

Abstract
POLG is the human gene that encodes the catalytic subunit of DNA polymerase γ (Pol γ), the replicase for human mtDNA. A POLG Y955C point mutation causes human chronic progressive external ophthalmoplegia (CPEO), a mitochondrial disease with eye muscle weakness and mtDNA defects. Y955C POLG was targeted transgenically (TG) to the murine heart. Survival was determined in four TG (+/−) lines and wild type (WT) littermates (−/−). Left ventricle (LV) performance (echocardiography and MRI), heart rate (electrocardiography), mtDNA abundance (real time PCR), oxidation of mtDNA (8-OHdG), histopathology and electron microscopy (EM) defined the phenotype.
Cardiac targeted Y955C POLG yielded a molecular signature of CPEO in the heart with cardiomyopathy, mitochondrial oxidative stress, and premature death. Increased LV cavity size and LV mass, bradycardia, decreased mtDNA, increased 8-OHdG, and cardiac histopathological and mitochondrial EM defects supported and defined the phenotype.
This study underscores the pathogenetic role of human mutant POLG and its gene product in mtDNA depletion, mitochondrial oxidative stress and cardiomyopathy as it relates to the genetic defect in CPEO. The transgenic model pathophysiologically links human mutant Pol γ, mtDNA depletion and mitochondrial oxidative stress to the mtDNA replication apparatus and to cardiomyopathy.
INTRODUCTION
Pol γ is part of an assembly of proteins and enzymes responsible for replication of mtDNA (1, 2). It is composed of a catalytic subunit responsible for polymerase and exonuclease activity and a small accessory subunit that enhances binding and processivity (3). Exonuclease activity allows for proofreading of the growing DNA strand and increases faithful copying of mtDNA (4). Other investigators employed transgenic mouse (TG) models of defective mtDNA replication that expressed exonucleolytic proofreading-deficient Pol γ and that yielded increased mtDNA mutations (5) and cardiomyopathy (CM) (6, 7), but oxidative stress was not operative in those studies (8, 9). Those experimentally constructed, engineered Pol γ mutations lacked a pathophysiologically based counterpart in human disease. In contrast, studies here used a TG generated with cardiac-targeted human mutant POLG that encodes Y955C Pol γ. This mutation causes Chronic Progressive External Ophthalmoplegia (CPEO), an authentic human mitochondrial illness that rarely causes cardiomyopathy (CM) (10). This clinical fact enabled us to use the cardiac targeted Y955C TG in structural and functional evaluations of the target again without confounding effects from other tissues. TGs here developed CM, mtDNA depletion and oxidative stress.
CPEO is an inherited disorder (either autosomal dominant or recessive) with features of mtDNA depletion and accumulation of mtDNA mutations. The illness is characterized by adult onset, bilateral ptosis, progressive weakness of the external ocular muscle, blepharoptosis, ophthalmoparesis, skeletal myopathy with ragged red fibers, wasting, exercise intolerance, and depressed activity of respiratory chain enzymes (11). Large-scale deletions of mtDNA occurred in muscle biopsies obtained from members of kindreds with autosomal dominant CPEO (12). This suggested a specific autosomal gene could be responsible for mtDNA instability. Positional cloning linked four genes to the observation that included the adenine nucleotide translocator (ANT1) at locus 4q34-35 (13), mitochondrial helicase (Twinkle) at locus 10q24 (14), thymidine phosphorylase (associated with recessive mitochondrial neurogastrointestinal encephalomyopathy; [MNGIE], a form of CPEO) (15) at locus 22q13.32-qter, and the gene encoding the catalytic subunit of Pol γ, POLG, at locus 15q25 (16). One recently reported patient with autosomal dominant CPEO harbored a mutation in the gene for the Pol γ accessory subunit (17). Importantly, candidate genes encode products that replicate mtDNA (2) or are involved with metabolism of nucleotide precursors for mtDNA replication (18).
Y955C is the most common and severe autosomal dominant mutation in the POLG gene (16, 19). It is associated with CPEO and Parkinsonism (20) and recently described to cause premature ovarian failure (21). Tyr955 interacts with the incoming dNTP in the active site within the polymerase domain to maintain high fidelity of DNA synthesis (22). Recombinant Y955C Pol γ exhibits <1% wild-type activity and severely decreased processivity. This stalling of mtDNA synthesis with minimal catalytic activity may explain the profound clinical changes in Y955C heterozygotes (23), but has not been defined pathophysiologically in a murine transgenic model. Substitution of Y955 to cysteine also increases nucleotide misinsertion errors 1–2 orders of magnitude in absence of exonucleolytic proofreading (22). Of note, other POLG point mutations are associated with CM (24), albeit uncommonly.
We published biochemical data on the mutant Y955C Pol γ protein in CPEO (2, 22, 23, 25), however the Y955C POLG mutation has not been investigated in transgenic murine models. The effect of human Y955C Pol γ on the murine heart was explored using transgenically targeted Y955C POLG driven by the alpha myosin heavy chain promoter (α-MyHC). This transgenic approach was pioneered by Robbins (26), and was adapted and used successfully by us in the past (22, 27–29) and in studies here. Cardiac targeted Y955C POLG yielded expression of abundant but enzymatically defective Pol γ polypeptide in TG hearts. This resulted in inefficient mtDNA replication, mitochondrial oxidative stress, and structural and functional features of CM. TG experiments demonstrated that inefficient mtDNA replication and mitochondrial oxidative stress are bona fide contributors to CM and that mtDNA depletion results from a Pol γ mutation.
MATERIALS AND METHODS
Generation of α-MyHC/Y955C transgenic mice (TG)
Established methods and approaches with the α-myosin heavy chain promoter (30) were applied to the Y955C Pol γ TG in ways that resembled those used previously (27, 29, 31, 32).
Genotyping
For the TG line α-MyHC/Y955C, the presence of the mutant transgene was detected in the founders and their offspring using Southern blotting and real-time PCR essentially as recently described (27, 29, 32).
TG gene copy analysis
Four transgenic lines were established for the targeted overexpression of mutant POLG. To determine the relative copy number in each, the level of human Y955C POLG was analyzed semi-quantitatively from tail DNA extracts using real-time PCR and Light Cycler TaqMan Master kit. Target genes were amplified using specific primers for human POLG (forward: 5′-CCTCGGCCTGGTATTCCT-3′ and reverse: 5′-TGGTCCAAGAGTAAC GCTCTTC-3′, and Universal Probe Library probe #71; Roche Diagnostics Corp., Indianapolis, IN) and “housekeeping” gene, GAPDH (forward: 5′-GATGCTACAAGCAGGCCTTT-3′ and reverse: 5′-GCAGAAAGCAAGGGCAAA-3′, and Universal Probe Library probe #4; Roche Diagnostics Corp.). DNA amplification was performed using LightCycler 480 (Roche Diagnostics Corp.) on individual tissues extracted from at least 7 mice within each line. Relative copy number dosage was normalized to GAPDH (single copy gene) from WT.
