

Abstract
Human UDP-glucuronosyltransferase 1A10 has been identified as the major isoform involved in the biotransformation of a wide range of phenolic substrates, including native estrogens and their oxidized metabolites. Our recent studies point to the F90-M91-V92-F93 amino acid motif of UGT1A10, which was identified using photoaffinity labeling followed by LC-MS/MS analysis, as a key determinant of the binding of phenolic substrates. In this report, we have evaluated the role of F90, V92, and F93 in the recognition of estrogens by UGT1A10 using site-directed mutagenesis. Kinetic studies using 5 mutants revealed that F90 and F93 are critical residues for the recognition of all estrogen substrates. The substitution of F90 with alanine totally abolished the activity of this enzyme toward all the estrogens investigated. Overall, sequential removal for the aromatic (FtoL) and of the hydrophobic chain (FtoA and VtoA) from amino acids 90, 92, and 93 position effectively alters estrogen recognition. This demonstrates that individual features of the native and hydroxylated estrogens determine the specific binding properties of the compound within the binding site of the human UGT1A10 and the mutants. The resulting activities are completely abolished, unchanged, increased or decreased depending on the structures of both the mutant and the substrate. The novel identification of UGT1A10 as the major isoform involved in the glucuronidation of all estrogens and the discovery of the importance of the FMVF motif in the binding of steroids will help to elucidate the molecular mechanism of glucuronidation, resulting in the design of more effective estrogen-based therapies.
Supplementary Key Words: UGTs, UGT1A10, estrogens, binding-site, site-directed mutagenesis
INTRODUCTION
UDP-glucuronosyltransferases (UGTs) are a family of membrane-bound glycoproteins that biotransform the largest number of endogenous and exogenous compounds of any phase II enzyme [1]. Glucuronidation reactions are catalyzed by UDP-glucuronosyltransferases (UGTs), which comprise two families, UGT1 and UGT2 [2]. The isoforms of the UGT1A subfamily are encoded by a single gene located on chromosome 2 and exhibit distinct, but overlapping, substrate specificities [2]. UGTs from both families are of particular interest due to their direct involvement in the biotransformation of estradiol (E2), and the 4-hydroxylated catechol-estrogens (4-OH-CEs), which are know genotoxic [3].
Other drug metabolizing enzymes involved in the biotransformation and detoxification of estrogens include cytochromes P450 (CYP450s) [4], sulfotransferases (SULTs) [5,6], and catechol-O-methyltransferases [7]. CYP450s are capable of generating oxidative metabolites of estrogens. The role of SULT1E1 in the formation of sulfated estrogens in target tissue is the most recent finding regarding the conjugation of estrogen and its metabolites, and a potential role for these enzymes in preventing cancer is clear. SULTs are abundant is estrogen target tissues; however, they biotransform only very low concentrations of estrogens [8]. Analysis of circulating estrogens in the human body reveals that sulfates and glucuronides are the most abundant estrogen derivatives [8]. Under normal physiological conditions, these estrogen metabolizing enzymes control the concentrations of these compounds. However, it has been demonstrated that, under certain conditions, abnormal accumulation of estrogens in target tissues may lead to the development of breast cancer [9].
The study of estrogen glucuronidation and the characterization of the specific UGTs involved in the process is a relatively new undertaking [8,10–12]. Although several human UGTs have been identified as being involved in the conjugation of various estrogen compounds in the liver [13], intestine [12–14], and estrogen target tissues [15], the involvement of individual UGT isoforms, specifically the extrahepatic UGT1A10, in the biotransformation of estrogens is a more recent development [10,12,16,17]
Hydroxylated forms of native estrogens have been implicated in the initiation and development of breast cancer. Specifically, the 4-hydroxylated metabolites of estrogen have been shown to be associated with the development of estrogen-sensitive cancers in estrogen target tissues [18]. A recent study showed that the concentration of 4-OH-CEs in breast cancer patients was significantly higher than that of 2-OH-CEs, which are mainly found in normal breast tissue [19]. 4-OH-CEs cause mutations in DNA by undergoing metabolic redox cycling to generate electrophilic metabolites that react with DNA to form depurinating DNA adducts, leading to mutations and ultimately to cancer [20,21]. UGTs, which have a higher capacity to metabolize estrogens than do SULTs, can prevent the generation and accumulation of reactive estrogen metabolites by facilitating their removal from the body [22].
Our laboratory has carried out extensive kinetic characterization of human UGT1A10 and identified it as the major UGT isoform involved in the in vitro glucuronidation of all native and CYP450-oxidized estrogens due to its high affinity and capacity for these substrates. Moreover, since there is no information available on the structure of the estrogen binding site of this enzyme, or of other enzymes and/or nuclear receptors that bind these compounds, identification of the estrogen binding site of UGT1A10 was undertaken. Using photoaffinity labeling, specific phenol probes, and LC-MS/MS sequencing, the F90-M91-V92-F93 amino acid motif was identified as the phenol binding site of UGT1A10 [23]. Several mutations of this motif were carried out by site-directed mutagenesis to confirm the importance of this site in phenol binding, and in the present studies, these same mutated enzymes were investigated for their ability to bind 7 different estrogens. As was the case for simple phenols, F90 and F93 were found to be essential for the glucuronidation of estrogens by UGT1A10. The identification of this estrogen binding motif in UGT1A10 is a novel discovery, which can be extrapolated to other UGT isoforms and to other estrogen binding proteins.
MATERIALS AND METHODS
Materials
[14C]UDP-glucuronic acid ([14C]UDP-GlcUA) (specific activity, 196 mCi/mmol) was purchased from Perkin-Elmer Life and Analytical Sciences (Boston, MA). Estrone (E1), estradiol (E2), estriol (E3), 2-OH-E2, 4-OH-E1, 4-OH-E2, and saccharolactone (saccharic acid-1,4-lactone) were obtained from Sigma-Aldrich (St. Louis, MO). 2-OH-E1 was purchased from Steraloids (Newport, RI). All other chemicals and solvents were of the highest quality commercially available.
