

Abstract
To investigate the role of the Fhit gene in carcinogen induction of neoplasia, we have inactivated one Fhit allele in mouse embryonic stem cells and produced (129/SvJ × C57BL/6J) F1 mice with a Fhit allele inactivated (+/−). Fhit +/+ and +/− mice were treated intragastrically with nitrosomethylbenzylamine and observed for 10 wk posttreatment. A total of 25% of the +/+ mice developed adenoma or papilloma of the forestomach, whereas 100% of the +/− mice developed multiple tumors that were a mixture of adenomas, squamous papillomas, invasive carcinomas of the forestomach, as well as tumors of sebaceous glands. The visceral and sebaceous tumors, which lacked Fhit protein, were similar to those characteristic of Muir–Torre familial cancer syndrome.
Keywords: esophageal, gastric cancer, N-nitrosomethylbenzylamine, carcinogen-induced tumorigenesis, Fhit knockout mice, tumor suppressor gene
Since it was first noted that human chromosomal fragile sites mapped to chromosome bands that were nonrandomly altered by translocations or deletions in neoplasias, it has been proposed that the recombinogenicity of fragile sites, possibly enhanced by environmental carcinogens, could lead to altered expression of oncogenes or tumor suppressor genes at fragile sites (1). The corollary of the proposal is that alterations to expression of genes at fragile sites contribute to clonal expansion of the neoplastic cells. FHIT is thus far the only example of a gene at a constitutive fragile region and shows many hallmarks of a tumor suppressor gene (2). The FHIT gene is altered by deletion or translocation in a large fraction of many types of cancer, including lung, cervical, gastric, and pancreatic (2–8). Fhit protein is lost or reduced in the majority of these cancers, in a large fraction of other cancer types (9–11), and preneoplastic lesions in the lung (12). Nevertheless, acceptance of Fhit as a tumor suppressor has not been universal (13), with some reports suggesting that fragility of the locus alone could account for the occurrence of clonal or oligoclonal genetic alterations at FHIT in cancers. The murine Fhit locus is similar to its human homolog (14, 15), encompasses a common fragile site, and is altered in murine cancer cell lines. To define the role of Fhit protein in cancer development, we have established a strain of Fhit +/− mice and have compared the frequency of carcinogen-induced tumor formation in Fhit +/+ and +/− mice by using the established N-nitrosomethylbenzylamine (NMBA) esophageal/gastric cancer model (16). On bioactivation, NMBA produces benzaldehyde and an electrophilic methylating agent (17) that methylates DNA, resulting in the formation of the promutagenic adduct, O6-methylguanine (18). NMBA was reported to induce both esophageal and forestomach tumors when administered by gavage or in the drinking water (16, 19). In a series of studies on esophageal tumor induction by NMBA in rats and mice, Fong and colleagues (16, 20, 21) have developed a model system that requires low doses of NMBA. We have used this model system to test the effects of NMBA administration on Fhit +/− mice. By 10 wk after NMBA exposure, all of the Fhit +/− mice developed a spectrum of visceral and skin tumors similar to those observed in a human cancer syndrome, Muir–Torre syndrome (MTS), caused by deficiency in a mismatch repair gene.
Materials and Methods
Immunoblot Analysis of Murine Fhit Protein.
A glutathione S-transferase (GST) gene-fused murine Fhit cDNA recombinant was cloned into a bacterial expression vector. In the resulting construct, pGEX4T1-mFhit, the murine Fhit protein coding sequence was placed downstream of the GST gene. GST-mFhit fusion protein was produced in the BL21 bacterial strain (22), and, after purification, GST-mFhit was cleaved with thrombin protease. Polyclonal antiserum against purified mouse Fhit protein was raised commercially (Cocalico Biologicals, Reamstown, PA) and used at 1:8,000 dilution in immunoblot and 1:4,000 in immunohistochemistry experiments. Specificity was tested on protein lysates from murine cells and tissues with and without endogenous or exogenous Fhit protein expression.
Immunohistochemistry.
After antigen retrieval, endogenous peroxidase was inhibited with 3% hydrogen peroxide, and nonspecific binding sites were blocked with normal goat serum (21). Slides were incubated with primary rabbit anti-murine Fhit (1:4,000 dilution, overnight), followed by incubation with biotinylated goat anti-rabbit antibody. Slides were then incubated with strepavidin horseradish peroxidase (Dako; 1:1,000 dilution). Fhit protein was localized by a final incubation with 3,3′-diaminobenzidine tetrahydrochloride (Sigma). Slides were counterstained with hematoxylin, dehydrated, and coverslipped.
Carcinogenicity Study.
(C57BL/6J × 129/SvJ) F1 mice (B6129F1s) that were Fhit +/+ or +/− were produced in the Kimmel Cancer Center (KCC) animal facility. Eighteen Fhit +/+ and 22 Fhit +/− mice (30–46 wk) were given eight intragastric doses of NMBA (Ash Stevens, Detroit, MI) over the course of 3 wk at 2 mg/kg body weight. About 50% of the mice were killed 6 wk after the final NMBA dose and the remaining mice at 10 wk. Tumor incidence differences were analyzed by two-tailed Fisher's exact test (23). For comparison, four untreated Fhit +/− mice (54–59 wk old) and one untreated Fhit +/+ mouse (59 wk old) were similarly autopsied. At autopsy, whole esophagi and stomachs were removed and opened longitudinally. Other tissues with apparent tumors were also examined. The number of animals bearing tumors in the esophagus, forestomach, squamocolumnar junction with the glandular stomach, and other tissues were scored. Tissues were fixed in buffered formalin and examined histologically after hematoxylin and eosin (H&E) staining for the presence of hyperkeratosis, parakeratosis, dysplasia, papillomas, adenomas, and carcinomas.