SDS PAGE Coomassie Blue staining and Western Blotting
To quantitate protein abundance in the mitochondrial lysate, proteins were resolved on SDS PAGE in Criterion 4–12% Bis-Tris gels followed by Coomassie Blue staining. To quantitate Pol γ, a similar gel (5 fold overload of mitochondrial lysate) was prepared and transferred to Imobilon-P for Western Blotting using anti-human Pol γ antibody (DPg) as described (33).
Mortality of Y955C Pol γ TG lines
Kaplan Meier survival curves were generated and calculated according to standard methods on all TG lines and on WT littermates (pooled; N=16).
Mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) quantitation in heart tissue from Y955C Pol γ TGs using real time PCR
Methods employed were based on modifications of those used by others (34, 35) and detailed by us recently (32). Briefly, DNA sequences for primers and probes used for quantization of mitochondrial and nuclear DNA with real-time PCR technique were adapted from others 25. The accessory subunit of murine mitochondrial DNA polymerase gamma (ASPG) and mouse mitochondrial cytochrome oxidase subunit 1 (COX) were the subjects of separate quantization of mouse nuclear and mitochondrial DNA, respectively. The mitochondrial forward primer (5′- TCGTTGATTATTCTCAACCAATCA-3′) and reverse primer (5′- GCCTCCAATTATTATTGGTATTACTATGA-3′) were used to amplify the target segment of COXI gene. Hybridization probes were 3′-flouresceine (5′-AACCAGGTGCACTTTTAGGAGATGACCF3′) and 5′-LC Red 640 3′-phosphate –blocked (5′L- AATTTACAATGTTATCGTAACTGCCCATGCP3′) for this gene. The nuclear forward primer (5′-GGAGGAGGCACTTTCTCAGC-3′) and reverse primer (5′- GAAGACCTGCTCCCTGAACAC-3′) were used to amplify the nuclear ASPG gene. A 3′-flouresceine labeled oligonucleotide (5′-GCGCTTTGGACCTTTGGGTGTAG-F3′) and a 5′-LC Red 640 3′–phosphate blocked (5′L-GTTACGAAAGAACCTAGCCTCACA GTGGT-P3′) oligonucleotide were used as hybridization probes for the nuclear gene. Amplification was performed in a LightCycler 480 (Roche) and consisted of a denaturation step (8 minutes), 50 cycles of amplification (95°C for 8 sec, 55°C for 10 sec followed by single fluorescence acquisition, and 72°C for 10 sec), a melting curve (95°C for 30 sec with a ramping rate of 4.8°C/sec, 45°C for 61 sec with a ramping rate of 2.5°C/sec and 95°C for 0 sec), and a cooling cycle (40°C). Temperature ramping rates were 2.5°C/sec unless noted.
Standard DNA curves for quantization of the LC products were employed. Both mitochondrial and nuclear target sequences were PCR amplified and cloned into the pCR2.1-TOPO vector (Invitrogen) following the manufacturer’s protocol. Each plasmid was linearized by digestion with Hind III restriction dendonuclease (Roche) following the manufacturer’s protocol. Each fragment insert was then PCR amplified using their corresponding original primers and ran out on 2% agarose gels to ensure quality and proper size of the inserts.
Analysis of Mitochondrial DNA for 8-hydroxy-2-deoxyguanosine (8-OHdG) as a marker of mitochondrial oxidative stress
Mitochondrial DNA from TG (C line) and WT myocardium was hydrolyzed to nucleosides using nuclease P1 and alkaline phosphatase as described previously (36). Cardiac mtDNA samples were isolated (mtDNA Extractor CT Kit, Wako Chemicals USA, Inc., Richmond, VA ), and analyzed for abundance of 8-OHdG (as a marker for oxidative stress to mtDNA; (37, 38)) relative to abundance of 2-deoxyguanosine (2dG; x10−5) by HPLC coupled with coulometric electrochemical and spectrophotometric detection (CoulArray Model 5600; ESA Inc., Chelmford, MA). Nucleoside concentrations were calculated from multipoint standard curves generated daily with freshly prepared standards at concentrations that encompassed those observed in the samples. 8-OHdG levels were expressed as a mean molar ratio and standard error of the mean (SEM) to 10−5 2dG (N=6).
Echocardiography in Y955C Pol γ TGs and WT
Echocardiography was performed on age- and gender-matched WT and TGs as described previously (39). For ECHO observations, Y955C Pol γ TG line B, N=8; WT, N=7; for Y955C Pol γ TG line C, TG N=6; WT N=6. For Y955C Pol γ TG line D, TG N=8; WT N=4. For Y955C Pol γ TG line E, TG N=8, WT N=7.
MRI of the heart Y955C Pol γ TGs and WT littermates
Y955C Pol γ line D TG (n=3) and WT littermates (n=3) were evaluated via MRI at 16–20 weeks as has been recently described (32). MRI was accomplished on a 4.7T Varian/INOVA (200/33) Spectroscopy and Imaging System (Varian, Palo Alto, CA).
Histopathological features from Y955C TG hearts
Hearts from Y955C ( C line, n=3 per cohort; 60 days old) and equal numbers of WT littermates were rapidly removed, immersion fixed in 10% neutral buffered formalin at ambient temperature for 24 h, and carefully bi-valved to demonstrate views of the ventricular chambers and to document cardiomegaly, or cytological changes such as hypertrophy. Samples were processed, sectioned (6 μm), stained individually with hematoxylin and eosin or with Masson’s Trichrome and examined microscopically on a Nikon 800 Ultraphot Microscope (Nikon, Melville, NY). All images were saved electronically to enable histopathological changes in hearts of TGs to be compared to those of WTs.
Ultrastructural pathological evaluations (EM) of cardiac mitochondria in Y955C POLG TGs
Samples from TG (C line) and WT murine hearts (N=12) were evaluated using EM to define mitochondrial structural changes. Sections (approximately 1 mm cubes) were rapidly fixed in diluted Karnovsky’s fixative and processed for EM as in the past (39). Embedded sections (0.5 μ) were cut with a glass knife and stained with Toluidine Blue for orientation. Ultrathin (900 Å) sections were cut with a diamond knife, stained with uranyl acetate and lead citrate and viewed on a Philips Morgagni electron microscope (Philips, Amsterdam NL). Each EM image was reviewed independently by two investigators for the presence of mitochondrial proliferation or structurally abnormal mitochondria (e.g., intramitochondrial lamellar bodies, cristae reduplication) (40). Structurally damaged mitochondria were operationally defined as having loss or dissolution of ≥25% of cristae along with the above features.
