blocking experiment suggest that reactivity of A4ac and A4cd with MEF is primarily due to MOG present in the MEF preparation.
Figure 4.
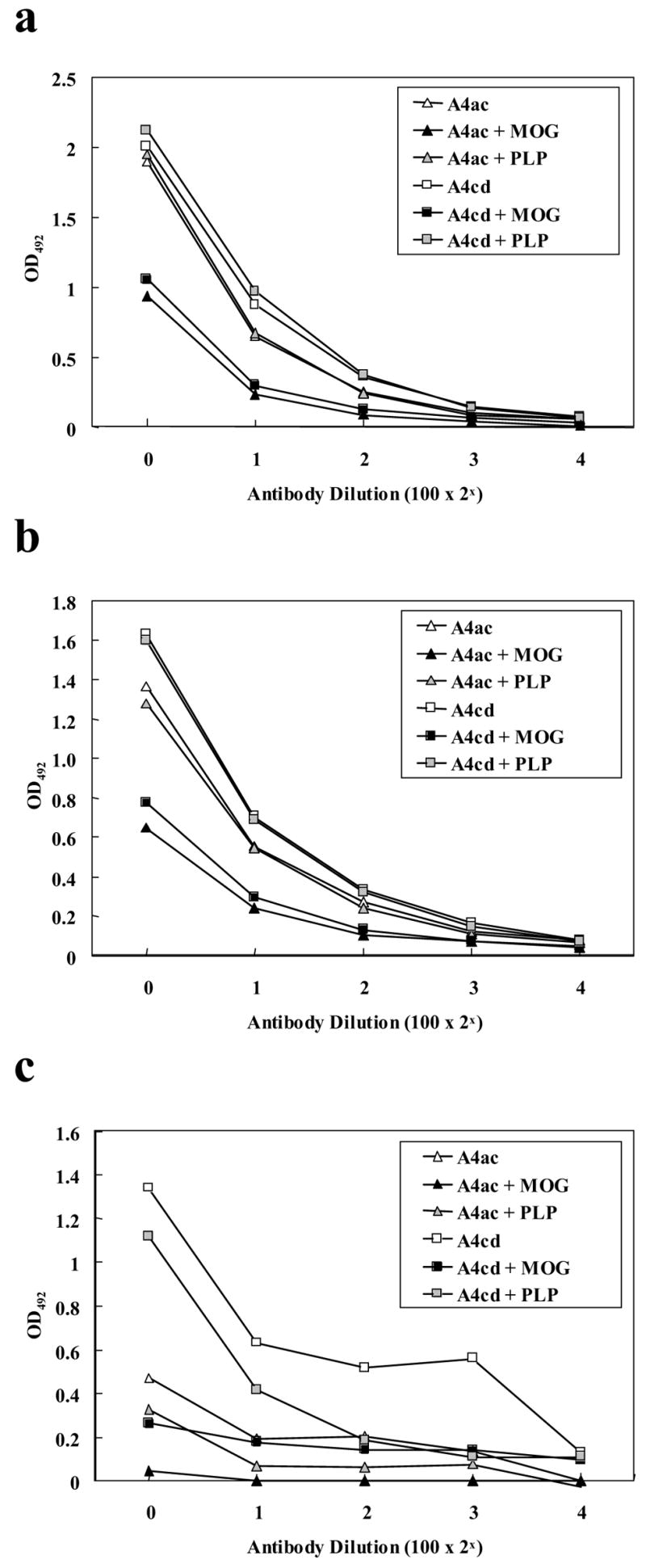
Inhibition of MOG92–106 mAb binding to dsDNA (a), liver (b) and MEF (c) by pre-incubating with MOG, but not with PLP, prior to ELISA analyses. A4a and A4cd were incubated with 20 μg (a,b) or 5 μg (c) MOG92–106 or PLP139–151 prior to ELISA analyses. Significant blocking was seen only when the antibodies were preincubated with MOG.
3.4 Lymphocyte Binding and Cytotoxicity
Cold-reactive lymphocytotoxic antibodies have been identified in patients with MS, and have been found to correlate with Ig levels in the CSF (Weiner and Schocket, 1979) and disease progression (Scott and Spitler, 1983). To determine whether the polyreactivity of the MOG antibodies could have functional affects, we performed a lymphocytotoxicity assay. MOG belongs to the B7 family and MOG92–106 contains the consensus motif of the Ig superfamily (Gardinier et al., 1992). Since MOG92–106 reactive antibodies can also bind to protein isolated from blood, we examined their affects on lymphocytotoxicity in vitro. Spleen MNCs were incubated with complement plus mAbs, control Ab or media at 4°C, 15°C, 25°C and 37°C to determine whether they possess complement-dependent lymphocytotoxic activity. A4ac and A4cd have complement-dependent lymphocytotoxic activity at all of the temperatures tested; however, lymphocytotoxic activity was optimal at 15°C (Figure 5).
Figure 5.
