

Abstract
Inhibition of translation plays a role in apoptosis induced by a variety of stimuli, but the mechanism by which it promotes apoptosis has not been established. We have investigated the hypothesis that selective degradation of anti-apoptotic regulatory protein(s) is responsible for apoptosis resulting from translation inhibition. Induction of apoptosis by cycloheximide was detected within 2 – 4 h and blocked by proteasome inhibitors, indicating that degradation of short-lived protein(s) was required. Caspase inhibition and overexpression of Bcl-xL blocked cycloheximide-induced apoptosis. In addition, cycloheximide induced rapid activation of Bak and Bax, which required proteasome activity. Mcl-1 was degraded by the proteasome with a half-life of approximately 30 min following inhibition of protein synthesis, preceding Bak/Bax activation and the onset of apoptosis. Overexpression of Mcl-1 blocked apoptosis induced by cycloheximide, while RNAi knockdown of Mcl-1 induced apoptosis. Knockdown of Bim and Bak, downstream targets of Mcl-1, inhibited cycloheximide-induced apoptosis, as did knockdown of Bax. Apoptosis resulting from inhibition of translation thus involves the rapid degradation of Mcl-1, leading to activation of Bim, Bak and Bax. Because of its rapid turnover, Mcl-1 may serve as a convergence point for signals that affect global translation, coupling translation to cell survival and the apoptotic machinery.
Most signals that control survival of mammalian cells modulate the activity of Bcl-2 family members, which regulate the mitochondrial pathway of apoptosis (1, 2). Anti-apoptotic members of the Bcl-2 family, including Bcl-2, Bcl-xL, and Mcl-1, maintain cell survival by inhibiting the pro-apoptotic Bcl-2 proteins Bak and Bax through protein-protein interactions. Bak and Bax are typically activated by a second set of pro-apoptotic Bcl-2 proteins called BH3-only proteins, which associate with anti-apoptotic Bcl-2 proteins through interactions that displace and activate Bak and Bax. Once activated, Bak and Bax permeabilize the mitochondrial outer membrane, resulting in the release of cytochrome c and other pro-apoptotic factors that induce caspase activation and cell death.
Signaling pathways that regulate apoptosis can directly modify Bcl-2 family proteins, as well as alter the expression of Bcl-2 family members at both the transcriptional and translational levels. Many signaling pathways that regulate apoptosis target specific BH3-only proteins. For example, p53-mediated apoptosis involves transcriptional induction of the BH3-only proteins PUMA (3, 4) and Noxa (5), whereas PI 3-kinase/Akt signaling inhibits apoptosis through transcriptional repression of the BH3-only protein Bim (8) and phosphorylation of the BH3-only protein Bad, resulting in its sequestration by 14-3-3 proteins (6, 7).
In addition to regulating Bcl-2 family proteins, many of the signaling pathways that control apoptosis affect global translational activity, generally by regulation of the initiation factors eIF2, eIF2B, and eIF4E (9-11). A variety of stimuli that induce cell stress inhibit translation via phosphorylation of eIF2, which brings the initiating methionyl-tRNA to the ribosome. Inhibition of eIF2 is mediated by four eIF2α kinases that are activated in response to different stress stimuli: the dsRNA-activated protein kinase PKR, which is activated during viral infection; GCN2, which is activated under conditions of amino acid starvation; PERK, which is activated by accumulation of unfolded proteins in the ER; and HRI, which couples globin synthesis to heme availability in reticulocytes. While inhibition of translation can promote cell survival under conditions of ER stress or amino acid starvation, the phosphorylation of eIF2α by PKR plays a proapoptotic role in response to viral infection. Activation of PKR plays a central role in the antiviral response, which includes induction of apoptosis in response to interferon and dsRNA (12). The best characterized substrate of PKR is eIF2α, and its phosphorylation leads to inhibition of protein synthesis in virus-infected cells. This inhibition of global translation is critical to induction of apoptosis by PKR, since expression of mutant non-phosphorylatable S51A-eIF2α blocks apoptosis induced by PKR overexpression (13) as well as apoptosis induced by several stress stimuli that activate PKR, including dsRNA, interferon, TNFα, serum deprivation, and LPS (14-16).
While activation of PKR induces apoptosis through eIF2 inhibition, growth factor signaling through the PI 3-kinase/Akt pathway promotes cell survival in part by maintaining eIF2 activity through regulation of its guanine nucleotide exchange factor, eIF2B (17). One of the targets of PI 3-kinase/Akt signaling involved in regulation of cell survival is the pro-apoptotic protein kinase GSK-3β, which is inhibited by Akt phosphorylation (18-21). The substrates of GSK-3β include eIF2B, which is inhibited as a result of GSK-3β phosphorylation (22-24). Growth factor deprivation and inhibition of PI 3-kinase leads to activation of GSK-3β, which then phosphorylates and inhibits eIF2B, resulting in inhibition of translation initiation. Expression of nonphosphorylatable eIF2B mutants suppresses apoptosis induced by GSK-3β overexpression, PI 3-kinase inhibition, or growth factor deprivation, indicating that inhibition of eIF2B contributes to apoptosis resulting from inhibition of PI 3-kinase/Akt signaling (17).