Cloning and expression of human recombinant His-tagged UGTs
Details of the cloning and expression of UGT1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, and 1A10 in baculovirus-infected Sf9 insect cells as His-tagged proteins and the preparation of enriched membrane fractions were reported previously [24,25].
Microsome Preparation
Total membrane fractions from insect cells expressing UGT1A10 and its mutants were prepared as previously described [25]. Briefly, frozen baculovirus-infected insect cells were disrupted by osmotic shock, followed by centrifugation at 40,000 x g at 4 ºC for 60 min. Cells were then suspended in cold distilled water and centrifuged under the same conditions. The final membrane pellets were suspended in washing buffer (25 mM Tris-HCl, pH 7.8 and 0.5 mM EDTA), homogenized with a glass tissue homogenizer, divided into aliquots and stored at −70 ºC until use.
Activity Assays
UGT assays (30 μl total volume) contained 5 μl of buffer (1 M Tris, pH 8.0, 5 mM MgCl2, 5 mM saccharolactone), 5 μg of membrane protein, 250 μM unlabeled substrate (substrate structures are shown in Fig. 1) and 4 mM [14C]UDP-GlcUA. Substrates were added, dissolved in dimethyl sulfoxide (maximum final concentration: 2%). Controls omitting substrate were run with each assay. Reactions were initiated by adding radiolabeled UDP-glucuronic acid, and reaction mixtures were incubated at 37 ºC for 20 min, terminated by adding 30 μl ethanol, and cooled on ice.
Figure 1.
Structures of native estrogens and their hydroxylated metabolites.
Aliquots (40 μl) of each sample were then applied to the preabsorbent layer of channeled silica gel TLC plates (Baker Si250-PA, 19C, VWR, Sugarland, TX). Products and unreacted substrate were separated by development of the plates in chloroform-methanol-acetic acid-H20 (65:25:2:4, v/v). Glucuronides present on silica gel were identified by autoradiography of the TLC plates for 3–5 days at −80 ºC. Silica gel containing labeled glucuronides or in corresponding areas in control lanes was transferred to scintillation vials, and radioactivity was quantified by liquid scintillation counting (Packard TRI-CARB 2100TR, Perkin-Elmer) as previously described [26].
Enzyme Kinetics
For determination of Vmax and Km values, human recombinant UGT1A10 and its mutants were incubated in the presence of varying estrogen concentrations (5 to 500 μM) at a fixed concentration of [14C]UDP-GlcUA (4 mM) for 20 min. All incubations were performed in duplicate, and glucuronidation activities are expressed in nanomoles per milligram of protein per minute. Kinetic parameters were determined using Prism4 software (GraphPad Software, San Diego, CA).
Site-directed mutagenesis
The structures of the amino acids involved are shown in Fig. 2. A new derivative of the mammalian expression vector pcDNA 3.1 (+) (Invitrogen), called pcDNA 3.39, was prepared by replacing the Nhe1-Apa1 fragment of the multi-cloning site of pcDNA 3.1 (+) with the similarly-digested fragment from the multi cloning site of Litmus 39 (New England BioLabs, Ipswich, MA). The UGT1A10 gene was transferred from the pFastBac derivative pFB-XHC [24] into pcDNA 3.39 and site-directed mutagenesis was performed on this construct, pcDNA3.39-1A10, using the QuickChange Site-directed Mutagenesis Kit (Stratagene, La Jolla, CA). Single point mutations were introduced into pcDNA3.39-1A10 according to manufacturer instructions. A list of the mutagenic primers follows, written in the 5’ to 3’ direction, with the mutated nucleotides underlined:
Figure 2.
Structures of amino acids used to create mutants.
F90A-for: 5’-GATCAGAACCGGGAAGCCATGGTTTTCGCCCATGC-3’
F93A-for: 5’-CGGGAATTCATGGTTGCCGCCCATGCTCAATGGAA-3’
F90L-for: 5’-CAGAACCGGGAATTGATGGTTTTCGCCC-3’
F93L-for: 5’-CGGGAATTCATGGTGCTAGCCCATGCTCAATG-3’
V92A-for: 5’-GAACCGGGAATTCATGGCCTTCGCCCATGCTCAAT-3’
Reverse complementary primer pairs of each mutagenic primer were also used in each reaction. Constructs were then transformed into E. coli XL1-Blue and sequenced to verify the desired mutation.
RESULTS
Glucuronidation of estrogens and their hydroxyl derivatives by human UGTs
Eleven human recombinant UGT proteins were evaluated for their ability to glucuronidate the native estrogens, E1, E2, and E3, and their 2- and 4-hydroxy derivatives. Microsomal protein (5 μg) from baculovirus-infected Sf9 insect cells, which were engineered to express individual UGT enzymes, was used for all the 1A family proteins. This same set of enzymes has been used in previous studies from our lab and have been show to have a high catalytic capacity for a large variety of compounds, allowing for kinetic analyses be carried out under the optimal conditions for each isoform [23,25,27,28]. UGT2B7 was over-expressed in HK293 cells.
In these experiments, all estrogens (200 μM) were screened using the standard procedures presented in the Methods section. The results showed that E1 is glucuronidated primarily by two UGT isoforms, UGT1A1 and 1A10, with rates of 1 and 28 nmol/mg protein/min, respectively (Figure 3). E2 was conjugated mainly by UGT1A10 and UGT1A3, and, in both cases, the rates (0.1 and 4.8 nmol/mg protein/min), were significantly lower than for E1. E3, the major estrogen produced in pregnant women, was conjugated with the highest activity by UGT1A10 (5 nmol/mg protein/min) and 2B7 (3 nmol/mg protein/min). Small but consistent levels of glucuronidation of E3 by UGT1A1, 1A7, and 1A9 were also noted.