Statistical analysis
For mtDNA quantitation, data were expressed as the ratio of mean value of the mtDNA measurement to the mean value of nDNA divided by 1000 and the resultant values are expressed as mean ± standard error. A value of p<0.05 (determined by student’s unpaired T-test) was considered statistically significant. Echocardiographic determinations from all groups were compared by ANOVA (39).
RESULTS
Polypeptide blotting of Pol γ in cardiac mitochondrial extracts
Mitochondria isolated from Y955C TG and WT mouse hearts were assayed for relative abundance of human Y955C Pol γ polypeptide. Western blots containing equal amounts of cardiac mitochondrial protein were probed with the DPg polyclonal antibody (Figure 1A), which cross-reacted with mouse Pol γ (data not shown). Western blot analyses of mitochondrial extracts from both TG and WT hearts revealed that Y955C TG heart samples (lanes 2, 4, and 6) displayed increased signal density of the Pol γ band (140 kDa). The bands from TG heart samples co-migrated with the authentic Pol γ control (lane 1, Figure 1A). In contrast, Pol γ was not seen in extracts of mitochondria from WT hearts, indicating the native murine Pol γ polypeptide was below the threshold of detection. Figure 1B demonstrates equivalent protein amounts were loaded.
Figure 1.
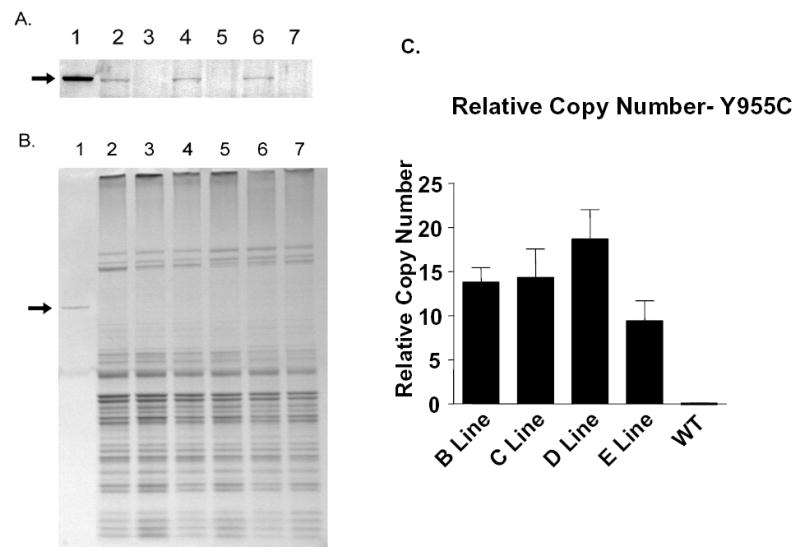
Y955C TG Pol γ protein expression and gene copy dosage: Protein from purified mitochondrial lysates of TG and control hearts were separated on a 4–12% poly-acrylamide gel and either transferred to Immobilon-P for Western blot analysis or Coomassie Blue staining. A) Western blot analysis of the heart mitochondrial extracts. Lane 1, Purified recombinant human Pol γ as control, Lanes 2, 4 and 6, mitochondrial lysates from Y955C TG hearts of 33, 25 and 10 week old mice, respectively. Lanes 3, 5 and 7, mitochondrial lysates from control TG negative hearts of 33, 25, and 10 week old mice, respectively. Arrow indicates pol γ. B) Coomassie Blue stained gel to demonstrate that equivalent portions of heart mitochondrial proteins were analyzed by Western blot. Lanes 1–7 are identical to that in panel A except that 5 times the amount of protein was loaded and separated in the gel for detection by Coomassie Blue staining. C. Relative gene copy number of mutant Y955C Pol γ was determined from cardiac tissues for each of the 4 lines generated. Arrow indicates pol γ.C) Relative gene copy number of human mutant Y955C Pol γ was determined from cardiac tissues for each of the 4 lines generated. Levels of POLG were analyzed semi-quantitatively from murine tail DNA extracts using real-time PCR and Light Cycler TaqMan Master kit and normalized to GAPDH from WT.
Transgenic, cardiac targeted Y955C Pol γ mice
Four viable Y955C Pol γ TG lines were created. They were operationally labeled Y955C Pol γ TG line B, C, D, and E. Relative gene copy number was determined for each of the TG Y955C lines generated (Figure 1C). In general, transgenesis revealed robust expression of the TG. Animals bred true for 6 generations before experimental use. No gross, external phenotype was recognized in TGs or littermates. No changes in behavior, growth, maturation, breeding, or Mendelian distribution of TG were found in any of the lines.
Mortality of Y955C Pol γ TG lines
Kaplan Meier survival curves were generated based on survival postpartum (in days) for over 600 days and calculated according to standard methods on all TG lines and on their WT littermates (WT pooled). Results showed different survival in the different TG lines (Figure 2A). Median survival was lowest in line D (90d), intermediate in lines C (210d) and E (>420d), and longest in line B (>600d) whose survival most resembled that found in the WT. To help define events in line D TGs that contributed to shortened lifespan, TGs were terminated at 21 days. Hemi-sections of the thorax revealed massive cardiomegaly with massive bilateral atrial enlargement, consistent with congestive heart failure (Figure 2B).
Figure 2.