PI3-kinase/Akt signaling also activates mTOR, which promotes the activity of multiple proteins involved in translation (25). mTOR regulates the activity of eIF4E (which binds to the 5′ cap of mRNAs) by phosphorylating eIF4E binding protein 1 (4E-BP1). In the absence of mTOR signaling, 4E-BP1 binds to eIF4E and inhibits translation initiation. Phosphorylation of 4E-BP1 by mTOR prevents its interaction with eIF4E, coupling PI 3-kinase/Akt and mTOR signaling to translation of capped mRNAs. Overexpression of eIF4E has been shown to inhibit apoptosis induced by several stimuli (26-28), while overexpression of 4E-BP1 can promote apoptosis (29, 30). Inhibition of mTOR with rapamycin also induces apoptosis of some tumor cells (31), although recent studies suggest that this may result from inhibition of Akt activation rather than from direct effects of mTOR on translation (32). mTOR also stimulates translation by phosphorylation of p70 S6 kinase (25) and regulation of eEF2 kinase (33), thereby maintaining activity of the elongation factor eEF2. While roles for the S6 kinases and eEF2 in apoptosis have not been established, the regulation of these additional factors by mTOR demonstrates the complexity of translation regulation by this pathway.
Despite its role in apoptosis induced by several stimuli, the mechanism by which translation inhibition contributes to apoptosis has not been established. One effect of inhibition of translation initiation factors is a selective increase in the translation of some mRNAs containing internal ribosome entry sites (IRES) or upstream ORFs (9, 10, 34). Many proteins that are selectively induced by this mechanism during cell stress either inhibit apoptosis, such as caspase inhibitors XIAP (35) and HIAP2 (36), or initiate an adaptive program to the cellular stress, such as the transcription factor ATF4 in response to oxidative stress (37). In addition, some proapoptotic proteins have been reported to be induced during translation inhibition, such as Fas and Bax during translation inhibition mediated by PKR (38), suggesting that selective translation of pro-apoptotic proteins may be one mechanism by which global translation inhibition promotes apoptosis.
However, apoptosis is also induced by inhibition of protein synthesis with cycloheximide in a variety of cells (17, 39-42). Since protein synthesis is completely inhibited by cycloheximide, increased translation of proapoptotic proteins can not be responsible for cell death under these conditions. One possibility is that translation inhibition could induce apoptosis by activating a cell stress pathway. Another possibility is that general turnover of cell constituents might lead to deterioration of the cell in an apoptosis-like manner. Alternatively, apoptosis could result from the selective loss of rapidly degraded anti-apoptotic regulatory protein(s).
In the present study, we have focused on induction of apoptosis by cycloheximide in order to analyze the events responsible for apoptosis resulting from inhibition of protein synthesis. We show that translation inhibition activates the mitochondrial pathway of apoptosis due to the loss of a regulatory protein(s) via proteasome-mediated degradation, and identify the anti-apoptotic Bcl-2 family member Mcl-1 as a key regulatory protein that couples cell survival to translational regulation.
EXPERIMENTAL PROCEDURES
Cell culture
Rat-1 fibroblasts were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% calf serum. PC12 rat pheochromocytoma cells were grown in DMEM supplemented with 10% fetal bovine serum and 5% horse serum. HeLa cells were grown in DMEM supplemented with 10% fetal bovine serum. T98G human glioblastoma cells were grown in Eagle's Minimal Essential Medium (MEM) supplemented with 10% fetal bovine serum. U937 human promyelocytic leukemia cells were grown in suspension in RPMI 1640 supplemented with 10% fetal bovine serum.
Apoptosis assay by DNA fragmentation
Rat-1, PC12, and T98G cells (5×106 cells/100 mm dish) were plated and U937 cells (3×106 cells/10 ml) suspended 24 h before treatment with cycloheximide (10 μg/ml) and other inhibitors as indicated in Figure legends. Cells were harvested, washed once with phosphate-buffered saline (PBS), and cytosolic nucleic acids isolated as previously described (43). Resulting nucleic acids were electrophoresed through a 1.5% agarose gel containing ethidium bromide. Gels were treated with 20 μg/ml RNase A for 3 h at 37°C prior to visualization by UV-transillumination.
Apoptosis assay by sub-G1 analysis
Following treatment, cells were harvested (adherent cells were trypsinized to provide a single cell suspension) and centrifuged. Cell pellets were washed twice in 1 ml PBS, and fixed in 1 ml pre-chilled 70% ethanol (in PBS) overnight at 4°C. The following day, cells were washed once with 1 ml PBS and suspended in 0.5 ml propidium iodide solution (50 μg/ml propidium iodide, 40 μg/ml RNase A in PBS). Cell suspensions were incubated 30 min at 37°C in the dark, followed by DNA content analysis by flow cytometry.
Immunoblot analysis
Proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred to PVDF membranes. Antibodies included anti-Bak NT (06-536, Upstate), anti-Bax (2772, Cell Signaling), anti-Mcl-1 (554103, PharMingen), anti-Bcl-2 (610538, BD Transduction Laboratories), anti-Bcl-x (610746, BD Transduction Laboratories), anti-Bcl-xL(2762, Cell Signaling), anti-Bim (4582, Cell Signaling), anti-PARP (9542, Cell Signaling), and anti-β-actin (A5441, Sigma). Western blots were developed using a chemiluminescence reagent (Perkin Elmer).