MTS Cases.
Archival paraffin blocks for two cases of MTS were available from the Surgical Pathology archives of Thomas Jefferson University Hospital (case 1) and the Christiana Hospital (case 2). For case 1, paraffin blocks for two sebaceous tumors were available, and, for case 2, one sebaceous tumor block. Normal and tumor cells were microdissected from the paraffin blocks, and DNA was prepared. Tissue sections were analyzed for Fhit expression by immunohistochemistry as described (9). Germline DNA was prepared from peripheral blood lymphocytes of the MTS patients.
Analysis of Tumor DNAs.
Microsatellite instability analysis (MSI).
Portions of the large sebaceous tumors were lysed in buffer containing 0.6% SDS and 50 μg/ml proteinase K and tumor DNAs prepared by standard phenol-chloroform extraction and ethanol precipitation. MSI was assayed by PCR amplification with primers for D1Mit4, D2Mit13, D3Mit1, D3Mit203, D6Mit59, D8Mit14, D10Mit2, D14Nds1, D17Mit123, and D19Mit36 for murine alleles (24) and primers D2S123, D3S1298, D18S35, BAT25, and BAT26 (25, 26) for human alleles. Primers were purchased from Research Genetics (Huntsville, AL) or the KCC Nucleic Acid Facility. Samples were amplified in a reaction mixture containing 50 ng template DNA, 10 mM Tris⋅HCl (pH 8.3), 50 mM KCl, 0.1 mg/ml gelatin, 1.5 mM MgCl2, 12.5 μM each dNTP, 0.5 unit Taq polymerase, 20 ng primers, and 1 μCi [32P]dCTP, for 30 cycles of 94°C for 30 s, 57°C for 30 s, and 72°C for 30 s. PCR product (1 μl) was mixed with 9 μl sequencing stop buffer (95% formamide/0.05% bromophenol blue/0.05% xylene cyanol FF/10 mM NaOH) and denatured at 94°C for 8 min. Then, 7 μl of this mixture were loaded onto a 6% acrylamide:bis (19:1), 8 M urea gel for electrophoresis at 80 W for 2–3 h. The gel was dried and exposed to x-ray film overnight.
Fhit sequence analysis.
Primer pairs flanking each of the human FHIT exons (22) and the mouse Fhit coding exons were used in PCR amplification of DNA from MTS cases or mouse tumors, respectively. Primer pairs surrounding the mouse Fhit exons were previously published (14) for exons 5 and 6 or newly designed for exon 4 (mfiex4F: GTGTTCTTCACAGTTACG; mfiex4R: CAATTCTATACATTCTTTGC), exon 7 (mfiex7F: GGCCTGCTGGATAATTCATA; mfiex7R: AGATAACATAATGAAAGAGC), exon 8 (mfiex8F: CACTGTCAAGTCAAAATATAG; mfiex8R (2): GGCCTTGTGACTAAATAATAA), and exon 9 (mfiex9F: CTCTCTCCTCCAATGTTAT; mfiex9R: AAGGTTAGCAGAAAGAGG). The products were purified with a PCR purification kit (Qiagen, Chatsworth, CA) before sequencing with Taq DyeDeoxy Terminator Cycle Sequencing Kits (Applied Biosystems). Sequencing reaction products were electrophoresed and recorded on a 377 DNA sequencer (Applied Biosystems).
Southern blot analysis.
To examine the integrity of the murine Fhit alleles in tumors, DNA from several sebaceous tumors was digested with restriction enzyme XbaI, electrophoresed on 0.8% agarose gels, and transferred to nylon membranes. After drying, membrane-bound DNAs were hybridized to 32P-labeled full-length murine Fhit cDNA or to exons 1–4, 4–10, or 7–9, to determine whether portions of Fhit alleles were deleted. Densitometry analysis of specific lanes of Southern blot autoradiographs was performed. Quantitation of signals was performed by using imagequant software (Molecular Dynamics).
Results
Production of Fhittm2KCC mice.
A 129/SvJ mouse genomic fragment encompassing Fhit exon 5 was cloned, and a termination codon was introduced into the exon 5 coding region. Exon 5 is the first protein coding exon, so that the termination codon prevents translation of a protein. There are no downstream Met codons that can initiate translation of a stable protein (27). This altered genomic clone was inserted into a derivative of the Mc1-TK vector along with the PGK Neo bpa gene (Fig. 1). RW4 ES cells (Genome Systems, St. Louis) were transfected with this Fhit targeting vector, and embryonic stem (ES) cell clones were selected with the vector integrated through homologous recombination into an endogenous Fhit allele (Fig. 1). The targeted ES cell clones were introduced into 3.5-day blastocysts to generate chimeras. Each of the chimeras transmitted the defective Fhit allele to offspring, as determined by Southern blot analysis of tail DNA from agouti pups. Progeny from one chimera (+/Fhittm2KCC referred to as Fhit +/− mice) were intercrossed, and genotyping revealed that all three genotypes were represented, with a ratio close to the expected Mendelian distribution. Disruption of the Fhit locus in the knockout mice was further verified by PCR analysis, as illustrated in Fig. 1. To confirm absence of a functional Fhit gene in Fhit −/− mice, weanling mice were sacrificed and organs removed for assessment of Fhit protein expression by immunoblot analysis and immunohistochemistry. Immunoblot analysis showed that Fhit −/− mouse tissues were entirely negative for Fhit protein (see Fig. 2); immunohistochemical detection of Fhit protein in Fhit +/+, +/−, and −/− kidney sections showed absence of Fhit protein in Fhit −/− sections and reduced expression in Fhit +/− sections (data not shown).