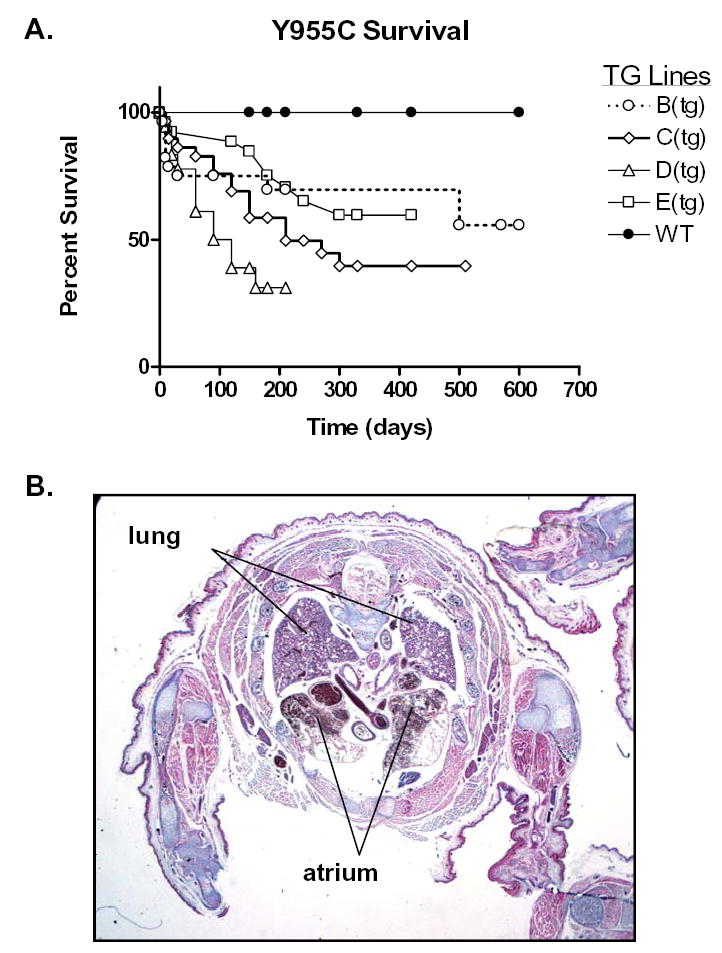
Kaplan-Meier survival curve and histopathologic assessment of Y955C TG: A) Differential survival in the different TG lines was found. Median survival was 90d in line D, 210d in line C, and >420d in line E, >600d in line B which most resembled the WT. B) Histopathological (Masson’s Trichrome stain) correlate with death in TG line D: Autopsy performed on 21 day old TG from line D. Hemi-section of thorax reveals massive atrial enlargement (A) suggesting heart failure (arrows) and compressing the lungs (L).
mtDNA Copy Number
Depletion of mtDNA is one hallmark of mtDNA dysfunction, and of CPEO. The accessory subunit of murine mitochondrial DNA polymerase gamma (ASPG) and mouse mitochondrial 17kDa subunit of COX 1 were the subjects of separate quantization of mouse nuclear and mitochondrial DNA, respectively (Figure 3A). In both young and older cohorts, Y955C TG mice exhibited reduced mtDNA abundance consistently (mtDNA/nDNC ratio) compared to their respective WT cohorts. These suggest Y955C transgene expression causes substantial mtDNA depletion in the heart and could account for CM, CHF and increased mortality.
Figure 3.
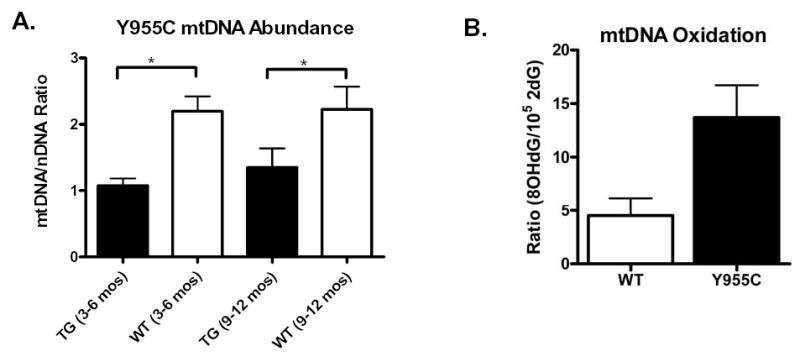
Real time PCR mtDNA/nDNA ratios and steady state abundance of 2dG and 8-OHdG in mitochondrial extracts from WT and Y955C TG mice: A) WT and TG cohorts’ cardiac samples (N ≥ 8) were analyzed using real time PCR. Decrease in mtDNA/nDNA ratio was found with Y955C Pol γ TG (C line) (*p<0.01). B) Mitochondria were isolated according to standard methods and mtDNA extracted. Nucleotide abundance was expressed as a mean molar ratio and standard error of the mean (SEM) of 8-OHdG to 2dG x10−5. Steady state abundance of 8-OHdG in Y955C hearts was 3-fold that found in WT (p< 0.05), (N=6).
8-OHdG in mitochondria from Y955C TG hearts
Mitochondria from Y955C heart samples were assessed for oxidative stress by isolating them from heart tissue (to remove nuclear DNA) followed by analyzing abundance of 8-OHdG in comparison to 2dG x 10−5. 8-OHdG levels in mtDNA from mitochondria of Y955C hearts were elevated 3-fold over those found in WT control hearts (p<0.05; Figure 3B) consistent with our studies on other murine models of mitochondrial oxidative stress (42).
Echocardiographic data from Y955C Pol γ TGs and WT controls
The Y955C Pol γ TG exhibited an ECHO phenotype of CM. ECHO from TGs at 60 days were performed on 3 lines and revealed increased LV mass (with >60% increase over respective WTs; normalized, mg/g body weight) (data not shown). Y955C line B was 1.66± 0.1 compared to 0.92± 0.07, line C was 1.82, and line D was 1.66± 0.2. These represented increase in LV mass of 70 to 111% (p<0.01) compared to WT littermates. At 120 days, LV mass in TG line C and D remained elevated at 1.31± 0.07 and 1.38± 0.1, respectively. These constituted a relative doubling of LV mass at 120 days (Figure 4).
Figure 4.
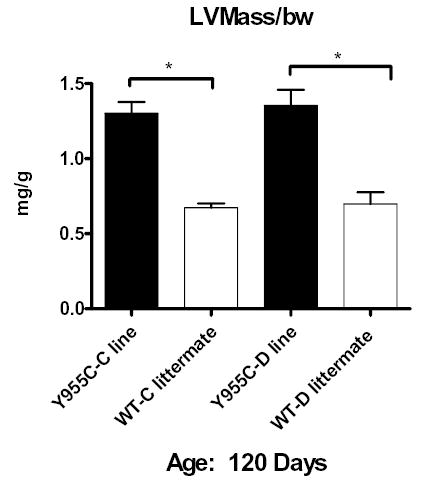
Quantitative analysis of ECHO images in Y955C Pol γ TG: LV mass was calculated in a blinded fashion, code was broken, and data tabulated from the Y955C Pol γ TG lines at 120 days. Data were normalized to body weight (mg/g) and plotted as mean ± SEM. At 120 days, line C and D had survivors. They exhibited cardiac mass that was >100% increased above that of WT (*p<0.01).