Bak & Bax activation assays
U937 cells (20 × 106 per 50 ml) were suspended in growth medium 24 h before treatment. Following treatment with cycloheximide, cells were washed once in PBS, suspended in 1 ml hypotonic RSB buffer (10 mM NaCl, 1.5 mM MgCl2, 10 mM Tris-HCl, pH 7.5) and incubated on ice for 10 min. Cells were ruptured with 20-30 strokes with a tight-fitting pestle in a Dounce homogenizer, 0.8 ml of 2.5× MS buffer (525 mM mannitol, 175 mM sucrose, 12.5 mM Tris-HCl, pH 7.5, 2.5 mM EDTA, pH 7.5) was added, and mitochondria were isolated by differential centrifugation. Samples were centrifuged at 1,300 × g for 10 min at 4°C, supernatants were transferred to clean tubes, and centrifugation was repeated twice. The resulting supernatants were then centrifuged at 17,000 × g for 20 min at 4°C to pellet mitochondria. The mitochondrial pellet was washed once with 0.5 ml 1× MS buffer and re-centrifuged. Cross-linking reactions were then performed on mitochondrial pellets suspended in 675 μl PBS by treatment with 10 mM bismaleimidohexane (BMH) (Pierce) for 30 min at room temperature (44). Mitochondria were pelleted and suspended in sample buffer supplemented with 10 mM DTT to quench the cross-linking reaction. Samples were then subjected to immunoblot analysis to detect oligomeric Bak and Bax.
Transient transfections and TUNEL assays
HeLa cells (5 × 105 per 60 mm plate) were plated 24 h before transfection. Transfections were performed using 6 μl TransIT-LT1 reagent (Mirus) per 60 mm plate according to the manufacturer's instructions. Cells were transfected with 1 μg pcDNA3 or the indicated Mcl-1, Bcl-xL, or Bcl-2 expression construct plus 1 μg pCMV-DsRed (BD Biosciences), which expresses a red fluorescent protein. Cells were treated with cycloheximide 24-48 h after transfection, and TUNEL assays were performed using the Fluorescein FragEL DNA Fragmentation Detection Kit (Calbiochem, catalog number QIA39). Cells were trypsinized and processed according to the manufacturer's recommended protocol for “Fluorescein-FragEL of cell suspensions for flow cytometry.” Transfected cells were identified by expression of the DsRed protein with a flow cytometer using the FL2 channel, and the percent TUNEL-positive cells quantified using the FL1 channel.
RNA interference
RNA interference transfections were conducted using HiPerfect reagent (Qiagen) and indicated pre-designed siRNAs (10 nM final concentration) from Ambion, Inc. For Mcl-1 RNAi experiments, transfection mixtures were prepared in 400 μl total volume by adding 24 μl HiPerfect and either nonspecific siRNA (Ambion negative control siRNA #1, catalog number 4611) or indicated Mcl-1 siRNA (Ambion siRNA ID#'s 6126, 6314, and 42844) to serum-free medium, mixed, and incubated at room temperature for 10 min. The transfection mixtures were then transferred to 60 mm plates and overlaid with 4.6 ml of a 105/ml cell suspension in serum-containing medium. Cells were then placed at 37°C, 5% CO2 for the indicated times before harvest for immunoblot analysis, DNA fragmentation assays, or sub-G1 analysis. For Bim, Bax, and Bak RNAi experiments, transfections were conducted as described above, using 200 μl total volumes and 12 or 24 μl HiPerfect reagent for the transfection mixtures, which were overlaid with 2.3 ml of a 105/ml cell suspension on 35 mm plates.
All siRNAs were purchased from Ambion. Each siRNA consisted of 19-nucleotide double-stranded RNA with two 3′-dT overhangs on each strand. siRNA sense sequences and Ambion ID numbers were as follows. Mcl-1 ID 6126: 5′-GGACACAAAGCCAAUGGGCTT-3′; Mcl-1 ID 6314: 5′-GGACUUUUAGAUUUAGUGATT-3′; Mcl-1 ID 42844: 5′-GGAGGCCUCGGCCCGGCGATT-3′; Bim ID 262307: 5′CCUUCUGAUGUAAGUUCUGTT-3′; Bak ID 120199: 5′-CCCAGAGAUGGUCACCUUATT Bak ID 120201: 5′-GCUUUAGCAAGUGUGCACUTT-3′ Bax ID 213259: 5′-GAGGUCUUUUUCCGAGUGGTT-3′.