Figure 1.

Murine Fhit genomic locus, targeting and screening strategy. The top line represents the Fhit genomic locus surrounding exon 5. The middle line depicts the targeting vector with a 6.6-kb HindIII (H)–PstI (P) fragment with a termination codon introduced into exon 5. The targeted locus after homologous recombination is shown at the bottom with the probe used for Southern blot screening of ES colony and progeny DNA after BamHI (B) cleavage. Positions of the primers used for PCR amplification of progeny DNA, to identify wild-type (F,R) and targeted (N,R) alleles, are shown. Restriction enzyme sites are shown for EcoRV (EV), EcoRI (E1), SphI (Sp), SacI (S), NotI (N), NcoI (Nc), and Pme1 (Pm). The 5′→3′ sequences of the three primers F, R, and N are, respectively: CTTGAATCTAGGCTGCATTCTAGCGAG, GATTCCTTGCTTACCTTTTGGGGATGG, and TGGGCTCTATGGCTTCTGAGGC. The first reaction product is a wild-type fragment of ≈450 bp containing exon 5; the second product is a mutant fragment of ≈280 bp spanning from the Neo selection gene to intron 5. PCR conditions were: denaturation 94°C, 30 s; annealing 62°C, 30 s; elongation 72°C, 30 s; 35 cycles.
Figure 2.

Absence of Fhit protein in the Fhit −/− mice. Lysates from tissues of Fhit −/− mice were tested for expression of Fhit by immunoblot analysis of mouse tissue lysates: lane 1, Fhit +/+ lung; lane 2, +/+ liver; lane 3, +/+ kidney; lane 4, Fhit −/− liver; lane 5, −/− kidney.
NMBA Induction of Tumors.
At 6 wk after the final NMBA dose, there was no visible difference in the Fhit +/+ and +/− mice. By 10 weeks after the final dose, three of the Fhit +/− mice showed tumors in the subcutis of the abdomen. On autopsy at 10 wk, more than 50% of the Fhit +/− mice exhibited one or more of these tumors in the abdominal, mammary, or axial area, sometimes invading muscle tissue. The tumors were removed for fixation before examination of the esophagus/forestomach. On inspection of whole esophagus and stomach tissues at 6 wk after treatment, 7 of 10 Fhit +/− mice showed one or more small tumors, whereas 2 of 10 Fhit +/+ mice showed a very small tumor of the esophagus or forestomach. At 10 wk, 11 of 12 Fhit +/− mice showed apparent tumors, usually multiple, in the forestomach, the squamocolumnar junction with the hind stomach, and/or in other tissues (Table 1); 2 of 8 Fhit +/+ mice exhibited tumors. An untreated Fhit +/+ mouse (59 wk) showed no abnormalities of skin, esophagus, forestomach, or junction. Three of four untreated Fhit +/− mice (54–59 wk) showed a small abdominal tumor in the skin.
Table 1.
Tumor induction in Fhit +/+ and +/− mice at 10 wk after NMBA treatment
| Id#, Sex | Phenotype
|
||||
|---|---|---|---|---|---|
| Esoph | Fst | SCJ | Subcutis | Histology comments | |
| ++ | |||||
| 40M | — | sm. T | — | — | adenoma |
| 37F | — | — | — | — | |
| 35F | — | — | — | — | |
| 29M | — | — | thick | — | |
| 25F | — | — | 7Ts | — | sq pap |
| 24F | thick | thick | — | — | hyperplasia |
| 22M | — | — | — | — | |
| 23M | — | — | — | — | |
| +/− | |||||
| 41M | — | 2Ts | Ts | — | sq ca fst, sq pap SCJ |
| 39M | — | T | Ts | — | sq pap fst |
| 38M | — | Ts | — | — | sq pap, adenoma fst |
| 31M | — | Ts | — | T,8×6 | sq pap, sq ca fst |
| 36F | T | Ts | Ts | T | sq pap esoph, fst, SCJ |
| 30M | — | T | — | — | sq pap fst |
| 34M | — | — | — | T,9×5 | seb T |
| 33M | T | Ts | — | T,7×6 | sq pap esoph, fst; seb T |
| 32M | — | T | — | T,5×6 | sq pap fst; seb T |
| 28M | — | — | Ts | — | |
| 27M | — | lg. T | Ts | 2Ts | sq pap SCJ; seb Ts |
| 21M | — | lg. T | — | T,7×4 | fst sq pap; seb T |
Some tumors were photographed, measured, or samples taken for DNA. A total of 92% of +/− animals and 25% of +/+ animals showed evidence of tumorigenesis by visual inspection. Average age of +/+ group 38 wk (range from 30 to 46 wk), average age of +/− group 39.9 wk (range from 34 to 46 wk); T, tumor; sq, squamous; pap, papilloma; seb, sebaceous; SCJ, squamocolumar junction; esoph, esophagus; fst, forestomach.