MRI of the heart Y955C Pol γ TGs and WT littermates
Y955C Pol γ line D TG (n=3) and WT littermates (n=3) were evaluated via MRI at 16 weeks as we have done in the recent past (32). Figure 5 is a representative 4 chamber view from a gender matched TG and WT littermates at 16 weeks of age. The image from the Y955C TG heart reveals cardiomegaly, biventricular dilation, and atrial enlargement compared to similar MRI views of hearts of WT littermates. This corroborates echocardiographic findings and confirms CM.
Figure 5.
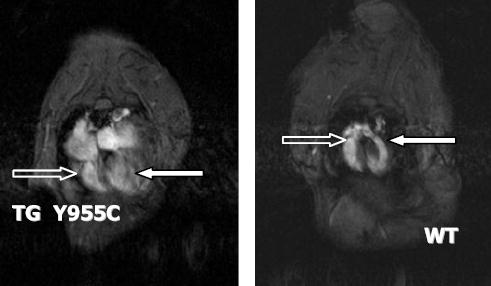
MRI of TG and WT hearts: MRI images of the murine heart were performed using methods as described previously. Four chamber enlargement in TG Y955C (left) and WT littermate (right) at 9 months of age. TG demonstrates 4 chamber enlargement of the heart with both right ventricle (open arrow) and left ventricle (filled arrow) enlargement.
Photomicrographs of Y955C TG hearts and WTs
To identify cardiac structural changes at the microscopic level in TGs, whole mount photomicrographs and high-power light photomicrographs were taken of TGs and WT littermates (Figure 6A, B). Cardiomegaly was observed in TGs with increased cavity dimensions of LV and RV, and free wall and septal thickening in TGs. These findings corroborated observations from both ECHO and MRI studies above that indicated CM. Histopathological examination revealed myocytolysis and cardiomyocyte hypertrophy, with multiple cytoplasmic granular figures that were consistent with mitochondria. Together, these non-specific histopathological features support the CM diagnosis.
Figure 6.
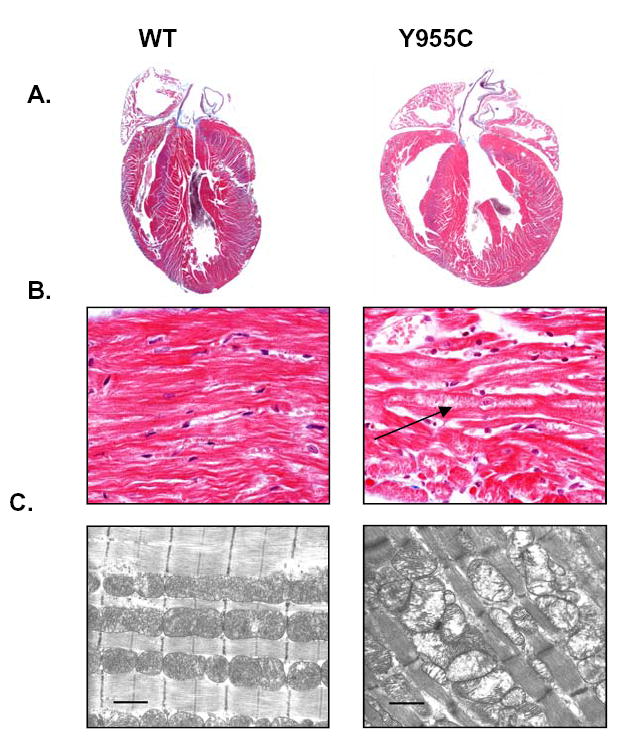
Histopathological (A, B) and EM (C) images of hearts and of mitochondria from Y955C Pol γ TGs: Gender matched littermates expressing Y955C TG or WT controls were used. TG over-expression in the heart caused cardiomyopathy with bi-ventricular thickening and cardiomegaly. Arrow points to lytic change with increased granularity. (Y955C: A, original magnification 1X, and B, original magnification 600 X). Histopathological results (hematoxylin and eosin staining) were supported by mitochondrial damage and enlargement as seen by EM (C, original magnification on EM: 26,000. Marker is 1μ).
Ultrastructural features of mitochondria in TG hearts
EM photomicrographs of Y955C Pol γ TG hearts reinforced histopathological data and correlated with changes in mtDNA. Findings of mitochondrial cristae dissolution and swelling and conspicuous defects in matrix density were commonly seen in Y955C Pol γ TG hearts compared to the age and gender matched WT controls (Figure 6C).
DISCUSSION
Pol γ is responsible for replication of mtDNA (2). Mutations in POLG, the gene for the catalytic subunit cause CPEO, Alper’s syndrome, ataxia-neuropathy, Parkinsonism (2, 43), and other heritable conditions (24).
Targeted cardiac transgenic mice were used to define features of cardiac dysfunction and CM (26), and were applied to explore features of CM in AIDS (44) where nucleoside reverse transcriptase inhibitors (NRTIs) cause mitochondrial dysfunction in the heart (45). Transgenic targeting of human Pol γ harboring the Y955C pathogenic mutation here resulted in depleted mtDNA, oxidative stress, pathological cardiomegaly, cardiac mitochondrial ultrastructural damage, CM with LV dysfunction, bradycardia, and premature death. These findings forge a pathogenetic link between defective mtDNA replication and cardiac dysfunction with overwhelming depletion of mtDNA and mutant human Pol γ expressed in the murine heart.
TG experiments here causally linked transgenically targeted cardiac expression of Y955C Pol γ enzyme (derived from a known pathogenic mutation in POLG) to mtDNA depletion, oxidative stress, and dysfunction (in the form of CM). In the targeted TG, it is reasonable that Y955C Pol γ affected mtDNA replication at the level of enzyme:template interface by reducing relative availability of native Pol γ compared to the mutant. The Y955C mutant polypeptide was sufficiently abundant in this model to disrupt mtDNA homeostasis by overwhelming native Pol γ and becoming the dominant enzyme in the system but with substantially diminished enzyme activity. The subcellular outcome was mitochondrial oxidative stress documented by increased abundance of 8-OHdG.
Homeostasis of the mtDNA replicative machinery is regulated (18). In the present model, genetic disruption with expression of this mutant Pol γ resulted in organ dysfunction and premature death (Figure 7). Based on mass action of the mutant Y955C Pol γ, a “dominant negative” phenotype was the first step in organellar dysfunction and ultimately cardiac dysfunction. It also may be inferred that, at least at the level of mtDNA replication machinery in the mouse, human Y955C mutant Pol γ may substitute effectively for native murine Pol γ or interfere with the murine mtDNA replicon. Transgenic substitution here allows mutant human Pol γ to participate directly in mtDNA replication with resultant depletion. Data here do not directly explain the organ specific nature of mtDNA defects in CPEO. However, they can apply to and reinforce the “OXPHOS paradigm” of Wallace in which organs that require significant energy from oxidative phosphorylation may be principal targets for genetic mitochondria diseases in which oxidative phosphorylation is limited (46).