RESULTS
Apoptosis induced by translation inhibition requires proteasome activity
If apoptosis in response to translation inhibition occurs due to either the degradation of a specific regulatory protein(s) or non-specific degradation of cellular constituents, then inhibition of proteolysis should block apoptosis. As the proteasome is responsible for degradation of most proteins within the cell, the effect of proteasome inhibition on apoptosis resulting from inhibition of protein synthesis was examined in four cell lines: Rat-1 fibroblasts, PC12 rat pheochromocytoma cells, U937 human promyelocytic leukemia cells, and T98G human glioblastoma cells. Rat-1, PC12, and T98G cells were studied as models of growth factor-dependent rodent and human cells (45, 46), whereas U937 cells were used because they rapidly undergo apoptosis in response to a variety of stimuli and are therefore advantageous for biochemical analysis (47). Protein synthesis was inhibited by treatment with cycloheximide at a concentration (10 μg/ml) that inhibited [35S]-methionine incorporation >95% (data not shown). Cells were pretreated with proteasome inhibitor MG132 for 30 min prior to treatment with cycloheximide for 0 – 6 h, after which fragmented cytosolic DNA was isolated and detected by agarose gel electrophoresis. Cycloheximide treatment resulted in rapid and time-dependent apoptosis, detected as early as 2 h, which was blocked by proteasome inhibition with MG132 in all cell lines tested (Figure 1A).
Fig. 1.
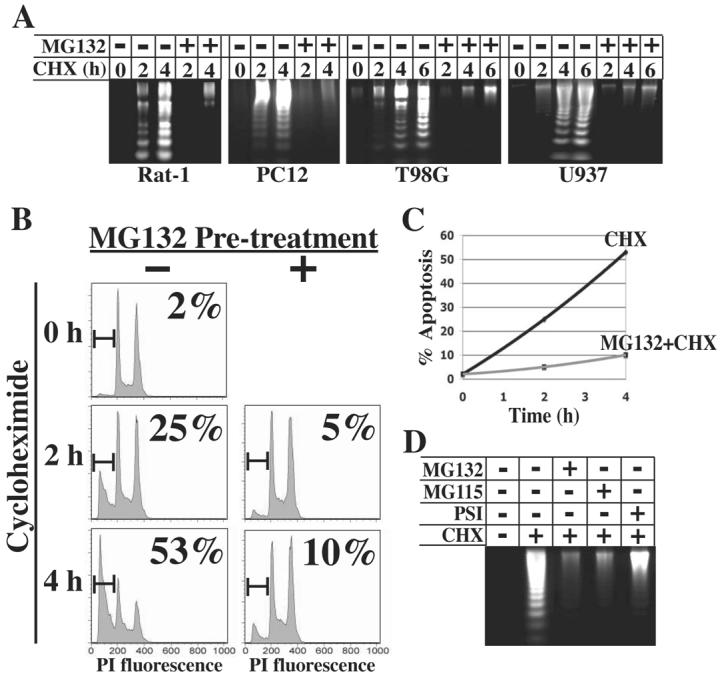
Inhibition of the proteasome blocks apoptosis induced by cycloheximide. A, Cultures of Rat-1, PC12, U937, or T98G cells were left untreated or treated with 10 μM MG132 for 30 min prior to treatment with 10 μg/ml cycloheximide (CHX). Cytosolic nucleic acids were isolated after the indicated time of cycloheximide treatment and DNA fragmentation assessed by gel electrophoresis. B, U937 cells were harvested at the indicated times after cycloheximide treatment, stained with propidium iodide, and analyzed for DNA content by flow cytometry. The percentage of cells with sub-G1 DNA content is indicated in each panel. C, The percentage of apoptotic cells from panel B is plotted as a function of time of cycloheximide treatment. D, U937 cells were treated with 10 μM MG132, 30 μM MG115, or 30 μM PSI for 30 min prior to treatment with cycloheximide. DNA fragmentation was assayed after 6 h of cycloheximide treatment.
DNA content analysis was performed on U937 cells by flow cytometry in order to quantify the percentage of cells undergoing apoptosis. As shown in Figures 1B and C, cycloheximide treatment resulted in 25% and 53% cells within the sub-G1 population at 2 and 4 h, respectively. However, pretreatment with MG132 largely blocked apoptosis, as only 5% and 10% cells were in the sub-G1 population at 2 and 4 h, respectively.
To ensure that the effects of MG132 were due to its inhibition of the proteasome, U937 cells were pretreated with two other proteasome inhibitors, MG115 and PSI (Proteasome Inhibitor I) prior to cycloheximide, both of which also blocked apoptosis through 6 h (Figure 1D). Collectively, these studies show that apoptosis induced by cycloheximide requires proteasome activity, indicating that the loss of one or more proteins via proteasome-mediated degradation is responsible for apoptosis induced by translation inhibition.
Translation inhibition activates the mitochondrial pathway of apoptosis
Protein degradation upon translation inhibition could cause cell death due to a non-specific global loss of protein, resulting in deterioration of the cell in an apoptosis-like manner. Alternatively, cell death could occur through caspase-dependent apoptosis due to the loss of a specific protein, or set of proteins, whose activity normally maintains cell survival by blocking caspase activation directly or indirectly. Treatment of U937 cells with cycloheximide for 6 h resulted in cleavage of the caspase substrate PARP, which was blocked by the general caspase inhibitor zVAD-fmk (Figure 2A), indicating that translation inhibition induces caspase activation. To test whether cell death is dependent on caspase activity, the effect of zVAD-fmk on cycloheximide-induced DNA fragmentation was examined. Pretreatment of Rat-1, U937, and T98G cells with zVAD-fmk blocked apoptosis through 4 h of cycloheximide treatment (Figure 2B), indicating that translation inhibition activates an apoptotic program leading to caspase-dependent cell death.
Fig. 2.