Figure 3. Glucuronidation activities of human recombinant UGTs toward native estrogens and their hydroxylated derivatives.
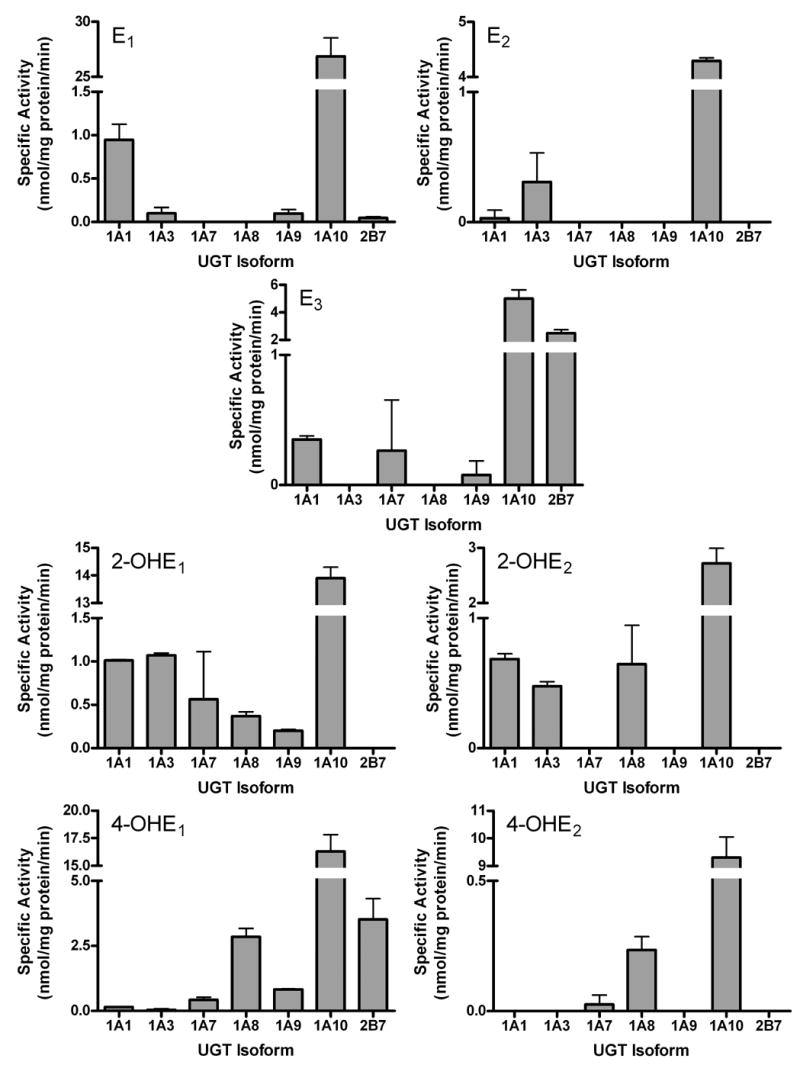
Glucuronidation activities of UGT1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, and 1A10 were measured using membrane fractions of recombinant UGTs expressed as His-tag proteins in baculovirus infected Sf9 insect cells (10 μg). The substrate and co-substrate (UDP-GlcUA) concentrations were 0.25 mM and 4 mM, respectively. The estrogen substrates were dissolved in DMSO (final concentration 2%) and the reaction was incubated for 20 min. Reactions were stopped by the addition of ethanol and products were separated by TLC and identified by autoradiography. Specific activities are expressed in nmol/mg protein/min and shown with standard deviations based on (2–6) experiments.
The product of the CYP450-catalyzed 2-hydroxylation of E1, was glucuronidated by the largest number of recombinant UGTs with UGT1A10, 1A1, 1A3, 1A7, 1A8, and 1A9 displaying activity towards 2-OH-E1. UGT1A10 glucuronidation of this hydroxylated derivative by was approximately 14 nmol/mg protein/min. 2-OH-E2 was a much poorer overall substrate, being glucuronidated only by UGT1A10, 1A1, 1A3, and 1A8, all of which displayed only modest activity toward this substrate. UGT1A10, 2B7, and 1A8 were found to be the most effective isoforms in the glucuronidation of the genotoxic 4-OH-E1, while only UGT1A10 effectively glucuronidated 4-OH-E2.
The most significant observation from these studies is that, under the same experimental conditions, UGT1A10 exhibited the highest activity towards all the estrogens tested, with E1 being glucuronidated at much higher rates than any other estrogen evaluated. UGT1A10 also catalyzed the conjugation of the 2- and 4-hydroxy metabolites. 2- and 4-OH-E1 and 4-OH-E2 were glucuronidated at similar rates but the rate of 2-OH-E2 glucuronidation was significantly lower.
Evaluation of the glucuronidation potency of a series of UGT1A10 mutants towards estrogens substrates and comparison to wild type UGT1A10
In these experiments, 5 mutants were evaluated for their activity toward native estrogens and their 2- and 4-hydroxyl derivatives (Figure 4). Removal of the aromatic ring from the side-chain of F90 totally abolished the activity of this enzyme toward all estrogens investigated. The F93A mutant showed significantly lower activity toward E1, E2, E3 and 2-OH-E1: however with 4-OH-E1 and -E2, activities were unchanged while 2-OH-E2 glucuronidation was increased slightly but significantly. In contrast, the more conservative mutation of F90 to L (Figure 2) formed a mutant that displayed very low activity with E1, E2, E3, and 2-OH-E2, but increased activity toward 2-OH-E1 and the 4-OH-E2. The F93L mutant had almost no activity toward the three native estrogens and significantly lower activity toward all the hydroxylated derivatives as compared to wild type UGT1A10. The catalytic activity of the V92A mutant was significantly higher toward all hydroxylated estrogens. However, E1 and E2 were glucuronidated at rates similar to those of the wild type enzyme, and with E3 a small decrease in activity was detected.