Figure 7.
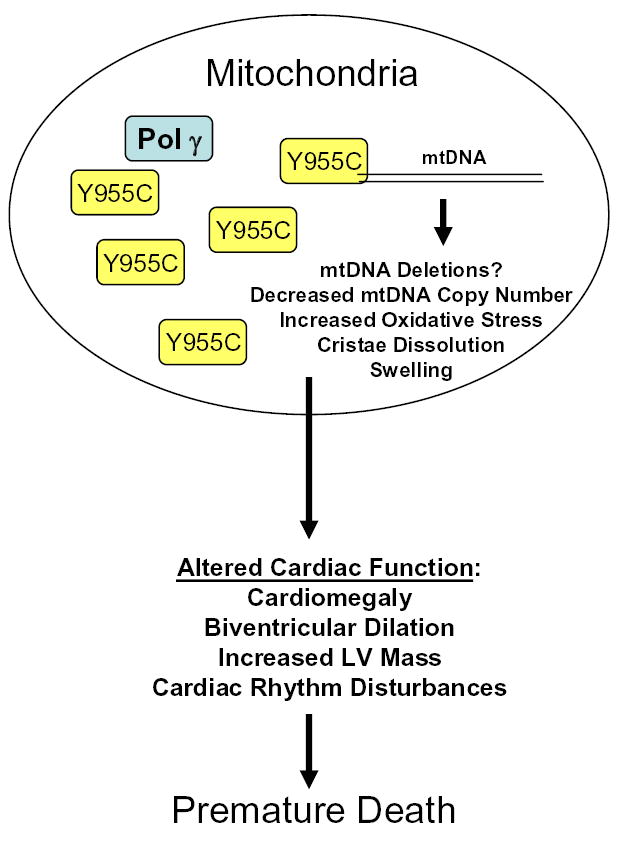
Schematic Summary of Effects of Y955C Pol γ in murine model. Cardiac targeted over-expression of Y955C mutant of Pol γ results in altered mtDNA biogenesis leading to cardiac dysfunction and ultimately premature death. Demonstrated are presence of both native Pol γ and over-expressed Y955C mutants.
The present work extends previous studies that focused on disruption of mtDNA biogenesis at the level of the mitochondrial nucleotide pools. In those studies, TGs were treated with NRTIs that compete with native nucleotides for intramitochondrial phosphorylation and transport (31, 32, 39, 47, 48). Data indicated transport of nucleotides into mitochondria may be disrupted by NRTIs.
Genetic “mtDNA depletion syndromes” (49, 50) offer support for the reasoning used in generating these experimental models and for the data obtained from them. In those illnesses, mitochondrial and cytoplasmic nucleotide pools are disturbed by mutations of kinases, transporters, and enzymes involved in nucleotide pool homeostasis. Such mutations yield mtDNA depletion. “Acquired mtDNA depletion” from administration of NRTIs for AIDS (31, 32) offers a pharmacological model to deplete mtDNA in vivo. NRTI chemical structure, dose, and duration of therapy each impacted the extent of mtDNA depletion and tissue target (31, 32).
This TG model strengthens the pathogenetic link between defects in mtDNA replication and cardiac dysfunction, particularly CM (51). Although not proven here (owing to the severity of the TG phenotype), it is conceivable that subtle genetic mutations in Pol γ (already described or yet to be uncovered) may offer further mechanistic insights if examined transgenically. Presently there are over 70 documented disease mutations and a handful of single nucleotide polymorphisms in the POLG gene (http://dir-apps.niehs.nih.gov/polg/index). Such Pol γ mutations may exhibit only mild enzyme dysfunction without a phenotype in the native, undisturbed condition. However, a mitochondrial dysfunction phenotype could occur in selected tissues if a challenge with NRTIs that deplete mtDNA (32) and cause oxidative stress (36) were added. If these events (mutation of polymerase and disturbed nucleotide pools from NRTIs) were to coincide, significant side effects could result. The combination of genetic predisposition and environmental effects serves as a cornerstone of the threshold effect seen in mitochondrial genetic diseases and related systems, as articulated by Wallace and colleagues (46, 52).
In summary, this study utilized TGs that expressed human Y955C Pol γ in the murine heart to define defective mtDNA replication in vivo at the level of the enzyme machinery that replicates mtDNA. Targeted cardiac transgenic expression of Y955C Pol γ was sufficient to yield a molecular phenotype of mtDNA depletion, a biochemical phenotype with increased abundance of the mutant enzyme in the target, increased 8-OHdG, histopathological and mitochondrial ultrastructural changes in cardiac cells of TGs, and organ dysfunction with cardiomegaly, increased ventricular volume, increased cardiac mass on ECHO and MRI. Together, these interrelated findings underscore mtDNA replication as the nexus for dysfunction. It is reasonable to suggest that when a threshold of genetic mtDNA replication defects occurs, particularly from ineffective Pol γ function, cardiac dysfunction and pathological features of CM is an outcome. The role of defective mtDNA replication in various CMs merits deeper investigation in the future.
Acknowledgments
This work was supported by DHHS, NIH, NHLBI R01 HL059798 and in part by HL072707 to WL and by NIH intramural research funds to WCC.