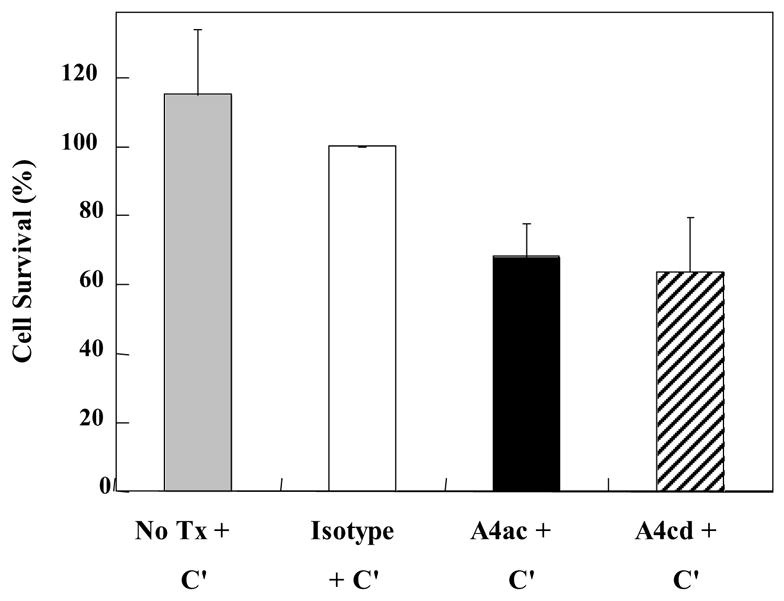
Affects of MOG antibodies on lymphocytes in vitro. Lymphocytotoxicity of the antibodies was assessed by incubating splenic mononuclear cells (MNCs) with the antibodies (A4ac, A4cd) plus complement (C′), an isotype control antibody (Isotype) plus complement or complement alone (NoTx+C′) at 15°C for 3.5 hours and comparing the percentage of cells surviving as detected by trypan blue exclusion. Incubation with MOG antibodies, A4ac and A4cd, plus complement resulted in MNC death. All experiments were performed in triplicate. Data shown are mean ± SD.
3.5 In Vivo Deposition of MOG Antibodies
To determine whether MOG antibody binds myelin in vivo, we injected naïve SJL/J mice intracerebrally with MOG antibody producing hybridoma cell lines, A4ac or A4cd, or a control hybridoma, XMMEN-OE5. One week after injection, mice were euthanized and CNS tissues were collected. Luxol fast blue staining and Ig immunohistochemistry were performed to examine the effects of hybridoma cells on myelin and determine whether the antibodies bound myelin in vivo. We found demyelination in three of 13 mice (23%) injected with A4ac or A4cd (Fig 6a, c, e). Demyelination was seen the areas such as the lateral optic tract, the fimbria hippocami, the internal capsule, and the midbrain, which are distant from the site of injection. Immunohistochemistry against Ig showed that the demyelinating lesions were always accompanied with Ig deposition (Fig 6d, f). In some mice injected with A4ac or A4cd, we found Ig deposition, specific for the white matter, including the anterior commisure, corpus callosum, optic nerve and the internal capsule (Figure 7b–e), while other mice had only tumor growth without demyelination or Ig deposition in the white matter. We did not see demyelinating lesions or Ig deposition in the white matter of nine mice injected with control hybridoma cells, XMMEN-OE5 (Fig 6a,b).
Figure 6.
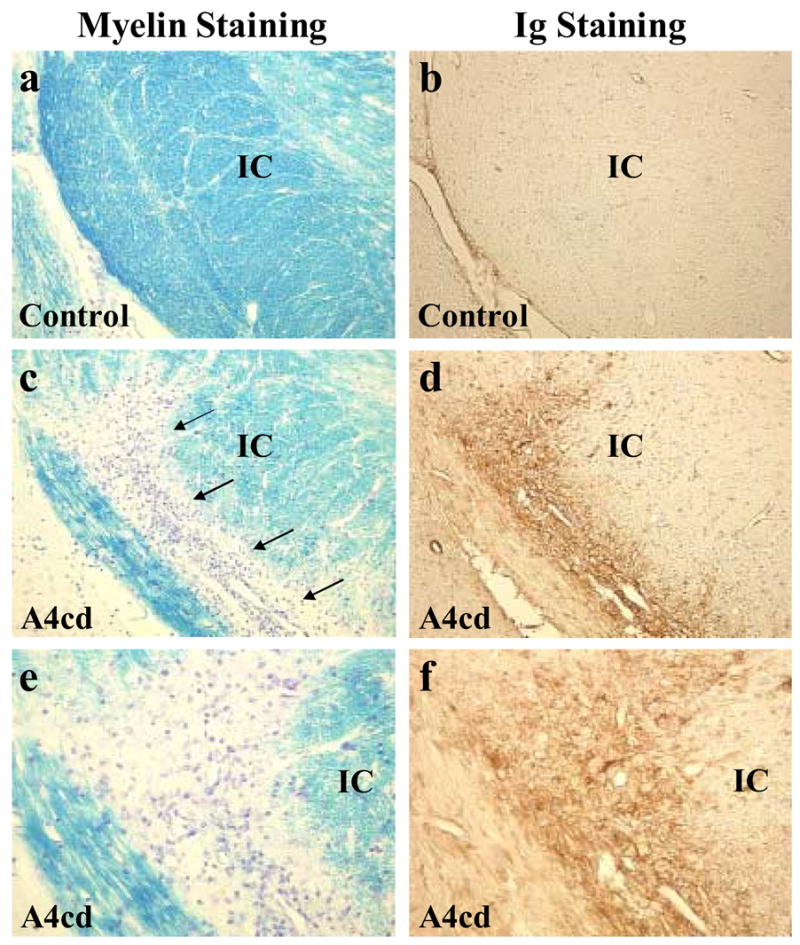
Demyelination induced by intracerebral injection of MOG antibody producing hybridomas. A4ac or A4cd hybridoma cells were injected intracerebrally into naïve SJL/J mice to examine the affects of hybridoma cells on myelination. Mice were killed one week later. (ab) We did not detect any demyelinating lesions or antibody deposition in areas of the brain distant from the site of injection in mice injected with hybridoma cells secreting nonmyelin IgM κ isotype control antibody (6a–b). (c,e) We detected demyelination (arrows) in the internal capsule (IC) of a mouse injected with A4cd, with antibody deposition in the demyelinated lesion (d,f). (a–d) Maginification × 110. (e–f) Magnification × 220.