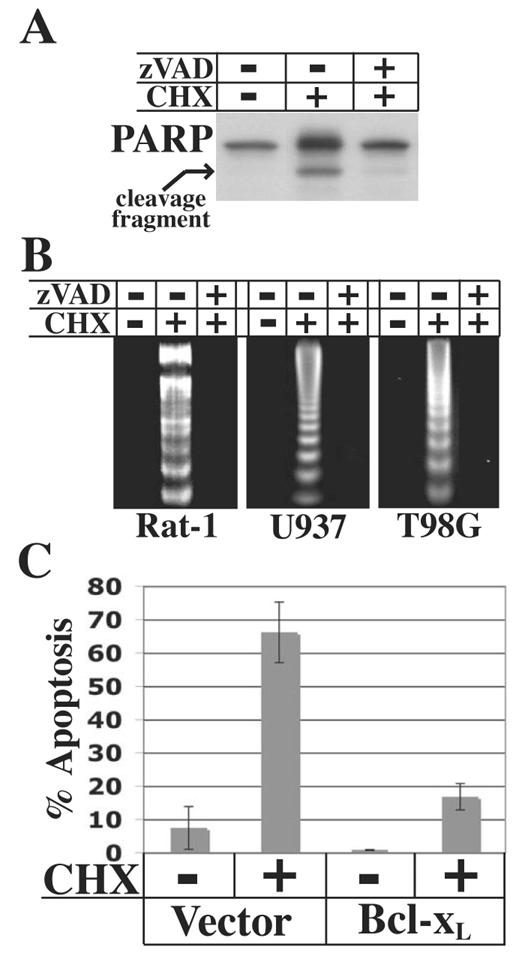
Apoptosis induced by translation inhibition is dependent on caspase activity and blocked by Bcl-xL overexpression. A, U937 cells were left untreated or treated with 10μM zVAD-fmk for 30 min prior to treatment with 10 μg/ml cycloheximide for 6 h. Cells were harvested for immunoblot analysis to detect PARP. B, Cells were treated with zVAD-fmk (Rat-1 and T98G cells, 100 μM; U937 cells, 10 μM) or left untreated for 30 min prior to treatment with 10 μg/ml cycloheximide for 4 h. Cytosolic nucleic acids were isolated and DNA fragmentation assessed by gel electrophoresis. C, HeLa cells were transfected with 1μg pCMV-DsRed expression construct and 1 μg pcDNA3 empty vector or pSG5-Bcl-xL expression contruct. 48 h after transfection, cells were treated with 10 μg/ml cycloheximide for 18 h, trypsinized, and apoptosis was assessed by TUNEL analysis. Transfected cells were identified by flow cytometry and data are presented as the percentage of transfected cells that were TUNEL-positive. The data represent average values of two independent cultures ± SD.
Caspase activation during apoptosis typically occurs due to mitochondrial outer membrane permeabilization (MOMP) and release of pro-apoptotic factors including cytochrome c into the cytosol (1). Pap and Cooper (17) previously reported that cycloheximide induces cytochrome c release, suggesting that translation inhibition activates apoptosis through the mitochondrial pathway. In healthy cells, MOMP and cytochrome c release are prevented by anti-apoptotic Bcl-2 proteins. Therefore, to further establish whether translation inhibition activates the mitochondrial pathway of apoptosis, the effect of overexpression of the anti-apoptotic Bcl-2 protein Bcl-xL was examined. Due to their high transfectability, HeLa cells were used in this experiment. Cells were transfected with empty vector or a Bcl-xL expression construct. Forty-eight h after transfection, cells were treated with cycloheximide for 18 h followed by TUNEL analysis in concert with flow cytometry. While transfection with empty vector resulted in approximately 65% apoptotic cells following cycloheximide treatment, overexpression of Bcl-xL largely blocked apoptosis (Figure 2C). These results further indicate that translation inhibition activates the mitochondrial pathway of apoptosis, leading to caspase activation and cell death.
A critical early step in mitochondrial apoptosis is activation of the pro-apoptotic Bcl-2 proteins Bak and/or Bax, which then homo-oligomerize to form pores in the mitochondrial outer membrane (1). If apoptosis following translation inhibition occurs via the mitochondrial pathway, active Bak and/or Bax should be detected upon cycloheximide treatment. Since they homo-oligomerize when activated, active Bak and Bax can be detected by treating mitochondria with the cross-linker bismaleimidohexane (BMH) prior to SDS-PAGE and immunoblot analysis, resulting in higher molecular weight complexes of active Bak and Bax. As shown in Figure 3A, oligomeric complexes of both Bak and Bax were detected after 8 h cycloheximide treatment of U937 cells, indicating that both proteins are activated following translation inhibition. Furthermore, the anti-apoptotic protein(s) degraded by the proteasome appears to act upstream of Bak/Bax activation, as proteasome inhibition with MG132 blocked Bak and Bax activation (Figure 3B). Collectively, these results indicate that translation inhibition results in the loss of a protein(s) via proteasome-mediated degradation, resulting in activation of Bak and Bax, caspase activation, and subsequent cell death.
Fig 3.