Histological and Immunohistochemical Analyses.
Histological examination revealed an esophageal squamous papilloma in 1 of 10 Fhit +/+ mice at 6 wk after NMBA (data not shown) and in Fhit +/− mice 33 and 36 at 10 wk post NMBA (Table 1). At 6 wk post NMBA treatment, Fhit +/− mice had more tumors than Fhit +/+ mice (70% vs. 20%; summarized in Table 2). At 10 wk post NMBA treatment, the difference between tumor burden in Fhit +/− mice (100%) compared with +/+ mice (25%) was highly significant (Tables 1 and 2). Most of the tumors were in the forestomach and squamocolumnar junction with the glandular stomach, as observed previously in B6 mice (16). Histological examination of the abdominal tumors of the Fhit +/− mice showed that they derived from sebaceous glands (Table 1, and Fig. 3) and were identical to the sebaceous tumors that are the hallmark of MTS, a variant of hereditary nonpolyposis colorectal cancer (HNPCC) syndrome. The small abdominal tumors observed in three of four untreated Fhit +/− mice were also sebaceous tumors. Other tissues of the untreated mice were normal. Sections from the fixed tissues were analyzed by immunohistochemical detection of Fhit protein expression, to determine whether the remaining Fhit allele had been inactivated in tumors. Epithelial cells lining the esophagus, forestomach, and junction with the glandular stomach were positive for Fhit expression. In the esophagus, the basal epithelial cells stain less strongly than the overlying squamous cells (see Fig. 3A). All of the squamous papillomas and other tumors were Fhit-negative, as illustrated in the examples shown in Fig. 3 B–F. Note especially the lack of Fhit expression in the sebaceous tumors shown in Fig. 3 E and F. To compare the mouse sebaceous tumors to sebaceous tumors from MTS cases, sebaceous tumor sections from two MTS cases were analyzed for expression of human Fhit. Fhit protein was detected in normal human hair follicle and sebaceous gland (Fig. 4 A and B) from the MTS tumor sections. Fhit protein was not expressed in two human sebaceous tumors from case 1 (see Fig. 4D for example) but was expressed in the sebaceous tumor from case 2 (not shown).
Table 2.
Incidence of tumors induced by multiple low NMBA doses in Fhit +/+ and +/− mice
| Wk post-treatment | Fraction of tumor-bearing animals
|
|||||
|---|---|---|---|---|---|---|
| Fhit | Esoph* | Fst | SCJ | Seb | Tumor-bearing mice | |
| 6 | +/+ | 1/10 | 1/10 | 0/10 | ND | 2/10 |
| 6 | +/− | 0/10† | 5/10 | 4/10 | ND | 7/10 |
| P = 1.0 | P = 0.14 | P = 0.09 | P = 0.07 | |||
| 10 | +/+ | 0/8 | 1/8 | 1/8 | 0/8 | 2/8 |
| 10 | +/− | 2/12 | 10/12 | 5/12 | 7/12 | 12/12 |
| P = 0.49 | P = .005 | P = 0.32 | P = 0.015 | P = 0.0007 | ||
Number of mice with tumors/respective number of mice.
† One esophagus showed dysplasia. The tumors at 6 wk were mainly squamous papillomas. The tumors at 10 wk in the +/− mice were mostly squamous papillomas, but two Fhit +/− mice had squamous carcinomas of the forestomach or junction; one Fhit +/+ and one +/− mouse at 10 wk showed a small adenoma of the forestomach. ND, not detected.
Figure 3.
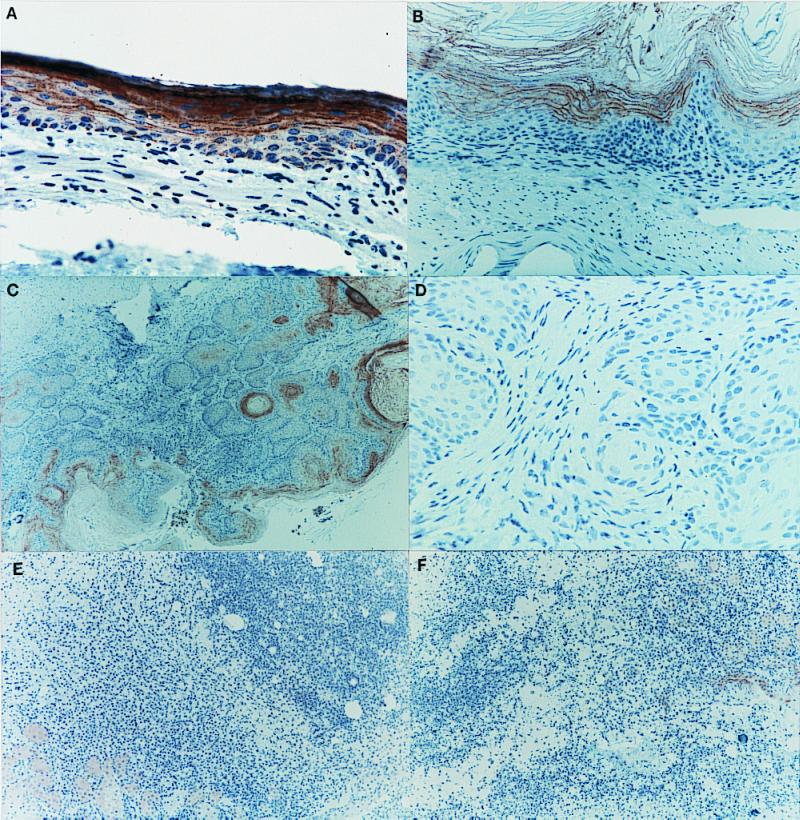
Immunohistochemical detection of Fhit expression. (A) Fhit expression in normal esophageal epithelium (×200) of Fhit +/+ mouse 23 at 10 wk post NMBA; the brown chromogen represents the Fhit protein. (B) Lack of Fhit expression in a squamous papilloma of the forestomach (×200) in Fhit +/− mouse 33 at 10 wk post NMBA; (C) Absence of Fhit expression in a squamous papilloma of the junction (×200) in Fhit +/+ mouse 25 at 10 wk post NMBA; (D) Lack of Fhit expression in an invasive squamous carcinoma of the forestomach (×100) in Fhit +/− mouse 31 at 10 wk post NMBA; (E) Lack of Fhit expression in a sebaceous tumor (×100) in Fhit +/− mouse 27 at 10 wk post NMBA; (F) Absence of Fhit protein in a sebaceous tumor (×100) in Fhit +/− mouse 21 at 10 wk post NMBA.