Figure 7.
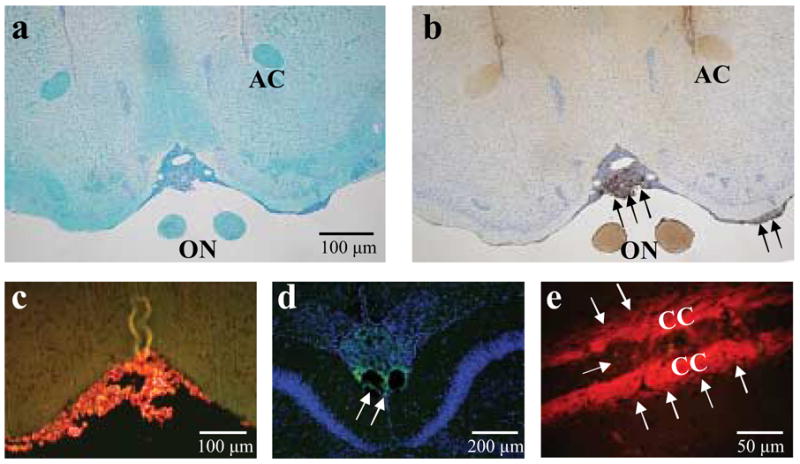
In vivo binding of MOG antibodies in the CNS. A4ac, A4cd or isotype control hybridoma cells were injected intracerebrally into naïve SJL/J mice to determine whether the antibodies bind myelin in vivo. Mice were killed six days later. (a) In many of the mice injected with MOG antibody producing hybridomas, we did not detect any demyelinating lesions in the white matter of the brain, including the anterior commissure (AC) and the optic nerve (ON), with Luxol fast blue staining. (b) However, we did find severe meningeal infiltration (arrows) of hybridoma cells and Ig deposition in areas of the white matter such as the optic nerve and anterior commissure in mice injected with A4ac or A4cd by means of anti-mouse IgG (H+L). Hybridoma cells in the meninges were also positive for Ig. (c,d) Using antibodies against mouse IgM, we demonstrated that the hybridoma cells expressed IgM on their surface (c), and we detected infiltration of hybridoma cells and IgM deposition (arrows) in the meninges (d). (e) Hybridoma cells infiltrated into the corpus callosum (CC) and Ig deposition (arrows) was seen in the adjacent area. (c,e) IgM is red. (d) IgM is green; TOPO-3 nuclear staining is blue.
Using antibodies against mouse IgM or heavy and light chains of IgG, we found that all three hybridoma cells infiltrated heavily in the meninges, the corpus callosum, ventricles and midbrain (Fig 7c–e). In all groups, most mice had Ig deposition in the meninges and vessels. We also detected diffuse antibody staining in both gray and white matter in the ipsilateral side of the hybridoma injection in a few mice in all groups (data not shown). We did not see antibody deposition in naïve mouse brains without injection of hybridoma cells producing MOG antibody. No inflammatory lesions were found in general organs of mice injected intracerebrally (data not shown). To determine whether MOG92–106 antibodies bound other tissues in vivo, we injected A4ac, A4cd or 8.18C5 hybridoma cells intraperitoneally into naïve mice. Antibody deposition was detected in the glomeruli of the kidney and in the liver in A4ac and A4cd injected mice (Figure 8a,c), but was not detected in mice injected with another MOG antibody producing hybridoma, 8.18C5(Figure 8b,d).
Figure 8.
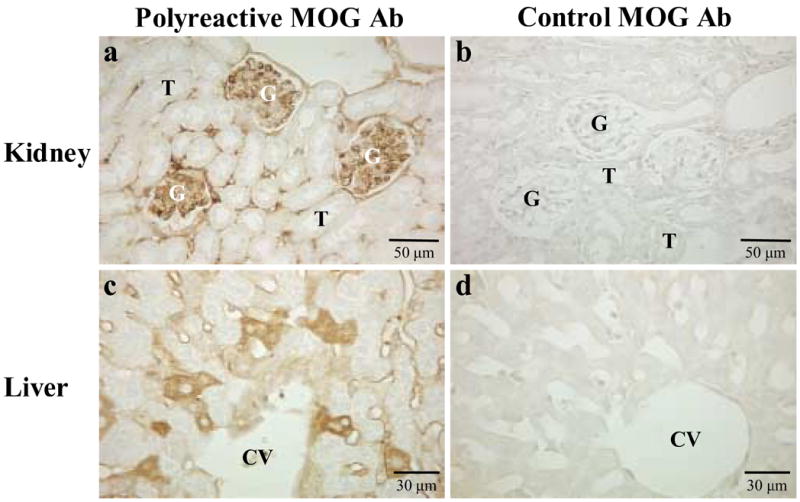
In vivo binding of MOG antibodies in general organs. Polyreactive MOG antibody (A4cd) or control MOG antibody (8.18C5) hybridoma cells were injected intraperitoneally into SJL/J mice to determine whether MOG92–106 antibodies bind other tissues in vivo. Mice were killed one month later and immunostaining for Ig deposition performed on general organs. No antibody deposition was detected in the glomeruli (G) of the kidneys (b) or hepatocytes in the liver (d) of mice injected with 8.18C5 hybridomas secreting antibody against a conformational epitope of MOG. In contrast significant amounts of antibody deposition were detected in the kidneys (a) and liver (c) of mice injected with A4cd hybridoma cells. T = tubule, CV = central vein.
4.0 Discussion
In this study we characterized two cloned hybridoma cell lines producing antibodies of the IgM κ isotype that react with MOG92–106. These antibodies also react with a variety of self-and nonself-antigens.