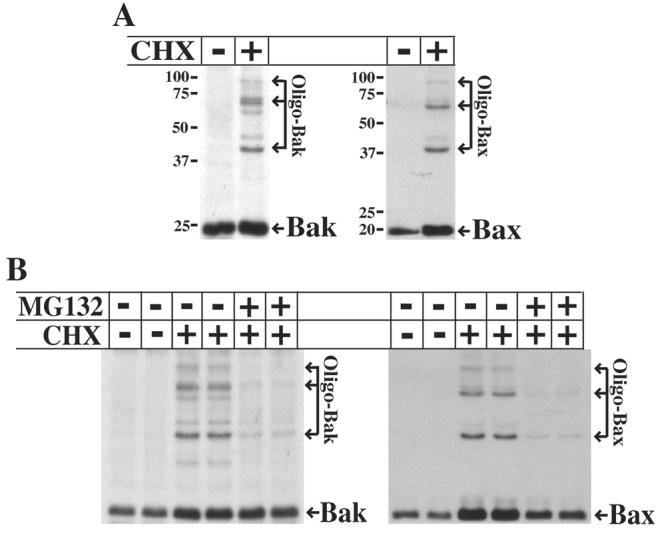
Translation inhibition induces Bak and Bax activation. A, U937 cells were left untreated or treated with 10 μg/ml cycloheximide for 8 h. Mitochondria were isolated and cross-linking reactions performed using the cross-linker BMH, followed by SDS-PAGE and immunoblot analysis to visualize oligomeric Bak and Bax. B, Duplicate cultures of U937 cells were left untreated or pretreated with 10 μM MG132 for 30 min prior to treatment with 10 μg/ml cycloheximide for 6 h. Cells were analyzed as described in A.
Proteasome-mediated degradation of Mcl-1 precedes Bak/Bax activation and apoptosis
Antiapoptotic Bcl-2 family proteins maintain cell survival by antagonizing Bak and Bax activation through protein-protein interactions (2). Therefore, the loss of one or more of these proteins could be responsible for the observed activation of Bak and Bax. Previous studies have shown that the anti-apoptotic Bcl-2 family protein Mcl-1 is rapidly turned over by proteasome-mediated degradation (48-51). Therefore, the stability of Mcl-1, as well those of Bcl-2 and BclxL, was assessed following cycloheximide treatment. Consistent with previous reports, Mcl-1 was rapidly lost with a half-life of less than 2 h in U937, T98G and HeLa cells (Figure 4), while both Bcl-2 and Bcl-xL remained largely stable.
Fig. 4.
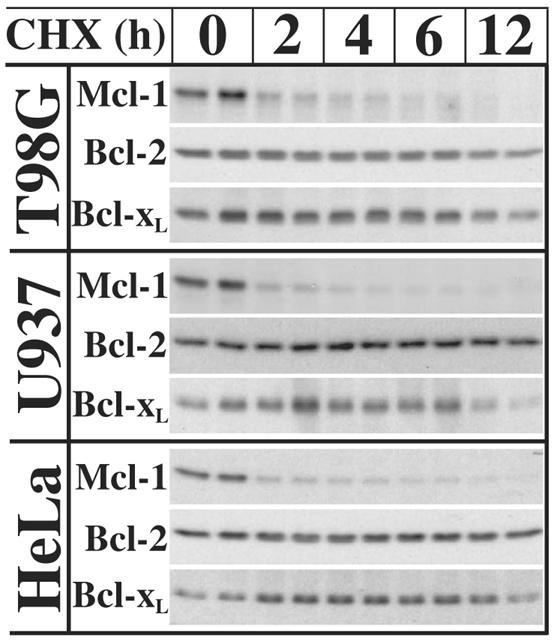
Mcl-1 is rapidly degraded following translation inhibition. Duplicate cultures of U937, T98G, and HeLa cells were harvested 0, 2, 4, 6, and 12 h after treatment with 10 μg/ml cycloheximide for immunoblot analysis of Mcl-1, Bcl-xL, and Bcl-2.
If the loss of Mcl-1 is responsible for the activation of Bak and Bax, its loss should precede both Bak/Bax activation and apoptotic events. Therefore, the kinetics of these events were compared in U937 cells following 0-6 h of cycloheximide treatment. As shown in Figure 5A (lanes 1-6), Mcl-1 degradation occurred rapidly with a half-life of approximately 30 min (Figure 5B). Importantly, the rapid drop in Mcl-1 preceded the detection of both DNA fragmentation and cleavage of the caspase substrate PARP, which were both first detected at 2 h (Figure 5A). Moreover, the time-dependent activation of Bak and Bax was also first detected at 2 h (Figure 5C), again following the early drop in Mcl-1 levels.
Fig. 5.