Figure 4. Glucuronidation activities of estrogen human recombinant wild type UGT1A10 and its mutants toward native estrogens and their hydroxylated derivatives.
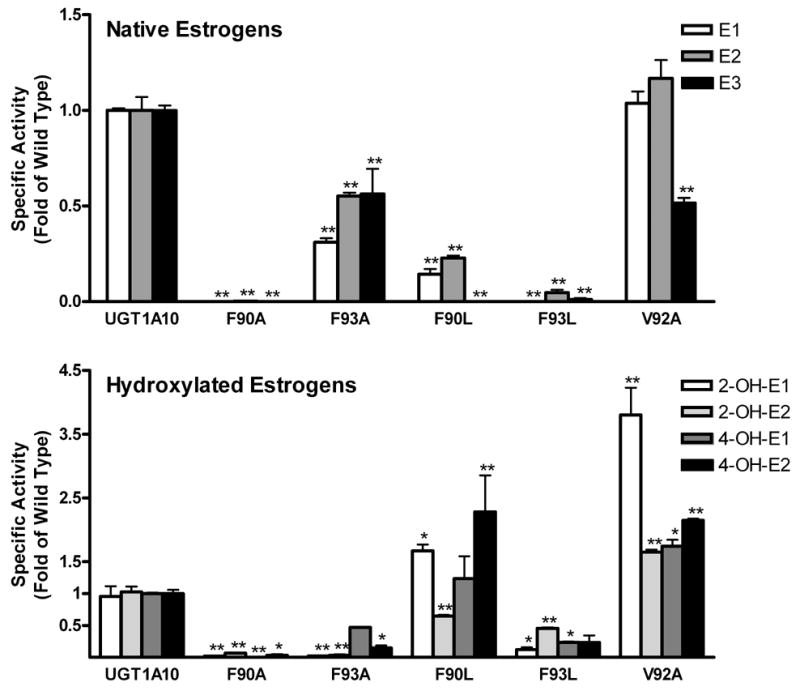
Glucuronidation activities of UGT1A10 and its mutants were assayed by incubating membrane fractions of recombinant UGTs expressed as His-tag proteins in baculovirus infected Sf9 insect cells (5 μg) ) with native and hydroxylated estrogens (250 μM for all but F93A, which was measured at 200 μM) and UDP-GlcUA (4 mM) for 20 min at 37 °C. Reactions were stopped by the addition of ethanol and products were separated by TLC and identified by autoradiography. Specific activities are expressed in nmol/mg protein/min. The results were analyzed by one way ANOVA followed by a Dunnett's Multiple Comparison Test. Measurements that vary significantly from wild type are indicated. (*: p<0.05; **: p<0.01).
Kinetic analysis of wild type and mutant UGT1A10 with E1, E2 and E3
Wild type UGT1A10 and its mutants were assayed in further detail to determine kinetic parameters (apparent Km and relative Vmax) toward E1, E2, and E3 (Figure 5). The F90A and F93L mutants exhibited little or no activity toward E1, E2, and E3 and were therefore excluded from kinetic analysis. The change of F93 to alanine produced an enzyme with a 75% decrease in activity (Vmax) toward E1 but a more than 6 fold increase in affinity (Km) (wt: 42.1 μM; F93A: 6.3 μM). The F90L mutant showed severely impaired activity toward E1 with a decrease in activity of 86%, and an increase in the affinity (12.3 μM).
Figure 5. Kinetic constants for E1, E2 and E3 by recombinant wild type UGT1A10 and mutants.
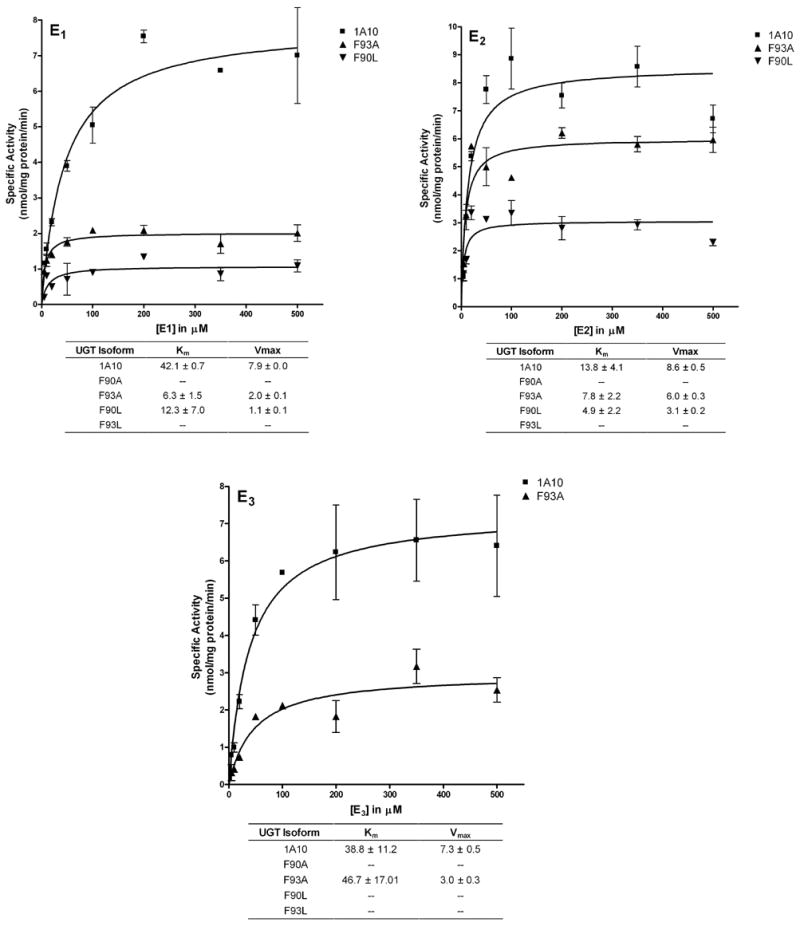
Glucuronidation activities of recombinant wild type UGT1A10 (■) and the F93A (▲) and F90L (▼) mutants were measured by incubating membrane fractions containing recombinant UGT1A10 and its mutants (5 μg) with increasing concentrations (shown in the figure) of the estrogens at a constant concentration of [14C]UDP-GlcUA (4 mM) for 10 min at 37°C. Reactions were stopped by the addition of ethanol and products were separated by TLC and identified by autoradiography. Curve fits and kinetic constants were determined using GraphPad Prism 4 software. The graphical fits of the data from each of the analyses with each substrate (mean ± SD of 4–6 determinations) are shown. Km values are given in μM, and Vmax values are given in nmol/mg protein/min.