A.SW mice sensitized with MOG92–106, which develop the progressive form of EAE, have antibody deposition in the CNS as well as other tissues; whereas SJL/J mice sensitized with MOG92–106, which develop RR-EAE, only display antibody deposition in the CNS and to a lesser extent than mice with P-EAE (Tsunoda et al., 2000). These results suggest that there is production of autoantibodies to non-CNS tissues in mice with P-EAE. Two MOG92–106 reactive antibodies that were previously shown to be of the IgM isotype and to be encoded by variable genes that are in the germline configuration (Libbey et al., 2006). We hypothesized that MOG92–106 reactive antibody itself might bind general organs, since MOG92–106 monoclonal antibodies, A4ac and A4cd, have characteristics of natural antibodies and MOG92–106 contains the consensus sequence for the Ig superfamily. In this study, we examined whether MOG92–106 mAbs had polyreactivity. A4ac and A4cd were found to have κ light chains and exhibit polyreactivity, yielding further evidence that the antibodies are indeed natural autoantibodies. Injection of the hybridoma cells secreting the MOG92–106 reactive natural antibodies resulted in demyelination and antibody deposition in the CNS as well as other tissues, recapitulating some of the pathology seen in A.SW mice with P-EAE.
Accumulating evidence implicates antibodies in demyelination and disease pathogenesis in MS. The evidence includes intrathecal Ig synthesis (Kabat and Freedman, 1950), the presence of oligoclonal IgG bands in the CSF (Johnson et al., 1977; Lowenthal et al., 1960), identification of plasma cells and Ig in MS lesions and Ig deposition at the border of actively demyelinating plaques (Esiri, 1977; Lumsden, 1971; Storch et al., 1998; Woyciechowska and Brzosko, 1977). Several studies have sought to identify the antigenic targets of the B cells and antibodies found in CSF and brain plaques in MS, yet no single target has been found which is directly responsible for the development or progression of MS. Several of the targets identified thus far are of interest to our study. Antibodies against MOG have been identified in the serum, CSF and brain parenchyma of patients with MS (O’Connor et al., 2005). Cold-reactive lymphocytotoxic antibodies have been identified in patients with MS, and have been found to correlate with Ig levels in the CSF (Weiner and Schocket, 1979) and disease progression (Scott and Spitler, 1983). Antibodies against dsDNA have been demonstrated to compose a significant fraction of the intrathecal IgG in patients with MS, and can be found in brain plaques and CSF of patients with MS. Two of the anti-dsDNA antibodies obtained from patients with MS were also able to react with the surface of oligodendrocytes and neuronal cells, suggesting that, similar to our antibodies obtained from a mouse with EAE, which also recognize dsDNA, these antibodies are polyreactive (Williamson et al., 2001). However, in contrast to the antibodies obtained from the mouse with P-EAE, the anti-dsDNA antibodies obtained from patients with MS did not react with MOG (Williamson et al., 2001).
Interestingly, MS patients also have higher levels of natural autoantibodies in their CSF compared to both patients with other neurological diseases and healthy controls (Matsiota et al., 1988). Organ-specific IgM autoantibodies to liver, heart, kidney and brain have been detected in sera of both normal individuals, and patients with MS (Daar and Fabre, 1981). Although involvement of general organs in MS has been reported, its implication in disease pathogenesis is not clear. In addition, demyelinating diseases similar to MS have been occasionally reported in systemic autoimmune diseases, such as autoimmune thyroid disease, myasthenia gravis, rheumatoid arthritis and systemic lupus erythematosus (SLE) (Achari et al., 1976; Aita et al., 1974; Meloff, 1980; Tanphaichitr, 1980). Hypocomplementemia and circulating immune complexes have also been demonstrated in MS (Tachovsky et al., 1976; Tanphaichitr, 1980). Ig and complement deposition in the glomerulus of the kidney have also been reported in MS (Whitaker et al., 1971). Renal function has been considered to be normal in MS; however, impaired renal function was reported in patients with PP- and SP-MS who were not treated with nephrotoxic drugs (Calabresi et al., 2002). Although renal damage can be seen in MS, it has been ascribed to either bladder dysfunction induced by damage to the CNS (neurogenic bladder) or renal toxicity of drugs. However, the autoimmune etiology has not been considered. It remains to be determined whether there is a transitional form between CNS (organ)-specific versus systemic autoimmune diseases, or whether RR-MS (organ-specific) transits to a more systemic disease resulting in SP-MS.
Autoimmune diseases can be divided into two categories: organ specific autoimmune diseases and systemic autoimmune diseases. The former is characterized by autoimmunity that is restricted to a single organ or organ system and includes diseases such as MS, diabetes and polymyositis. The latter is characterized by autoimmunity that is not restricted to a single organ or organ system and includes SLE and systemic sclerosis (Cohen, 2003). The antigenic targets of the autoimmune response also differ between the two forms of disease with the recognized autoantigens being expressed either only in specific organs or ubiquitously. Th1 mediated cellular autoimmunity has been associated with many organ specific immune responses, whereas Th2 mediated humoral immunity has been shown to play an important role in systemic autoimmune diseases (Froncek and Horwitz, 2002). Although in some disease models or patients, two autoimmune mechanisms seem to be linked or overlap. It is unclear whether the two mechanisms suppress each other (as seen in Th1 versus Th2) or both contribute to disease progression. For example, MS and EAE have been proposed to be Th1 mediated cellular autoimmune diseases. However, enhanced autoantibody responses with suppression of Th1 responses have been correlated with disease progression in MS and in our model of EAE (Tsunoda et al., 2000).