Footnotes
Grant Support: R01 HL 059798
References
- 1.Kaguni LS. DNA polymerase gamma, the mitochondrial replicase. Annu Rev Biochem. 2004;73:293–320. doi: 10.1146/annurev.biochem.72.121801.161455. [DOI] [PubMed] [Google Scholar]
- 2.Graziewicz MA, Longley MJ, Copeland WC. DNA Polymerase gamma in Mitochondrial DNA Replication and Repair. Chem Rev. 2006;106(2):383–405. doi: 10.1021/cr040463d. [DOI] [PubMed] [Google Scholar]
- 3.Lim SE, Longley MJ, Copeland WC. The mitochondrial p55 accessory subunit of human DNA polymerase gamma enhances DNA binding, promotes processive DNA synthesis, and confers N-ethylmaleimide resistance. J Biol Chem. 1999;274(53):38197–203. doi: 10.1074/jbc.274.53.38197. [DOI] [PubMed] [Google Scholar]
- 4.Longley MJ, Nguyen D, Kunkel TA, Copeland WC. The fidelity of human DNA polymerase gamma with and without exonucleolytic proofreading and the p55 accessory subunit. J Biol Chem. 2001;276(42):38555–62. doi: 10.1074/jbc.M105230200. [DOI] [PubMed] [Google Scholar]
- 5.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429(6990):417–23. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 6.Zhang D, Ezekiel UR, Chang SW, Zassenhaus HP. Gene expression profile in dilated cardiomyopathy caused by elevated frequencies of mitochondrial DNA mutations in the mouse heart. Cardiovasc Pathol. 2005;14(2):61–9. doi: 10.1016/j.carpath.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Zhang D, Mott JL, Chang SW, Stevens M, Mikolajczak P, Zassenhaus HP. Mitochondrial DNA mutations activate programmed cell survival in the mouse heart. Am J Physiol Heart Circ Physiol. 2005;288(5):H2476–83. doi: 10.1152/ajpheart.00670.2004. [DOI] [PubMed] [Google Scholar]
- 8.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309(5733):481–4. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 9.Mott JL, Zhang D, Stevens M, Chang S, Denniger G, Zassenhaus HP. Oxidative stress is not an obligate mediator of disease provoked by mitochondrial DNA mutations. Mutat Res. 2001;474(1–2):35–45. doi: 10.1016/s0027-5107(00)00159-7. [DOI] [PubMed] [Google Scholar]
- 10.Horvath R, Hudson G, Ferrari G, Futterer N, Ahola S, Lamantea E, et al. Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene. Brain. 2006;129(Pt 7):1674–84. doi: 10.1093/brain/awl088. [DOI] [PubMed] [Google Scholar]
- 11.Bohlega S, Tanji K, Santorelli FM, Hirano M, al-Jishi A, DiMauro S. Multiple mitochondrial DNA deletions associated with autosomal recessive ophthalmoplegia and severe cardiomyopathy. Neurology. 1996;46(5):1329–34. doi: 10.1212/wnl.46.5.1329. [DOI] [PubMed] [Google Scholar]
- 12.Zeviani M, Servidei S, Gellera C, Bertini E, DiMauro S, DiDonato S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature. 1989;339(6222):309–11. doi: 10.1038/339309a0. [DOI] [PubMed] [Google Scholar]
- 13.Kaukonen J, Juselius JK, Tiranti V, Kyttala A, Zeviani M, Comi GP, et al. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289(5480):782–5. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- 14.Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. 2001;28(3):223–31. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- 15.Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283(5402):689–92. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]
- 16.Van Goethem G, Dermaut B, Lofgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet. 2001;28(3):211–2. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- 17.Longley MJ, Clark S, Yu Wai Man C, Hudson G, Durham SE, Taylor RW, et al. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am J Hum Genet. 2006;78(6):1026–34. doi: 10.1086/504303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rampazzo C, Ferraro P, Pontarin G, Fabris S, Reichard P, Bianchi V. Mitochondrial deoxyribonucleotides, pool sizes, synthesis, and regulation. J Biol Chem. 2004;279(17):17019–26. doi: 10.1074/jbc.M313957200. [DOI] [PubMed] [Google Scholar]
- 19.Lamantea E, Tiranti V, Bordoni A, Toscano A, Bono F, Servidei S, et al. Mutations of mitochondrial DNA polymerase gammaA are a frequent cause of autosomal dominant or recessive progressive external ophthalmoplegia. Ann Neurol. 2002;52(2):211–9. doi: 10.1002/ana.10278. [DOI] [PubMed] [Google Scholar]
- 20.Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet. 2004;364(9437):875–82. doi: 10.1016/S0140-6736(04)16983-3. [DOI] [PubMed] [Google Scholar]
- 21.Pagnamenta AT, Taanman JW, Wilson CJ, Anderson NE, Marotta R, Duncan AJ, et al. Dominant inheritance of premature ovarian failure associated with mutant mitochondrial DNA polymerase gamma. Hum Reprod. 2006 doi: 10.1093/humrep/del076. [DOI] [PubMed] [Google Scholar]
- 22.Ponamarev MV, Longley MJ, Nguyen D, Kunkel TA, Copeland WC. Active site mutation in DNA polymerase gamma associated with progressive external ophthalmoplegia causes error-prone DNA synthesis. J Biol Chem. 2002;15:15. doi: 10.1074/jbc.C200100200. [DOI] [PubMed] [Google Scholar]
- 23.Graziewicz MA, Longley MJ, Bienstock RJ, Zeviani M, Copeland WC. Structure-function defects of human mitochondrial DNA polymerase in autosomal dominant progressive external ophthalmoplegia. Nat Struct Mol Biol. 2004;11(8):770–776. doi: 10.1038/nsmb805. [DOI] [PubMed] [Google Scholar]
- 24.Horvath R, Hudson G, Ferrari G, Futterer N, Ahola S, Lamantea E, et al. Phenotypic spectrum associated with mutations of the mitochondrial polymerase {gamma} gene. Brain. 2006 doi: 10.1093/brain/awl088. [DOI] [PubMed] [Google Scholar]
- 25.Copeland WC, Ponamarev MV, Nguyen D, Kunkel TA, Longley MJ. Mutations in DNA polymerase gamma cause error prone DNA synthesis in human mitochondrial disorders. Acta Biochim Pol. 2003;50(1):155–67. [PubMed] [Google Scholar]
- 26.Robbins J. Remodeling the cardiac sarcomere using transgenesis. Annu Rev Physiol. 2000;62:261–87. doi: 10.1146/annurev.physiol.62.1.261. [DOI] [PubMed] [Google Scholar]
- 27.Lewis W, Miller YK, Haase CP, Ludaway T, McNaught J, Russ R, et al. HIV viral protein R causes atrial cardiomyocyte mitosis, mesenchymal tumor, dysrhythmia, and heart failure. Laboratory Investigation. 2005;85(2):182–92. doi: 10.1038/labinvest.3700222. [DOI] [PubMed] [Google Scholar]