For E2, the F93A mutant showed a slight impairment of activity with a Vmax value that decreased by only 30%. The Km for this mutant decreased approximately 2 fold (wt: 13.8 μM; F93A: 7.8 μM). The change of F90 to L produced an enzyme with a decrease in catalytic activity toward E2 of 64%, and an almost 3 fold increase in affinity (4.9 μM).
E3 is the only substrate that was not glucuronidated by the F90L mutant. F93A was the only mutant to show measurable activity towards this estrogen, with a decrease in activity of 59% but a very similar Km value (wt: 38.8 μM; F93A: 46.7 μM).
Kinetic analysis of wild type and mutant UGT1A10 with 2-OH-E1 and 2-OH-E2
Analysis of the catalytic activity of the phenylalanine mutants with 2-OH-E1 and –E2 showed a complete loss of activity for the F90A mutant (Figure 6). The F93A mutant had severely impaired activity toward 2-OH-E1 with a decrease in activity of 99%. However, there was a large increase in the affinity of this mutant for these two estrogens (wt: 130.5 μM; F93A: 10.2 μM). The change of F90 to L produced an enzyme with a 72% increase in the catalytic activity toward 2-OH-E1 but a decreased affinity of more than 2 fold (291 μM), while the F93L mutant had an increased affinity (82.3 μM) and a decrease in activity of 88% toward this substrate.
Figure 6. Kinetic constants for 2-OH-E1, 2-OH-E2, 4-OH-E1 and 4-OH-E2 by recombinant wild type UGT1A10 and mutants.
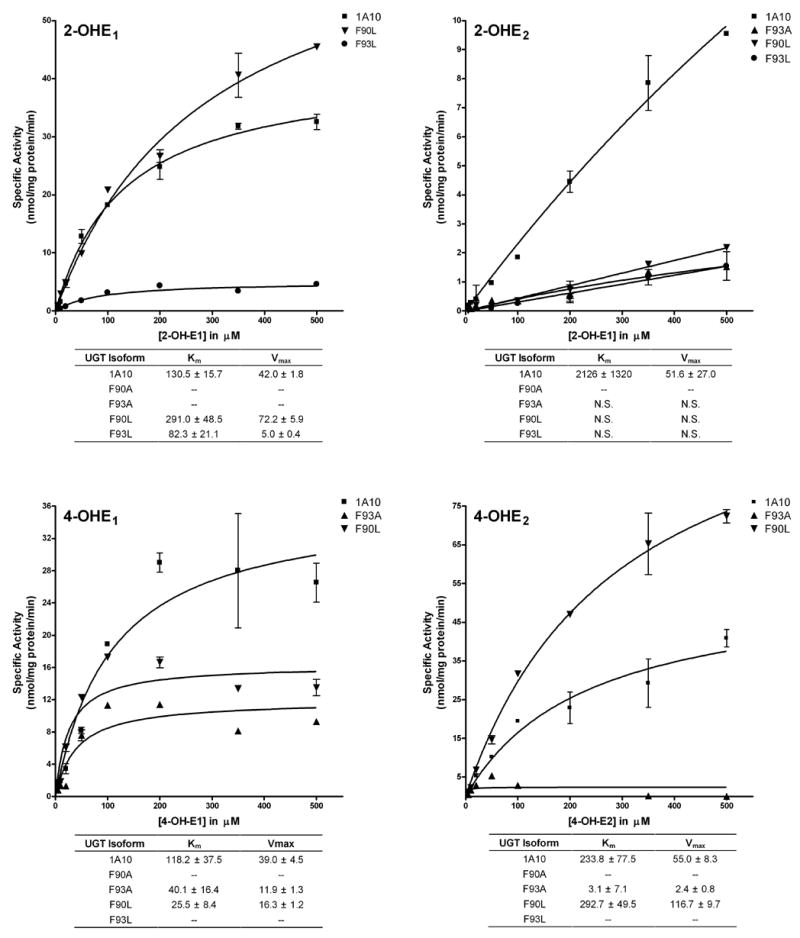
Glucuronidation activities of recombinant wild type UGT1A10 (■) and the F93A (▲), F90L (▼), and F93L (●) mutants were measured by incubating membrane fractions containing recombinant UGT1A10 and its mutants (5 μg) with increasing concentrations (shown in the figure) of 2-OH-E1, 2-OH-E2, 4-OH-E1 and 4-OH-E2 at a constant concentration of [14C]UDP-GlcUA (4 mM) for 10 min at 37°C. Reactions were stopped by the addition of ethanol and products were separated by TLC and identified by autoradiography. Curve fits and kinetic constants were determined using GraphPad Prism 4 software. The graphical fits of the data from each of the analyses with each substrate (mean ± SD of 4–6 determinations) are shown. Km values are given in μM, and Vmax values are given in nmol/mg protein/min.