Here we demonstrate that A.SW mice sensitized with MOG92–106 develop P-EAE with antibody deposition in the CNS. Interestingly, these animals also have antibody bound in other tissues. Two MOG92–106 reactive natural antibodies isolated from an A.SW mouse with P-EAE exhibit polyreactivity, which could potentially account for the antibody deposition and lesions detected outside the CNS in mice with P-EAE. The MOG92–106 reactive natural antibodies recognized not only tissue-restricted antigens such as MOG92–106, but also ubiquitously expressed antigens such as dsDNA and histone H1a, which are antigens associated with systemic autoimmunity. The MOG92–106 reactive antibodies were also found to or cause lymphocytotoxicity, providing another means by which they could contribute to systemic pathology. Finally, injection of the hybridoma cells secreting the MOG92–106 reactive antibodies into naïve mice resulted in demyelination and antibody deposition in tissues both inside and outside the CNS, such as kidney and liver, reminiscent of that seen in mice with P-EAE. Thus demonstrating that MOG92–106 reactive natural antibodies are a cause of systemic autoimmunity, since the disease manifestations can be recapitulated by passive transfer of the antibodies. Preliminary results show that mice with P-EAE have autoantibodies to dsDNA, blood and histone. Interestingly, autoantibody responses can be detected not only in sera from A.SW mice with P-EAE, but also in sera from SJL/J mice with SP-EAE induced with ultraviolet irradiation or apoptotic cell injections (Tsunoda et al., 2005a pg. 122–134 and 2005b pg.1631–1646). Preliminary analysis of tissues from SJL/J mice with SP-EAE supports the finding of Ig deposition in tissues outside the CNS in mice with P-EAE. The data collected thus far suggest that P-EAE induced with MOG92–106 exhibits characteristics of a systemic autoimmune disease. We propose that sensitization with MOG92–106 leads to activation and proliferation of natural antibody producing B1 cells that can bind and attack proteins in the CNS as well as a variety of other tissues resulting in demyelination and systemic autoimmune manifestations. Further studies need to be conducted to determine the actual contribution of MOG92–106 reactive natural antibodies to disease progression and whether P-EAE and progressive forms of MS are indeed systemic autoimmune diseases.
Acknowledgments
We thank Li-Qing Kuang, MD, Jane Libbey, MS, Tomoko Tanaka, MD and Yasunari Miyazaki, MD for many helpful discussions and Emily Jane Terry, Benjamin Marble, J. Wes Peterson, Aminatu Yusuf and Daniel G. Smith for excellent technical assistance. We are grateful to Ms. Kathleen Borick for outstanding preparation of the manuscript. This work was supported by NIH grant 5R01NS40350.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Achari AN, Trontelj JV, Campos DJ. Multiple sclerosis and myasthenia gravis. A case report with single fiber electromyography Neurology. 1976;26:544–546. doi: 10.1212/wnl.26.6.544. [DOI] [PubMed] [Google Scholar]
- Aita JF, Snyder DH, Reichl W. Myasthenia gravis and multiple sclerosis: An unusual combination of diseases. Neurology. 1974;24:72–75. doi: 10.1212/wnl.24.1.72. [DOI] [PubMed] [Google Scholar]
- Amor S, Groome N, Linington C, Morris MM, Dornmair K, Gardinier MV, Matthieu JM, Baker D. Identification of epitopes of myelin oligodendrocyte glycoprotein for the induction of experimental allergic encephalomyelitis in SJL and Biozzi AB/H mice. J Immunol. 1994;153:4349–4356. [PubMed] [Google Scholar]
- Asakura K, Miller DJ, Pease LR, Rodriguez M. Targeting of IgMκ antibodies to oligodendrocytes promotes CNS remyelination. J Neurosci. 1998;18:7700–7708. doi: 10.1523/JNEUROSCI.18-19-07700.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakura K, Miller DJ, Pogulis RJ, Pease LR, Rodriguez M. Oligodendrocyte-reactive O1, O4, and HNK-1 monoclonal antibodies are encoded by germline immunoglobulin genes. Mol Brain Res. 1995;34:283–293. doi: 10.1016/0169-328x(95)00190-4. [DOI] [PubMed] [Google Scholar]
- Barres BA, Hart IK, Coles HSR, Burne JF, Voyvodic JT, Richardson WD, Raff MC. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70:31–46. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- Berger T, Reindl M. Immunopathogenic and clinical relevance of antibodies against myelin oligodendrocyte glycoprotein (MOG) in multiple sclerosis. J Neural Transm Suppl. 2000;60:351–360. doi: 10.1007/978-3-7091-6301-6_25. [DOI] [PubMed] [Google Scholar]
- Calabresi PA, Austin H, Racke MK, Goodman A, Choyke P, Maloni H, McFarland HF. Impaired renal function in progressive multiple sclerosis. Neurology. 2002;59:1799–1801. doi: 10.1212/01.wnl.0000036618.68674.7a. [DOI] [PubMed] [Google Scholar]
- Cohen PL. Systemic Autoimmunity. In: Paul WE, editor. Fundamental Immunology. 5. Philadelphia: Lippincott Williams & Wilkins; 2003. pp. 1371–1399. [Google Scholar]