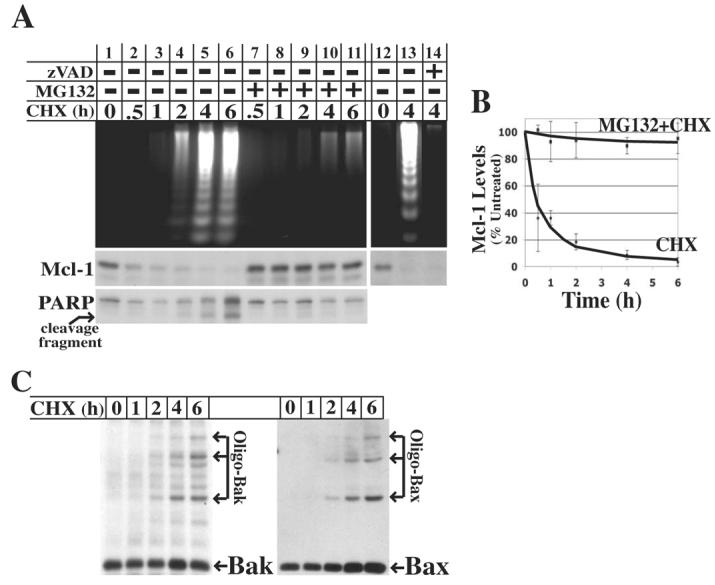
Mcl-1 degradation precedes several markers of apoptosis and is blocked by proteasome inhibition. A, U937 cells were either left untreated or pretreated with either 10 μM MG132 or 10 μM zVAD-fmk for 30 min prior to treatment with 10 μg/ml cycloheximide for 0 – 6 h. Each flask of cells was separated into two equal fractions, and processed to either assess DNA fragmentation by gel electrophoresis or Mcl-1 levels and PARP cleavage by SDS-PAGE and immunoblot analysis. B, The levels of Mcl-1 from three independent experiments with U937 cells were quantitated by densitometry and plotted (average ± SD) relative to untreated cells. C, U937 cells were left untreated or treated with 10 μg/ml cycloheximide for 1 – 6 h. Mitochondrial fractions were isolated and treated with cross-linker BMH followed by SDS-PAGE and immunoblot analysis to detect oligomeric Bak and Bax.
As expected, proteasome inhibition by pretreatment with MG132 blocked both apoptosis and the loss of Mcl-1 following cycloheximide treatment (Figure 5A, lanes 7-11 and Figure 5B). However, previous studies have shown that Mcl-1 is cleaved by caspases during apoptosis induced by some stimuli (52-56). While contrary to the observed kinetics, it is conceivable that Mcl-1 might be a target of early caspase activation during apoptosis induced by cycloheximide, in which case MG132 would block Mcl-1 loss indirectly by blocking caspase activation. To rule out this possibility, the effect of caspase inhibition on Mcl-1 stability was assessed. While caspase inhibition with zVAD-fmk blocked apoptosis, it did not block the loss of Mcl-1 (Figure 5A, lanes 12-14). Collectively, these observations further strengthen the hypothesis that the loss of Mcl-1 by proteasome-mediated degradation is, at least in part, responsible for Bak/Bax activation and apoptosis following translation inhibition.
Mcl-1 overexpression blocks apoptosis induced by translation inhibition
If the loss of Mcl-1 following translation inhibition is responsible for the subsequent induction of apoptosis, then maintenance of Mcl-1 levels should prevent apoptosis. To test this hypothesis, HeLa cells were transiently transfected with expression constructs for Mcl-1, Bcl-xL, or Bcl-2, or with empty vector followed by treatment with cycloheximide for 20 h. Approximately 55% of cells transfected with empty vector were apoptotic following cycloheximide treatment, while overexpression of Mcl-1 largely blocked apoptosis (Figure 6A). This further suggests that the loss of Mcl-1 is a contributing factor to apoptosis. As shown earlier, overexpression of Bcl-xL also blocked apoptosis, while overexpression of Bcl-2 provided only a partial block. Immunoblot analysis indicated that Bcl-xL and Bcl-2 were overexpressed at similar levels relative to the respective endogenous proteins (Figure 6B). Overexpression of Mcl-1 was modest compared to that of Bcl-xL and Bcl-2, likely due to the instability of Mcl-1 protein. However, significant levels of Mcl-1 were still detectable following treatment with cycloheximide, likely as a result of higher levels of overexpression in some transfected cells.
Fig. 6.
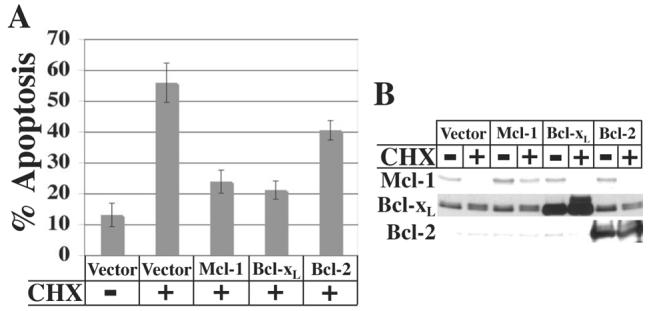
Effect of Mcl-1, Bcl-xL, and Bcl-2 overexpression on apoptosis induced by translation inhibition. A, HeLa cells were transfected with 1 μg pCMV-DsRed plus 1 μg pcDNA3 empty vector or pcDNA3-Mcl-1, -Bcl-xL, or –Bcl-2 expression constructs. 24 or 48 h after transfection, cells were left untreated or treated with 10 μg/ml cycloheximide for 20 h and harvested for TUNEL analysis by flow cytometry. Data represent the percentage of transfected cells that were TUNEL-positive and are averages ± SD from four independent experiments. B, Immunoblot analysis of Mcl-1, Bcl-xL and Bcl-2 expression in cells from one of the experiments shown in A.