Evaluation of the glucuronidation capacity of wild type UGT1A10 and its mutants toward 2-OH-E2 showed that, in contrast to the other estrogens evaluated, saturation conditions were not achieved at even at concentrations of 500 μM.
Kinetic analysis of wild type and mutant UGT1A10 with 4-OH-E1 and 4-OH-E2
Analysis of the glucuronidation activity of the phenylalanine mutants with 4-OH-E1 showed a complete loss of activity for the F90A and F93L mutants (Figure 6), and the F93A mutant showed a decrease in activity of 69%. Interestingly, there was a significant increase in the affinity for this mutant (wt: 118.2 μM; F93A: 40.1 μM). The change of F90 to L produced an enzyme with a decrease of catalytic activity toward 4-OH-E1 of 78%, but a decreased affinity of more than 4 fold (25.5 μM).
The F90A and F93L mutants exhibited little or no activity toward 4-OH-E2 and were excluded from kinetic analysis. The F93A mutant had a decrease in activity of 96%, and a significant increase in the affinity (wt: 233.8 μM; F93A: 3.1 μM). The change of F90 to L produced an enzyme with a significant increase of catalytic activity toward 4-OH-E2 112%, but with similar affinity (292.7 μM).
DISCUSSION
Human UGTs are one of the most important classes of Phase II detoxification enzymes, due to the fact that they accept the largest number of structurally diverse chemicals as substrates. Yet, despite the large number of biotransformation reactions mediated by UGTs, relatively little is known about the critical amino acids involved in the direct interactions of these enzymes with naturally occurring substrates, drugs, or environmental pollutants. In our previous report, the two phenylalanine residues of the F90-M91-V92-F93 motif located within the N-terminal domain of UGT1A10 were identified as being critical to the binding and glucuronidation of simple phenols [23]. This motif was identified using photoaffinity labeling followed by LCMS/MS sequencing and site-directed mutagenesis. In those studies, two mutations of this motif, F90A and F93A, were generated and evaluated with two specific phenolic compounds, 4-nitrophenol and 4-methylumbelliferone [23]. The current study was undertaken to further clarify the role of the previously identified FMVF motif of UGT1A10, and analysis of the two previously generated mutants as well as 3 new mutants, F90L, F93L, and V92A, revealed that the two amino acids, F90 and F93, are also critical for the glucuronidation of native estrogens and their hydroxylated derivatives, while V92 only decreases the activity of this enzyme toward E3.
In our previous work, we demonstrated that, in the UGT1A10 binding site created by the FMVF motif, the phenylalanine residues, which have a neutral aromatic side chain, could associate with the phenolic ring of pNP and 4-MU via hydrophobic aromatic ring stacking interactions [23]. We identified F90 as being crucial for these interactions. As is evident from the present studies with estrogens, the same amino acid residue is also indispensable for the binding of both native estrogens and their hydroxylated derivatives (Figures 4, 5, and 6). The F90A mutant had no activity toward any of the substrates investigated. On the other hand, the role of F93 seems to be slightly different for simple phenols and estrogen compounds, and the effect of mutation of this amino acid depends strongly on the structure of the substrate. The most striking observation is that, for both the 2-hydroxylated estrogens, the F93A mutant showed no detectable activity. Therefore, for these hydroxylated estrogens, both F90 and F93 are crucial for their activity. For simple phenols, the affinity of the F93A mutant decreased 3 and 10 fold for 4-MU and pNP, respectively. With the exception of E3 (Km practically unchanged), both native and hydroxylated estrogens showed an increase in affinity with F93A, from 2-77 fold (Figure 5 and 6). In all cases, the activity was significantly decreased, reducing the glucuronidation potency of this mutant, but the affinity for the substrates was increased.
For the present studies, we also generated another set of more conservative phenylalanine mutants, where the aromaticity of the side chain was removed and replaced with a medium length four carbon side chain (Figure 2). This leucine substitution results in a slightly reduced hydrophobicity and eliminates the potential ring-stacking interactions. Two very different phenomena were observed for these mutants. For all the estrogens investigated, the F93L mutation resulted either in either total elimination or severe reduction of glucuronidation activity. On the other hand, the effect of the F90L mutant varied depending on whether the substrate was a native or hydroxylated estrogen. With E3, the glucuronidation activity of this mutant was totally abolished and with E1 and E2, the activity was severely diminished (8–9 fold). Interestingly, with 2-OH-E1 and 4-OH-E2 a significant increase in glucuronidation potency was observed with this mutant.
Our previous sequence analysis of photolabeled UGT1A10 [23] suggested that V92, might also be involved in the binding of phenolic substrates. Therefore, we generated the V92A mutant, and evaluated its activity toward our estrogen substrates. Preliminary screening of this mutation demonstrated that, with the exception of E3, the activity of this enzyme was either unchanged, for E1 and E2, or significantly increased, for hydroxylated estrogens. Kinetic analysis of this mutant has not yet been carried out.
Thus, it appears that the entire motif plays a role in the binding of estrogens. The hydrophobic side chains at position 90 and 93 seems to be essential for specific binding of all estrogens. Individual features of the native and hydroxylated estrogens determine the specific binding properties of the compound within the binding site of the human UGT1A10 and the mutants, resulting in activities that are completely abolished, unchanged, increased or decreased depending on the structures of both the mutant and the substrate. Overall, these present studies with estrogens totally confirm the findings of our previous studies with phenols that the FMVF motif is most certainly the substrate binding site of UGT1A10 [23]. Studies with other UGT1A10 substrates are underway.