Figure 4.

Immunohistochemical detection of human Fhit in MTS tumors. (A) Fhit expression in normal hair follicle (×200); note that dense keratin horn shows nonspecific staining; (B) Fhit expression in normal sebaceous gland (×200); (C) H&E staining of an MTS case 1 sebaceous tumor; (D) Lack of Fhit expression in most cells of the case 1 sebaceous tumor.
Genotypic Analysis.
Murine tissues.
DNA was prepared from tail biopsies, as well as portions of the larger tumors of Fhit +/+ and +/− mice, to examine the integrity of the Fhit loci in tumors. To determine whether the wild-type Fhit allele was deleted or rearranged in tumors, the DNA was typed for the presence of wild-type or targeted Fhit alleles by PCR amplification and both exon 5 alleles were detected (not shown). Tail and tumor DNAs were also digested with restriction enzymes and typed for presence of wild-type or altered Fhit alleles by Southern blot. The results shown in Fig. 5 reveal the presence of both wild-type and targeted Fhit exons 5 in tumors 21, 27, and 31 (Fig. 5, lanes 1, 3, and 4). At least one copy of all other Fhit exons is retained in the tumors (compare lanes 1 and 2). Additional analyses of BamHI or XbaI digested tumor DNAs hybridized to probes for mouse exons 1–4, 7–9, and 4–10 did not reveal rearrangements or homozygous deletions of Fhit loci, although hemizygous deletions could not be ruled out. For example, densitometry analysis to compare intensity of bands for specific Fhit exons in lanes 1–3 of Fig. 5 showed that the signal for exon 1 in lane 1 (tumor from mouse 21) was half as strong as the signal for exon 1 in lanes 2 and 3 relative to other exons. The signal for exon 5 in lanes 1 and 3 (tumor from mouse 21 and 27) is split into two bands, one near the top and one at the bottom of the lanes, representing the wild-type and mutant exon 5, respectively. To determine whether NMBA had induced mutations in the wild-type Fhit allele, DNA from sebaceous tumors from mice 21, 27, and 31 and from squamous papillomas in mice 25 and 27 were also examined for mutations within Fhit exons 4–9. Primers flanking exons were used to amplify and sequence exons 4–9 in these tumors. No mutations were detected. Human MTS syndrome is usually, if not always, caused by inactivation of mismatch repair genes, and MTS tumors usually exhibit MSI. Tail and tumor DNAs were used as templates in PCR amplifications of 10 microsatellite loci in a search for MSI. Results for three of these loci are shown in Fig. 6. MSI was not observed at any of the mouse loci tested, demonstrating that the mouse MTS-like disease does not have an underlying mismatch repair defect. Thus, although Fhit protein is inactivated in all of the NMBA-induced tumors, presumably through alteration of wild-type Fhit alleles within the mouse fragile site, we do not know the mechanism of inactivation in these tumors. We conclude that the wild-type Fhit allele is probably altered through deletion of one or more exons and that the deletions are obscured by presence of the recombinant allele.
Figure 5.

Integrity of Fhit loci in murine tumors. DNA from tails and sebaceous tumors was cleaved with XbaI, electrophoresed, transferred to a membrane, and hybridized to a 32P-labeled full-length Fhit cDNA probe. Fhit exons are indicated on the left; the asterisk indicates the inactivated Fhit exon 5. Lanes 1, 3, and 4 contained DNAs from sebaceous tumors from Fhit +/− mice 21, 27, and 31; lane 2 contained DNA from the tail of Fhit +/+ mouse 25, and lane 5 contained DNA from a Swiss mouse 3T3 cell line, which exhibits a variant-sized exon 3 (obscured by another fragment) because of a polymorphism. The Fhit +/+ and +/− mice are B6129F1s, which exhibit two different alleles of exon 8. (Right) The agarose gel before blotting of the digested DNAs to the membrane; this gel illustrates that amounts of DNA loaded in individual lanes varied from ≈1 μg (lane 4) to ≈10 μg (lane 2).
Figure 6.