- Daar AS, Fabre JW. Organ-specific IgM autoantibodies to liver, heart and brain in man: generalized occurrence and possible functional significance in normal individuals, and studies in patients with multiple sclerosis. Clin Exp Immunol. 1981;45:37–47. [PMC free article] [PubMed] [Google Scholar]
- Detskey PZ, Bigbee JW, DeVries GH. Isolation and characterization of axolemma-enriched fractions from discrete areas of bovine CNS. Neurochem Res. 1988;13:449–454. doi: 10.1007/BF01268880. [DOI] [PubMed] [Google Scholar]
- Esiri MM. Immunoglobulin-containing cells in multiple-sclerosis plaques. Lancet. 1977;2:478. doi: 10.1016/s0140-6736(77)91603-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froncek MJ, Horwitz DA. Cytokines in the pathogenesis of systemic lupus erythematosus. In: Wallace DJ, Hahn BH, editors. Dubois’ Lupus Erythematosus. 6. Philadelphia, PA: Lippincott Williams & Wilkins; 2002. pp. 187–203. [Google Scholar]
- Fujinami RS, Oldstone MBA. Alterations in expression of measles virus polypeptides by antibody: molecular events in antibody-induced antigenic modulation. J Immunol. 1980;125:78–85. [PubMed] [Google Scholar]
- Gardinier MV, Amiguet P, Linington C, Matthieu JM. Myelin/oligodendrocyte glycoprotein is a unique member of the immunoglobulin superfamily. J Neurosci Res. 1992;33:177–187. doi: 10.1002/jnr.490330123. [DOI] [PubMed] [Google Scholar]
- Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med. 1999;5:170–175. doi: 10.1038/5532. [DOI] [PubMed] [Google Scholar]
- Greer JM, Sobel RA, Sette A, Southwood S, Lees MB, Kuchroo VK. Immunogenic and encephalitogenic epitope clusters of myelin proteolipid protein. J Immunol. 1996;156:371–379. [PubMed] [Google Scholar]
- Johnson KP, Arrigo SC, Nelson BJ, Ginsberg A. Agarose electrophoresis of cerebrospinal fluid in multiple sclerosis. A simplified method for demonstrating cerebrospinal fluid oligoclonal immunoglobulin bands Neurology. 1977;27:273–277. doi: 10.1212/wnl.27.3.273. [DOI] [PubMed] [Google Scholar]
- Kabat EA, Freedman DA. A study of the crystalline albumin, γ globulin and total protein in the cerebrospinal fluid of 100 cases of multiple sclerosis and in other diseases. Am J Med Sci. 1950;219:55–64. doi: 10.1097/00000441-195001000-00009. [DOI] [PubMed] [Google Scholar]
- Kirschning E, Jensen K, Dübel S, Rutter G, Hohenberg H, Will H. Primary structure of the antigen-binding domains of a human oligodendrocyte-reactive IgM monoclonal antibody derived from a patient with multiple sclerosis. J Neuroimmunol. 1999;99:122–130. doi: 10.1016/s0165-5728(99)00118-6. [DOI] [PubMed] [Google Scholar]
- Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- Kojima K, Berger T, Lassmann H, Hinze-Selch D, Zhang Y, Gehrmann J, Reske K, Wekerle H, Linington C. Experimental autoimmune panencephalitis and uveoretinitis transferred to the Lewis rat by T lymphocytes specific for the S100β molecule, a calcium binding protein of astroglia. J Exp Med. 1994;180:817–829. doi: 10.1084/jem.180.3.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen JP, Kvaale G, Riise T, Nyland H, Aarli JA. Multiple sclerosis--more than one disease? Acta Neurol Scand. 1985;72:145–150. doi: 10.1111/j.1600-0404.1985.tb00856.x. [DOI] [PubMed] [Google Scholar]
- Lassmann H, Brunner C, Bradl M, Linington C. Experimental allergic encephalomyelitis: The balance between encephalitogenic T lymphocytes and demyelinating antibodies determines size and structure of demyelinated lesions. Acta Neuropathol (Berl) 1988;75:566–576. doi: 10.1007/BF00686201. [DOI] [PubMed] [Google Scholar]
- Libbey JE, Peterson LK, Tsunoda I, Fujinami RS. Monoclonal MOG-reactive autoantibody from progressive EAE has the characteristics of a natural antibody. J Neuroimmunol. 2006;173:135–145. doi: 10.1016/j.jneuroim.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Linington C, Berger T, Perry L, Weerth S, Hinze-Selch D, Zhang Y, Lu HC, Lassmann H, Wekerle H. T cells specific for the myelin oligodendrocyte glycoprotein mediate an unusual autoimmune inflammatory response in the central nervous system. Eur J Immunol. 1993;23:1364–1372. doi: 10.1002/eji.1830230627. [DOI] [PubMed] [Google Scholar]
- Linington C, Morgan BP, Scolding NJ, Wilkins P, Piddlesden S, Compston DA. The role of complement in the pathogenesis of experimental allergic encephalomyelitis. Brain. 1989;112:895–911. doi: 10.1093/brain/112.4.895. [DOI] [PubMed] [Google Scholar]
- Linington C, Webb M, Woodhams PL. A novel myelin-associated glycoprotein defined by a mouse monoclonal antibody. J Neuroimmunol. 1984;6:387–396. doi: 10.1016/0165-5728(84)90064-x. [DOI] [PubMed] [Google Scholar]
- Lowenthal A, Van Sande M, Karcher D. The differential diagnosis of neurological diseases by fractionating electrophoretically the CSF G-globulins. J New Drugs. 1960;6:51–56. doi: 10.1111/j.1471-4159.1960.tb13448.x. [DOI] [PubMed] [Google Scholar]
- Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. [National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis] Neurology. 1996;46:907–911. doi: 10.1212/wnl.46.4.907. [DOI] [PubMed] [Google Scholar]
- Lumsden CE. The immunogenesis of the multiple sclerosis plaque. Brain Res. 1971;28:365–390. doi: 10.1016/0006-8993(71)90052-7. [DOI] [PubMed] [Google Scholar]
- Matsiota P, Blancher A, Doyon B, Guilbert B, Clanet M, Kouvelas ED, Avrameas S. Comparative study of natural autoantibodies in the serum and cerebrospinal fluid of normal individuals and patients with multiple sclerosis and other neurological diseases. Ann Inst Pasteur Immunol. 1988;139:99–108. doi: 10.1016/0769-2625(88)90134-1. [DOI] [PubMed] [Google Scholar]
- McDonald WI. Rachelle Fishman-Matthew Moore Lecture. The pathological and clinical dynamics of multiple sclerosis J Neuropathol Exp Neurol. 1994;53:338–343. doi: 10.1097/00005072-199407000-00003. [DOI] [PubMed] [Google Scholar]
- Meloff K. Multiple sclerosis and polyarteritis (Letter to the Editor) Arch Neurol. 1980;37:786. [Google Scholar]
- Mendel I, Kerlero de Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor Vβ expression of encephalitogenic T cells. Eur J Immunol. 1995;25:1951–1959. doi: 10.1002/eji.1830250723. [DOI] [PubMed] [Google Scholar]
- Miller DJ, Rodriguez M. A monoclonal autoantibody that promotes central nervous system remyelination in a model of multiple sclerosis is a natural autoantibody encoded by germline immunoglobulin genes. J Immunol. 1995;154:2460–2469. [PubMed] [Google Scholar]
- Miller DJ, Sanborn KS, Katzmann JA, Rodriguez M. Monoclonal autoantibodies promote central nervous system repair in an animal model of multiple sclerosis. J Neurosci. 1994;14:6230–6238. doi: 10.1523/JNEUROSCI.14-10-06230.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor KC, Appel H, Bregoli L, Call ME, Catz I, Chan JA, Moore NH, Warren KG, Wong SJ, Hafler DA, Wucherpfennig KW. Antibodies from inflamed central nervous system tissue recognize myelin oligodendrocyte glycoprotein. J Immunol. 2005;175:1974–1982. doi: 10.4049/jimmunol.175.3.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olerup O, Hillert J, Fredrikson S, Olsson T, Kam-Hansen S, Möller E, Carlsson B, Wallin J. Primarily chronic progressive and relapsing/remitting multiple sclerosis: two immunogenetically distinct disease entities. Proc Natl Acad Sci USA. 1989;86:7113–7117. doi: 10.1073/pnas.86.18.7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki S. Cell lines and hybridomas. In: Delves PJ, editor. Cellular Immunology LABFAX. Oxford, UK: Academic Press. BIOS Scientific Publishers Limited; 1994. pp. 65–93. [Google Scholar]
- Papenfuss TL, Rogers CJ, Gienapp I, Yurrita M, McClain M, Damico N, Valo J, Song F, Whitacre CC. Sex differences in experimental autoimmune encephalomyelitis in multiple murine strains. J Neuroimmunol. 2004;150:59–69. doi: 10.1016/j.jneuroim.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Pender MP, Csurhes PA, Wolfe NP, Hooper KD, Good MF, McCombe PA, Greer JM. Increased circulating T cell reactivity to GM3 and GQ1b gangliosides in primary progressive multiple sclerosis. J Clin Neurosci. 2003;10:63–66. doi: 10.1016/s0967-5868(02)00270-9. [DOI] [PubMed] [Google Scholar]
- Pham-Dinh D, Mattei MG, Nussbaum JL, Roussel G, Pontarotti P, Roeckel N, Mather IH, Artzt K, Lindahl KF, Dautigny A. Myelin/oligodendrocyte glycoprotein is a member of a subset of the immunoglobulin superfamily encoded within the major histocompatibility complex. Proc Natl Acad Sci USA. 1993;90:7990–7994. doi: 10.1073/pnas.90.17.7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reindl M, Linington C, Brehm U, Egg R, Dilitz E, Deisenhammer F, Poewe W, Berger T. Antibodies against the myelin oligodendrocyte glycoprotein and the myelin basic protein in multiple sclerosis and other neurological diseases: a comparative study. Brain. 1999;122(Pt 11):2047–2056. doi: 10.1093/brain/122.11.2047. [DOI] [PubMed] [Google Scholar]
- Rosenbluth J, Liang WL, Schiff R, Dou WK. Spinal cord dysmyelination induced in vivo by IgM antibodies to three different myelin glycolipids. Glia. 1997;19:58–66. doi: 10.1002/(sici)1098-1136(199701)19:1<58::aid-glia6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Rosenbluth J, Schiff R, Liang WL, Dou WK, Moon D. Antibody-mediated CNS demyelination: focal spinal cord lesions induced by implantation of an IgM anti-galactocerebroside-secreting hybridoma. J Neurocytol. 1999;28:397–416. doi: 10.1023/a:1007021916210. [DOI] [PubMed] [Google Scholar]
- Sadatipour BT, Greer JM, Pender MP. Increased circulating antiganglioside antibodies in primary and secondary progressive multiple sclerosis. Ann Neurol. 1998;44:980–983. doi: 10.1002/ana.410440621. [DOI] [PubMed] [Google Scholar]
- Schnell L, Schwab ME. Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature. 1990;343:269–272. doi: 10.1038/343269a0. [DOI] [PubMed] [Google Scholar]