In order to understand the significance of the identification of the estrogen binding site in UGTs, we need to take into consideration the importance of estrogen glucuronidation itself. At physiological concentrations, estrogens can be effectively excreted from the body after sulfation catalyzed by SULTs [29] and this mode of detoxification is very effective at low concentrations However, if estrogens accumulate in the body to levels above normal physiological conditions, the SULTs do not have the capacity to clear the increased levels. However, UGTs can, and do, effectively remove this excess. More importantly, the biotransformation of estrogens via glucuronidation is the only metabolic pathway that can effectively remove both native estrogens and their hydroxylated derivatives.
Therefore, glucuronidation of estrogens serves two different functions. First, by eliminating the excess of native steroids, especially E2, which serve as the ligands for the estrogen receptors (ERα and -β), glucuronidation can control the steady-state concentration of estrogenic ligands for these receptors. Second, glucuronidation and elimination of hydroxylated estrogens from circulation and from target tissues significantly protects against the development of breast cancer caused by an excess of 4-hydroxylated estrogens.
In summary, the novelty of this work is that it represents the first identification of UGT1A10 as the major isoform responsible for the glucuronidation of all native estrogens and their hydroxylated metabolites. This is also the first successful identification of crucial amino acids involved in the estrogen binding site of this or any UGT isoform. It is anticipated that this discovery will constitute a basis for the elucidation of the molecular mechanism of estrogen glucuronidation. Moreover, the localization of the estrogen binding site in UGT1A10 could allow better evaluation of glucuronidation activities of polymorphic variants of UGTA10 or other UGT isoforms involved in estrogen glucuronidation. This information can also be extended to help determine the architecture of the estrogen binding site in other proteins such as ER, CYP450s, SULTs, etc.
Acknowledgments
This work was supported in part by NIH grants DK56226 and DK60109 (to AR-P) and the Academy of Finland grant no. 207535 (to MF).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dutton GJ. Glucuronidation of Drugs and Other Compounds. Boca Raton, Fl: CRC Press Inc; 1980. [Google Scholar]
- 2.Mackenzie PI. The UDP-glucuronosyltransferase multigene family. Raleigh: Toxicology Communications Inc; 1995. [Google Scholar]
- 3.Guillemette C, Belanger A, Lepine J. Metabolic inactivation of estrogens in breast tissue by UDP-glucuronosyltransferase enzymes: an overview. Breast Cancer Res. 2004;6:246–54. doi: 10.1186/bcr936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu BT, Conney AH. Is 2-methoxyestradiol an endogenous estrogen metabolite that inhibits mammary carcinogenesis? Cancer Res. 1998;58:2269–77. [PubMed] [Google Scholar]
- 5.Brooks SC, Horn L. Hepatic sulfation of estrogen metabolites. Biochim Biophys Acta. 1971;231:233–41. doi: 10.1016/0005-2760(71)90272-4. [DOI] [PubMed] [Google Scholar]
- 6.Hobkirk R. Steroid sulfotransferases and steroid sulfate sulfatases: characteristics and biological roles. Can J Biochem Cell Biol. 1985;63:1127–44. doi: 10.1139/o85-141. [DOI] [PubMed] [Google Scholar]
- 7.Guldberg HC, Marsden CA. Catechol-O-methyl transferase: pharmacological aspects and physiological role. Pharmacol Rev. 1975;27:135–206. [PubMed] [Google Scholar]
- 8.Raftogianis R, Creveling C, Weinshilboum R, Weisz J. Estrogen metabolism by conjugation. J Natl Cancer Inst Monogr. 2000:113–24. doi: 10.1093/oxfordjournals.jncimonographs.a024234. [DOI] [PubMed] [Google Scholar]
- 9.Feigelson HS, Henderson BE. Estrogens and breast cancer. Carcinogenesis. 1996;17:2279–84. doi: 10.1093/carcin/17.11.2279. [DOI] [PubMed] [Google Scholar]
- 10.Lepine J, Bernard O, Plante M, Tetu B, Pelletier G, Labrie F, Belanger A, Guillemette C. Specificity and regioselectivity of the conjugation of estradiol, estrone, and their catecholestrogen and methoxyestrogen metabolites by human uridine diphospho-glucuronosyltransferases expressed in endometrium. J Clin Endocrinol Metab. 2004;89:5222–32. doi: 10.1210/jc.2004-0331. [DOI] [PubMed] [Google Scholar]
- 11.Cheng Z, Rios GR, King CD, Coffman BL, Green MD, Mojarrabi B, Mackenzie PI, Tephly TR. Glucuronidation of catechol estrogens by expressed human UDP-glucuronosyltransferases (UGTs) 1A1, 1A3, and 2B7. Toxicol Sci. 1998;45:52–7. doi: 10.1006/toxs.1998.2494. [DOI] [PubMed] [Google Scholar]
- 12.Cheng Z, Radominska-Pandya A, Tephly TR. Studies on the substrate specificity of human intestinal UDP-glucuronosyltransferases 1A8 and 1A10. Drug Metab Dispos. 1999;27:1165–70. [PubMed] [Google Scholar]