Assessment of MSI in tumors. DNA templates from mouse and human tumors and controls were amplified by using primers flanking microsatellite alleles. Labeled amplified products were run on PAGE gels, dried, and exposed. The D6Mit59, D19Mit36, and D17Mit123 panels represent murine alleles amplified from Fhit +/− mouse 27 forestomach tumor (lane 1), Fhit +/+ mouse 25 forestomach tumor (lane 2), Fhit +/+ mouse 25 tail (lane 3), Fhit +/− mouse 21 sebaceous tumor (lane 4), Fhit +/− mouse 27 sebaceous tumor (lane 5), Fhit +/− mouse 27 second sebaceous tumor (lane 6), Fhit +/− mouse 31 sebaceous tumor (lane 7), K1735 mouse melanoma cell line (lane 8), NP3 mouse cell line (lane 9), negative control (no DNA) (lane 10). No MSI was observed in the mouse tumors for the three markers shown. The D18S35 and D3S1295 panels represent germline and tumor DNA from a human MTS case: DNA from peripheral blood lymphocytes (lane 1), DNA from sebaceous tumor 185 from the same individual (lane 2), lymphocyte DNA (lane 3), and DNA from sebaceous tumors 185 (lane 4) and 9029 (lane 5) from the same individual. These sebaceous tumors showed MSI at each allele successfully amplified.
Human tissues.
Not all MTS cases have been shown to exhibit germline mutations of MSH2 or MLH1, nor do all MTS tumors exhibit MSI, the hallmark of mismatch repair deficiency. It is possible that some MTS cases with Fhit-negative tumors are caused by germline mutation in the FHIT gene. To address this question, we determined whether the MTS cases used in this study exhibited wild-type germline FHIT alleles. Restriction enzyme digestion of germline DNA from the two MTS cases did not reveal alterations of the FHIT locus (data not shown). Each FHIT exon was amplified from the two MTS cases and the products sequenced. All germline FHIT exons from both cases showed wild-type sequences. The majority of MTS cases are because of germline mutations of the mismatch repair gene MSH2. Thus, MTS tumors would be expected to show MSI. DNA from the two Fhit-negative sebaceous tumors of MTS case 1 and the Fhit-positive tumor from case 2 did exhibit MSI with several of the five markers tested (for examples, see Fig. 6, Bottom).
Discussion
The Fhit +/− Phenotype.
The purpose of this study was to determine whether inactivation of one Fhit allele in mice would cause a tumor phenotype, and to determine whether the phenotype would be influenced by carcinogen treatment. Observation of sebaceous tumors in three of four untreated Fhit +/− mice by 1 yr of age provides a partial answer to the first question. The full spectrum of tumors that will develop spontaneously in Fhit +/− and Fhit −/− mice is not yet known. A full 100% of NMBA-treated Fhit +/− mice exhibited tumors compared with 25% of the treated Fhit +/+ mice, a highly significant difference, and none of the +/+ mice developed sebaceous tumors. Thus, absence of one Fhit allele caused susceptibility to sebaceous tumors and carcinogen induction of gastric tumors. Whether the development of sebaceous tumors in Fhit +/− mice was affected by the NMBA treatment is less clear. As shown in Table 1, 5 of 12 Fhit +/− mice (under 1 yr of age) showed large (>5 × 5 mm) sebaceous tumors, 3 of which were noted before autopsy. Sebaceous tumors have not been observed in untreated mice, except by autopsy, which revealed small s.c. tumors (<2 × 2 mm) in three of four mice over 1 yr old. We know that the tumors in both mouse strains do not express Fhit protein; NMBA treatment has resulted in inactivation of both fragile Fhit alleles in the Fhit +/+ mice, but it was necessary to inactivate only one Fhit allele in the +/− mice, thus enhancing the frequency of tumor development, analogously to the 2-hits vs. 1-hit required in human sporadic vs. familial cancers. Because the only genetic difference between the Fhit +/+ and +/− mice is the targeted Fhit allele in the +/− mice, we believe that the second Fhit allele is the gatekeeper in tumor development, although the carcinogen has undoubtedly caused mutations of other suppressor genes in tumors of both mouse strains. Because Fhit +/− and −/− mice are fertile, long-lived, and sensitive to carcinogen, they will serve as useful models for carcinogen-induction of tumors of various organs.
The Role of NMBA.
Although carcinogen treatment increases the frequency of occurrence of tumors in Fhit +/− mice, spontaneous tumors do occur. Thus, the question arises—what is the role of the carcinogen, NMBA, in producing the MTS-like disease in Fhit haploinsufficient mice? We propose that the carcinogen fulfills a role similar to the role of mismatch repair deficiency in human MTS cases—both carcinogen and mismatch repair deficiency increase the frequency of alteration of the fragile Fhit locus, allowing selective growth of Fhit-negative tumors. Possibly, in the presence of the O6-methylguanine mispairs, the Msh2-Msh6 complexes delay the already late-replicating Fhit locus, so that replication is still incomplete in G2/M, leading to deletions in the fragile Fhit locus. The organ specificity of NMBA is because of the presence of esophageal cytochrome P450 enzymes that bioactivate the carcinogen (17). Although the N-7 position of guanine in DNA is the major site of alkylation, methylation at the O-6 position is more relevant for the biological activity (18), because the O-6-methylguanine adduct is associated with base mispairing and mutagenesis. As discussed above, Fhit +/− mice develop sebaceous tumors spontaneously, although the sebaceous tumors in the NMBA-treated Fhit +/− mice were larger and more numerous. How NMBA treatment affects development or progression of the sebaceous tumors will require further study.
Muir–Torre Syndrome.