- 28.Lewis W. Nucleoside reverse transcriptase inhibitors, mitochondrial DNA and AIDS therapy. Antivir Ther. 2005;10 (Suppl 2):M13–27. [PubMed] [Google Scholar]
- 29.Raidel SM, Haase C, Jansen NR, Russ RB, Sutliff RL, Velsor LW, et al. Targeted myocardial transgenic expression of HIV Tat causes cardiomyopathy and mitochondrial damage. Am J Physiol Heart Circ Physiol. 2002;282(5):H1672–8. doi: 10.1152/ajpheart.00955.2001. [DOI] [PubMed] [Google Scholar]
- 30.Subramaniam A, Jones WK, Gulick J, Wert S, Neumann J, Robbins J. Tissue-specific regulation of the alpha-myosin heavy chain gene promoter in transgenic mice. J Biol Chem. 1991;266(36):24613–20. [PubMed] [Google Scholar]
- 31.Lewis W, Haase CP, Miller YK, Ferguson B, Stuart T, Ludaway T, et al. Transgenic expression of the deoxynucleotide carrier causes mitochondrial damage that is enhanced by NRTIs for AIDS. Laboratory Investigation. 2005;85(8):972–81. doi: 10.1038/labinvest.3700301. [DOI] [PubMed] [Google Scholar]
- 32.Lewis W, Kohler JJ, Hosseini SH, Haase CP, Copeland WC, Bienstock RJ, et al. Antiretroviral nucleosides, deoxynucleotide carrier and mitochondrial DNA: evidence supporting the DNA pol gamma hypothesis. Aids. 2006;20(5):675–84. doi: 10.1097/01.aids.0000216367.23325.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan SS, Longley MJ, Naviaux RK, Copeland WC. Mono-allelic POLG expression resulting from nonsense-mediated decay and alternative splicing in a patient with Alpers syndrome. DNA Repair (Amst) 2005;4(12):1381–9. doi: 10.1016/j.dnarep.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 34.Cote HC, Yip B, Asselin JJ, Chan JW, Hogg RS, Harrigan PR, et al. Mitochondrial:nuclear DNA ratios in peripheral blood cells from human immunodeficiency virus (HIV)-infected patients who received selected HIV antiretroviral drug regimens. J Infect Dis. 2003;187(12):1972–6. doi: 10.1086/375353. [DOI] [PubMed] [Google Scholar]
- 35.Davani EY, Brumme Z, Singhera GK, Cote HC, Harrigan PR, Dorscheid DR. Insulin-like growth factor-1 protects ischemic murine myocardium from ischemia/reperfusion associated injury. [see comment] Critical Care (London) 2003;7(6):R176–83. doi: 10.1186/cc2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Velsor LW, Kovacevic M, Goldstein M, Leitner HM, Lewis W, Day BJ. Mitochondrial oxidative stress in human hepatoma cells exposed to stavudine. Toxicol Appl Pharmacol. 2004;199(1):10–9. doi: 10.1016/j.taap.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 37.Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci U S A. 1988;85(17):6465–7. doi: 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams MD, Van Remmen H, Conrad CC, Huang TT, Epstein CJ, Richardson A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. Journal of Biological Chemistry. 1998;273(43):28510–5. doi: 10.1074/jbc.273.43.28510. [DOI] [PubMed] [Google Scholar]
- 39.Lewis W, Haase CP, Raidel SM, Russ RB, Sutliff RL, Hoit BD, et al. Combined antiretroviral therapy causes cardiomyopathy and elevates plasma lactate in transgenic AIDS mice. Laboratory Investigation. 2001;81(11):1527–36. doi: 10.1038/labinvest.3780366. [DOI] [PubMed] [Google Scholar]
- 40.Dalakas MC, Illa I, Pezeshkpour GH, Laukaitis JP, Cohen B, Griffin JL. Mitochondrial myopathy caused by long-term zidovudine therapy [see comments] N Engl J Med. 1990;322(16):1098–105. doi: 10.1056/NEJM199004193221602. [DOI] [PubMed] [Google Scholar]
- 41.Chu V, Otero JM, Lopez O, Sullivan MF, Morgan JP, Amende I, et al. Electrocardiographic findings in mdx mice: a cardiac phenotype of Duchenne muscular dystrophy. Muscle Nerve. 2002;26(4):513–9. doi: 10.1002/mus.10223. [DOI] [PubMed] [Google Scholar]
- 42.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11(4):376–81. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 43.Longley DB, Wilson TR, McEwan M, Allen WL, McDermott U, Galligan L, et al. c-FLIP inhibits chemotherapy-induced colorectal cancer cell death. Oncogene. 2006;25(6):838–48. doi: 10.1038/sj.onc.1209122. [DOI] [PubMed] [Google Scholar]
- 44.Lewis W. Use of the transgenic mouse in models of AIDS cardiomyopathy. Aids. 2003;17 (Suppl 1):S36–45. doi: 10.1097/00002030-200304001-00006. [DOI] [PubMed] [Google Scholar]
- 45.Lewis W, Day BJ, Copeland WC. Mitochondrial toxicity of nrti antiviral drugs: an integrated cellular perspective. Nat Rev Drug Discov. 2003;2(10):812–22. doi: 10.1038/nrd1201. [DOI] [PubMed] [Google Scholar]
- 46.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lewis W, Grupp IL, Grupp G, Hoit B, Morris R, Samarel AM, et al. Cardiac Dysfunction Occurs in the HIV-1 Transgenic Mouse Treated with Zidovudine. Laboratory Investigation. 2000;80(2):187–97. doi: 10.1038/labinvest.3780022. [DOI] [PubMed] [Google Scholar]
- 48.Divi RL, Haverkos KJ, Humsi JA, Shockley ME, Thamire C, Nagashima K, et al. Morphological and molecular course of mitochondrial pathology in cultured human cells exposed long-term to Zidovudine. Environ Mol Mutagen. 2006 doi: 10.1002/em.20245. [DOI] [PubMed] [Google Scholar]
- 49.Hirano M, Marti R, Ferreiro-Barros C, Vila MR, Tadesse S, Nishigaki Y, et al. Defects of intergenomic communication: autosomal disorders that cause multiple deletions and depletion of mitochondrial DNA. Semin Cell Dev Biol. 2001;12(6):417–27. doi: 10.1006/scdb.2001.0279. [DOI] [PubMed] [Google Scholar]
- 50.Elpeleg O. Inherited mitochondrial DNA depletion. Pediatr Res. 2003;54(2):153–9. doi: 10.1203/01.PDR.0000072796.25097.A5. [DOI] [PubMed] [Google Scholar]
- 51.Kajander OA, Karhunen PJ, Jacobs HT. The relationship between somatic mtDNA rearrangements, human heart disease and aging. Human Molecular Genetics. 2002;11(3):317–24. doi: 10.1093/hmg/11.3.317. [DOI] [PubMed] [Google Scholar]
- 52.Loeb LA, Wallace DC, Martin GM. The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc Natl Acad Sci U S A. 2005;102(52):18769–70. doi: 10.1073/pnas.0509776102. [DOI] [PMC free article] [PubMed] [Google Scholar]