- 13.Mojarrabi B, Butler R, Mackenzie PI. cDNA cloning and characterization of the human UDP glucuronosyltransferase, UGT1A3. Biochem Biophys Res Commun. 1996;225:785–90. doi: 10.1006/bbrc.1996.1251. [DOI] [PubMed] [Google Scholar]
- 14.Czernik PJ, Little JM, Barone GW, Raufman JP, Radominska-Pandya A. Glucuronidation of estrogens and retinoic acid and expression of UDP-glucuronosyltransferase 2B7 in human intestinal mucosa. Drug Metab Dispos. 2000;28:1210–6. [PubMed] [Google Scholar]
- 15.Belanger G, Beaulieu M, Marcotte B, Levesque E, Guillemette C, Hum DW, Belanger A. Expression of transcripts encoding steroid UDP-glucuronosyltransferases in human prostate hyperplastic tissue and the LNCaP cell line. Mol Cell Endocrinol. 1995;113:165–73. doi: 10.1016/0303-7207(95)03627-j. [DOI] [PubMed] [Google Scholar]
- 16.Basu NK, Kubota S, Meselhy MR, Ciotti M, Chowdhury B, Hartori M, Owens IS. Gastrointestinally distributed UDP-glucuronosyltransferase 1A10, which metabolizes estrogens and nonsteroidal anti-inflammatory drugs, depends upon phosphorylation. J Biol Chem. 2004;279:28320–9. doi: 10.1074/jbc.M401396200. [DOI] [PubMed] [Google Scholar]
- 17.Chouinard S, Tessier M, Vernouillet G, Gauthier S, Labrie F, Barbier O, Belanger A. Inactivation of the pure antiestrogen Fulvestrant and other synthetic estrogen molecules by UGT1A enzymes expressed in the breast tissue. Mol Pharmacol. 2005 doi: 10.1124/mol.105.015891. [DOI] [PubMed] [Google Scholar]
- 18.Liehr JG. Antioxidant and prooxidant properties of estrogens. J Lab Clin Med. 1996;128:344–5. doi: 10.1016/s0022-2143(96)80002-3. [DOI] [PubMed] [Google Scholar]
- 19.Yue W, Santen RJ, Wang JP, Li Y, Verderame MF, Bocchinfuso WP, Korach KS, Devanesan P, Todorovic R, Rogan EG, Cavalieri EL. Genotoxic metabolites of estradiol in breast: potential mechanism of estradiol induced carcinogenesis. J Steroid Biochem Mol Biol. 2003;86:477–86. doi: 10.1016/s0960-0760(03)00377-7. [DOI] [PubMed] [Google Scholar]
- 20.Liehr JG. Role of DNA adducts in hormonal carcinogenesis. Regul Toxicol Pharmacol. 2000;32:276–82. doi: 10.1006/rtph.2000.1432. [DOI] [PubMed] [Google Scholar]
- 21.Stack DE, Byun J, Gross ML, Rogan EG, Cavalieri EL. Molecular characteristics of catechol estrogen quinones in reactions with deoxyribonucleosides. Chem Res Toxicol. 1996;9:851–9. doi: 10.1021/tx960002q. [DOI] [PubMed] [Google Scholar]
- 22.Guillemette C, Belanger A, Lepine J. Metabolic inactivation of estrogens in breast tissue by UDP-glucuronosyltransferase enzymes: an overview. Breast Cancer Res. 2004;6:246–54. doi: 10.1186/bcr936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiong Y, Bernardi D, Bratton S, Ward MD, Battaglia E, Finel M, Drake RR, Radominska-Pandya A. Phenylalanine 90 and 93 are localized within the phenol binding site of human UDP-glucuronosyltransferase 1A10 as determined by photoaffinity labeling, mass spectrometry, and site-directed mutagenesis. Biochemistry. 2006;45:2322–32. doi: 10.1021/bi0519001. [DOI] [PubMed] [Google Scholar]
- 24.Kuuranne T, Kurkela M, Thevis M, Schanzer W, Finel M, Kostiainen R. Glucuronidation of anabolic androgenic steroids by recombinant human UDP-glucuronosyltransferases. Drug Metab Dispos. 2003;31:1117–24. doi: 10.1124/dmd.31.9.1117. [DOI] [PubMed] [Google Scholar]
- 25.Kurkela M, Garcia-Horsman JA, Luukkanen L, Morsky S, Taskinen J, Baumann M, Kostiainen R, Hirvonen J, Finel M. Expression and characterization of recombinant human UDP-glucuronosyltransferases (UGTs). UGT1A9 is more resistant to detergent inhibition than other UGTs and was purified as an active dimeric enzyme. J Biol Chem. 2003;278:3536–44. doi: 10.1074/jbc.M206136200. [DOI] [PubMed] [Google Scholar]
- 26.Little JM, Williams L, Xu J, Radominska-Pandya A. Glucuronidation of the dietary fatty acids, phytanic acid and docosahexaenoic acid, by human UDP-glucuronosyltransferases. Drug Metab Dispos. 2002;30:531–3. doi: 10.1124/dmd.30.5.531. [DOI] [PubMed] [Google Scholar]
- 27.Luukkanen L, Taskinen J, Kurkela M, Kostiainen R, Hirvonen J, Finel M. Kinetic characterization of the 1A subfamily of recombinant human UDP-glucuronosyltransferases. Drug Metab Dispos. 2005;33:1017–26. doi: 10.1124/dmd.105.004093. [DOI] [PubMed] [Google Scholar]
- 28.Little JM, Kurkela M, Sonka J, Jantti S, Ketola R, Bratton S, Finel M, Radominska-Pandya A. Glucuronidation of oxidized fatty acids and prostaglandins B1 and E2 by human hepatic and recombinant UDP-glucuronosyltransferases. J Lipid Res. 2004;45:1694–703. doi: 10.1194/jlr.M400103-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki T, Miki Y, Nakata T, Shiotsu Y, Akinaga S, Inoue K, Ishida T, Kimura M, Moriya T, Sasano H. Steroid sulfatase and estrogen sulfotransferase in normal human tissue and breast carcinoma. J Steroid Biochem Mol Biol. 2003;86:449–54. doi: 10.1016/s0960-0760(03)00356-x. [DOI] [PubMed] [Google Scholar]