The coexistence of one or more sebaceous tumors with one or more visceral carcinomas was described by Muir; since then, more than 150 cases have been reported (25). MTS is familial (28) and has been found in families with HNPCC (29). The most frequently observed internal neoplasm is colorectal carcinoma; thus, the syndrome shares clinical and pathological characteristics with HNPCC. A large subgroup of MTS cases exhibit MSI and germline mutations in MSH2 or MLH1 genes (25). The Fhit-deficient mouse tumors do not show MSI, and loss of Fhit expression plays a role in their MTS-like disease; thus, it is unlikely that the mouse syndrome involves mismatch repair deficiency. We believe that, in the mouse tumors, the second Fhit allele was inactivated through deletion of one or more exons and that Fhit protein loss played a gatekeeper role in tumor formation. We know from studies of the human FHIT locus that biallelic deletions can be observed and characterized by scanning the ≈1.5-Mb locus by PCR amplification using primer pairs spaced at 10–50 kb pair intervals. In the case of the mouse tumors, we expect partial deletion of only the wild-type allele. This type of deletion is difficult to observe in DNA from small tumors with noncancerous cells intermixed. Thus, the mechanism of inactivation of the wild-type Fhit allele is not known. For a sebaceous tumor from Fhit +/− mouse 21, we observed that the exon 1 signal was diminished by half on Southern blot, suggesting the possible absence of one Fhit exon 1, presumably from the wild-type allele. This would be consistent with the suggestion that the epicenter of mouse Fhit fragility was shifted toward the 5′ end of the gene (15) rather than being centered between exons 3 and 6 as in the human FRA3B (27).
If human and mouse MTS cases arise through similar mechanisms, then the FHIT gene may be a target of damage in a fraction of mismatch repair deficient tumors, especially those with MSH2 deficiency, leading to Fhit protein loss and clonal expansion of Fhit-negative cells. If Fhit inactivation is a frequent result of mismatch repair deficiency, and a frequent pathway to MTS, then Fhit +/− mice could be considered predisposed to MTS. The frequency of inactivation of the FHIT gene in human mismatch repair deficiency syndromes could be determined by examination of colon and other tumors with MSI for Fhit protein expression. In fact, it was previously observed that two of three human pancreatic cancer cell lines with high MSI had homozygous deletions within FHIT (30). Interestingly, Msh2 (24) and Msh6 (31) null mice exhibit sebaceous tumors at a low frequency, suggesting that crossing Fhit-deficient mice with Msh2-deficient mice would lead to increased frequency of sebaceous and other tumors, compared with the spontaneous tumor frequency of either parental mouse strain.
Acknowledgments
We thank Karl Smalley for statistical analysis, Almeta Mathis for manuscript preparation, Dr. Tommaso Dragani for helpful discussion and critical reading of the manuscript, and the KCC transgenic facility for generation of chimeric founder mice. This work was supported by Grants CA21124 and CA56336 from the National Cancer Institute, by Grant 97B115-REV from the American Institute for Cancer Research, Grant ME99–105 from the Pennsylvania Department of Health, and by a generous gift from Mr. George Strawbridge.
Abbreviations
- NMBA
N-nitrosomethylbenzylamine
- H&E
hematoxylin and eosin
- MTS
Muir–Torre syndrome
- MSI
microsatellite instability
- ES
embryonic stem
- HNPCC
hereditary nonpolyposis colorectal cancer
- GST
glutathione S-transferase
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.080063497.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.080063497
References
- 1.Yunis J J, Soreng A L. Science. 1984;226:1199–1204. doi: 10.1126/science.6239375. [DOI] [PubMed] [Google Scholar]
- 2.Ohta M, Inoue H, Cotticelli M G, Kastury K, Baffa R, Palazzo J, Siprashvili Z, Mori M, McCue P, Druck T, et al. Cell. 1996;84:587–597. doi: 10.1016/s0092-8674(00)81034-x. [DOI] [PubMed] [Google Scholar]
- 3.Sozzi G, Veronese M L, Negrini M, Baffa R, Cotticelli M G, Inoue H, Tornielli S, Pilotti S, DeGregorio L, Pastorino V, et al. Cell. 1996;85:17–26. doi: 10.1016/s0092-8674(00)81078-8. [DOI] [PubMed] [Google Scholar]
- 4.Hendricks D T, Taylor R, Reed M, Birrer M J. Cancer Res. 1997;57:2112–2115. [PubMed] [Google Scholar]