- Scott CF, Spitler LE. Lymphocytotoxic antibody in multiple sclerosis: activity against T cell subsets and correlation with disease activity. Clin Exp Immunol. 1983;53:133–139. [PMC free article] [PubMed] [Google Scholar]
- Shulman M, Wilde CD, Köhler G. A better cell line for making hybridomas secreting specific antibodies. Nature. 1978;276:269–270. doi: 10.1038/276269a0. [DOI] [PubMed] [Google Scholar]
- Storch MK, Piddlesden S, Haltia M, Iivanainen M, Morgan P, Lassmann H. Multiple sclerosis: In situ evidence for antibody- and complement-mediated demyelination. Ann Neurol. 1998;43:465–471. doi: 10.1002/ana.410430409. [DOI] [PubMed] [Google Scholar]
- Sun J, Link H, Olsson T, Xiao BG, Andersson G, Ekre HP, Linington C, Diener P. T and B cell responses to myelin-oligodendrocyte glycoprotein in multiple sclerosis. J Immunol. 1991;146:1490–1495. [PubMed] [Google Scholar]
- Tachovsky TG, Lisak RP, Koprowski H, Theofilopoulos AN, Dixon FJ. Circulating immune complexes in multiple sclerosis and other neurological diseases. Lancet. 1976;2:997–999. doi: 10.1016/s0140-6736(76)90835-7. [DOI] [PubMed] [Google Scholar]
- Tanphaichitr K. Multiple sclerosis associated with eosinophilic vasculitis, pericarditis, and hypocomplementemia. Arch Neurol. 1980;37:314–315. doi: 10.1001/archneur.1980.00500540092017. [DOI] [PubMed] [Google Scholar]
- Thompson AJ, Polman CH, Miller DH, McDonald WI, Brochet B, Filippi M, Montalban X, De Sá J. Primary progressive multiple sclerosis. Brain. 1997;120:1085–1096. doi: 10.1093/brain/120.6.1085. [DOI] [PubMed] [Google Scholar]
- Tsunoda I, Iwasaki Y, Terunuma H, Sako K, Ohara Y. A comparative study of acute and chronic diseases induced by two subgroups of Theiler’s murine encephalomyelitis virus. Acta Neuropathol (Berl) 1996;91:595–602. doi: 10.1007/s004010050472. [DOI] [PubMed] [Google Scholar]
- Tsunoda I, Kuang LQ, Theil DJ, Fujinami RS. Antibody association with a novel model for primary progressive multiple sclerosis: Induction of relapsing-remitting and progressive forms of EAE in H2S mouse strains. Brain Pathol. 2000;10:402–418. doi: 10.1111/j.1750-3639.2000.tb00272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda I, Kuang LQ, Tolley ND, Whitton JL, Fujinami RS. Enhancement of experimental allergic encephalomyelitis (EAE) by DNA immunization with myelin proteolipid protein (PLP) plasmid DNA. J Neuropathol Exp Neurol. 1998;57:758–767. doi: 10.1097/00005072-199808000-00005. [DOI] [PubMed] [Google Scholar]
- Wang LY, Fujinami RS. Enhancement of EAE and induction of autoantibodies to T-cell epitopes in mice infected with a recombinant vaccinia virus encoding myelin proteolipid protein. J Neuroimmunol. 1997;75:75–83. doi: 10.1016/s0165-5728(96)00235-4. [DOI] [PubMed] [Google Scholar]
- Warrington AE, Asakura K, Bieber AJ, Ciric B, Van Keulen V, Kaveri SV, Kyle RA, Pease LR, Rodriguez M. Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proc Natl Acad Sci USA. 2000;97:6820–6825. doi: 10.1073/pnas.97.12.6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner HL, Schocket AL. Lymphocytes in multiple sclerosis: correlation with CSF immunoglobulins and cold-reactive lymphocytotoxic antibodies. Neurology. 1979;29:1504–1508. doi: 10.1212/wnl.29.11.1504. [DOI] [PubMed] [Google Scholar]
- Whitaker JN, Dowling PC, Cook SD. Immunofluorescent studies of the kidney in human neurologic disorders. J Neuropathol Exp Neurol. 1971;30:129–130. [PubMed] [Google Scholar]
- Willenborg DO, Staykova MA. Cytokines in the pathogenesis and therapy of autoimmune encephalomyelitis and multiple sclerosis. Adv Exp Med Biol. 2003;520:96–119. doi: 10.1007/978-1-4615-0171-8_7. [DOI] [PubMed] [Google Scholar]
- Williamson RA, Burgoon MP, Owens GP, Ghausi O, Leclerc E, Firme L, Carlson S, Corboy J, Parren PW, Sanna PP, Gilden DH, Burton DR. Anti-DNA antibodies are a major component of the intrathecal B cell response in multiple sclerosis. Proc Natl Acad Sci USA. 2001;98:1793–1798. doi: 10.1073/pnas.031567598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winfield JB, Winchester RJ, Wernet P, Fu SM, Kunkel HG. Nature of cold-reactive antibodies to lymphocyte surface determinants in systemic lupus erythematosus. Arthritis Rheum. 1975;18:1–8. doi: 10.1002/art.1780180101. [DOI] [PubMed] [Google Scholar]
- Woyciechowska JL, Brzosko WJ. Immunofluorescence study of brain plaques from two patients with multiple sclerosis. Neurology. 1977;27:620–622. doi: 10.1212/wnl.27.7.620. [DOI] [PubMed] [Google Scholar]
- Xiao BG, Linington C, Link H. Antibodies to myelin-oligodendrocyte glycoprotein in cerebrospinal fluid from patients with multiple sclerosis and controls. J Neuroimmunol. 1991;31:91–96. doi: 10.1016/0165-5728(91)90014-x. [DOI] [PubMed] [Google Scholar]
- Zhou L, Trapp BD, Miller RH. Demyelination in the central nervous system mediated by an anti-oligodendrocyte antibody. J Neurosci Res. 1998;54:158–168. doi: 10.1002/(SICI)1097-4547(19981015)54:2<158::AID-JNR4>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]