- 5.Greenspan D L, Connolly D C, Wu R, Lei R Y, Vogelstein J T C, Kim Y-T, Mok J E, Munoz N, Bosch X, Shah K, Cho K R. Cancer Res. 1997;57:4692–4698. [PubMed] [Google Scholar]
- 6.Baffa R, Veronese M L, Santoro R, Mandes B, Palazzo J P, Rugge M, Santoro E, Croce C M, Huebner K. Cancer Res. 1998;58:4708–4714. [PubMed] [Google Scholar]
- 7.Simon B, Bartsch D, Prasnika N, Munch K, Blum A, Arnold R, Goke B. Cancer Res. 1998;58:1538–1587. [PubMed] [Google Scholar]
- 8.Sorio C, Baron A, Orlandini S, Zamboni G, Pederzoli P, Huebner K, Scarpa A. Cancer Res. 1999;59:1308–1314. [PubMed] [Google Scholar]
- 9.Hadaczek P, Siprashvili Z, Markiewski M, Domagala W, Druck T, McCue P A, Pekarsky Y, Ohta M, Huebner K, Lubinski J. Cancer Res. 1998;58:2946–2951. [PubMed] [Google Scholar]
- 10.Ingvarsson S, Agnarsson B A, Sigbjornsdottir B I, Kallioniemi O-P, Barkardottir R B, Kovatich A, Schwarting R, Hauck W W, Huebner K, McCue P A. Cancer Res. 1999;59:2682–2689. [PubMed] [Google Scholar]
- 11.van Heerden W F P, Swart T J P, van Heerden M B, van Rensburg E J, Englebecht S, Dreyer L, Huebner K. J Oral Pathol Med. 1999;28:433–437. doi: 10.1111/j.1600-0714.1999.tb02102.x. [DOI] [PubMed] [Google Scholar]
- 12.Sozzi G, Pastorino U, Moiraghi L, Tagliabue E, Pezzella F, Ghirelli C, Tornielli S, Sard L, Huebner K, Pierotti M A, et al. Cancer Res. 1998;58:5032–5037. [PubMed] [Google Scholar]
- 13.Le Beau M M, Drabkin H, Glover T W, Gemmell R, Rassool F V, McKeithan T W, Smith D. Genes Chromosomes Cancer. 1998;21:281–289. doi: 10.1002/(sici)1098-2264(199804)21:4<281::aid-gcc1>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 14.Pekarsky Y, Druck T, Cotticelli M G, Ohta M, Shou J, Mendrola J, Montgomry J C, Buchberg A M, Manenti G, Fong L Y Y, et al. Cancer Res. 1998;58:3401–3408. [PubMed] [Google Scholar]
- 15.Glover T W, Hoge A, Miller D E, Ascara-Wilke J E, Adam A, Dagenais S L, Wilke C M, Dierick H A, Beer D G. Cancer Res. 1998;58:3409–3414. [PubMed] [Google Scholar]
- 16.Fong L Y Y, Magee P N. Cancer Lett (Shannon, Irel) 1999;143:63–69. doi: 10.1016/s0304-3835(99)00191-3. [DOI] [PubMed] [Google Scholar]
- 17.Labuc G E, Archer M C. Cancer Res. 1982;42:3181–3186. [PubMed] [Google Scholar]
- 18.Fong L Y Y, Lin H J, Lee C H. Int J Cancer. 1979;23:679–682. doi: 10.1002/ijc.2910230514. [DOI] [PubMed] [Google Scholar]
- 19.Sander J, Schweinsberg F. Z Krebsforsch. 1973;19:157–161. [PubMed] [Google Scholar]
- 20.Fong L Y Y, Lee J S K, Chan W C, Newberne P M. J Natl Cancer Inst. 1984;72:419–425. [PubMed] [Google Scholar]
- 21.Fong L Y Y, Lau K-M, Huebner K, Magee P N. Carcinogenesis. 1997;18:1477. doi: 10.1093/carcin/18.8.1477. 1484. [DOI] [PubMed] [Google Scholar]
- 22.Druck T, Hadaczek P, Fu T-B, Ohta M, Siprashvili Z, Baffa R, Negrini M, Kastury K, Veronese M L, Rosen D, et al. Cancer Res. 1997;57:504–512. [PubMed] [Google Scholar]
- 23.Armitage P, Berry G. Statistical Methods in Medical Research. Oxford: Blackwell Scientific; 1987. [Google Scholar]
- 24.Reitmair A H, Redston M, Cai J C, Chuang T C Y, Bjerknes M, Cheng H, Hay K, Gallinger S, Bapat B, Mak T W. Cancer Res. 1996;56:3842–3849. [PubMed] [Google Scholar]
- 25.Kruse R, Rutten A, Lamberti C, Hosseiny-Malayeri H R, Wang Y, Ruelfs C, Jungck M, Mathiak M, Ruzicka T, Hartschuh W, et al. Am J Hum Genet. 1998;63:63–70. doi: 10.1086/301926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bocker T, Diermann J, Friedl W, Gebert J, Holinski-Feder E, Karner-Harnusch J, von Knebel-Doeberitz M, Koelble K, Moeslein G, Schackert H-K, et al. Cancer Res. 1997;57:4739–4743. [PubMed] [Google Scholar]
- 27.Huebner K, Garrison P N, Barnes L D, Croce C M. Annu Rev Genet. 1998;32:7–31. doi: 10.1146/annurev.genet.32.1.7. [DOI] [PubMed] [Google Scholar]
- 28.Lynch H T, Lynch P M, Pester J, Fusaro R M. Arch Intern Med. 1981;141:607–611. [PubMed] [Google Scholar]
- 29.Lynch H T, Smyrk T C, Watson P, Lanspa S J, Lynch J F, Lynch P M, Cavalieri R J, Boland C R. Gastroenterology. 1993;104:1535–1549. doi: 10.1016/0016-5085(93)90368-m. [DOI] [PubMed] [Google Scholar]
- 30.Hilgers W, Kern S E. Genes Chromosomes Cancer. 1999;26:1–12. [PubMed] [Google Scholar]
- 31.Edelmann W, Yang K, Umar A, Heyer J, Lau K, Fan K, Liedtke W, Cohen P E, Kane M F, Lipford J R, et al. Cell. 1997;91:467–477. doi: 10.1016/s0092-8674(00)80433-x. [DOI] [PubMed] [Google Scholar]