Knockdown of Mcl-1 via RNA interference is sufficient to induce apoptosis
To determine if the loss of Mcl-1 is sufficient to induce apoptosis, the effect of Mcl-1 knockdown by RNA interference was tested in both T98G and HeLa cells. Initially, immunoblot analysis was performed 24 h post-transfection with three unique siRNAs designed to knockdown Mcl-1: siRNA 6314, 6126, and 42844 (Figure 7A). siRNA 6314 yielded >95% knockdown of Mcl-1 protein compared to nonspecific (NS) siRNA, while siRNA 6126 consistently yielded an approximate 50% knockdown, and siRNA 42844 had no effect. Importantly, all three siRNAs had no effect on Bcl-2 or Bcl-xL protein levels.
Fig. 7.
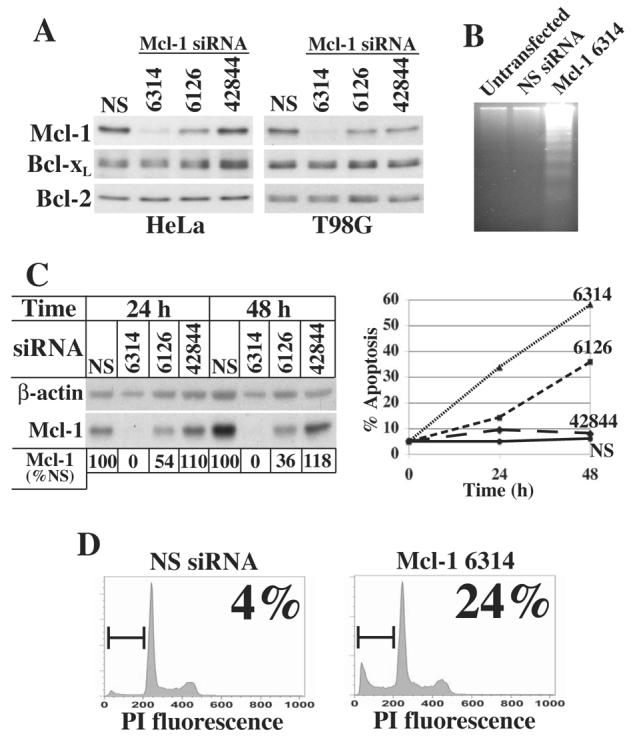
Mcl-1 knockdown induces apoptosis. A, HeLa and T98G cells were transfected with nonspecific (NS) siRNA or one of three siRNAs designed to knockdown Mcl-1, designated siRNAs 6314, 6126, and 42844. Cells were harvested 24 h after transfection and 20 μg protein subjected to SDS-PAGE and immunoblot analysis. B, T98G cells were left untransfected or transfected with nonspecific (NS) siRNA or Mcl-1 siRNA 6314. 48 h after transfection, cytosolic nucleic acids were isolated and subjected to gel electrophoresis to detect DNA fragmentation. C, T98G cells were transfected with NS siRNA or Mcl-1 siRNA 6314, 6126 or 42844. Cells were harvested 24 or 48 h after transfection for analysis of Mcl-1 protein levels by immunoblot and apoptosis by flow cytometry to quantitate the fraction of cells with sub-G1 DNA content. Mcl-1 levels following transfection with Mcl-1 siRNAs relative to transfection with nonspecific siRNA for the same durations were determined by densitometry and normalized to β-actin and shown below the immunoblot as % Mcl-1 remaining. D, HeLa cells were transfected with nonspecific (NS) siRNA or Mcl-1 siRNA 6314. Cells were harvested 24 h after transfection and analyzed by flow cytometry to determine the fraction of cells with sub-G1 DNA content.
Next, the effect of Mcl-1 knockdown with siRNA 6314 on apoptosis was examined in T98G cells. While transfection of T98G cells with a nonspecific siRNA did not induce apoptosis, knockdown of Mcl-1 with siRNA 6314 was sufficient to induce apoptosis as detected by DNA fragmentation 48 h after transfection (Figure 7B). A more thorough analysis of the effects of Mcl-1 knockdown in T98G cells was then conducted in which each of the three siRNAs were transfected for 24 and 48 h. Cells were harvested at each time point and examined for both Mcl-1 protein levels by immunoblot and percent apoptotic cells by sub-G1 analysis (Figure 7C). As expected, transfection with Mcl-1 siRNA 42844 neither knocked down Mcl-1 nor induced apoptosis. However, transfection with Mcl-1 siRNA 6314 yielded complete knockdown of Mcl-1 at 24 and 48 h, and resulted in a time-dependent apoptosis with 35% and 58% apoptotic cells at 24 and 48 h, respectively. Moreover, Mcl-1 siRNA 6126 yielded intermediate effects on both Mcl-1 protein levels and apoptosis. A 2- to 3-fold drop in Mcl-1 protein was observed at 24 and 48 h, respectively, yielding 15% and 37% apoptotic cells at the same time points. These results demonstrate the importance of maintaining a threshold level of Mcl-1 for cell survival, and suggest that drops in Mcl-1 levels following translation inhibition in response to various cellular stresses may be a critical factor contributing to the induction of apoptosis.
Lastly, the effect of near complete knockdown of Mcl-1 was tested in HeLa cells by transfection with Mcl-1 siRNA 6314, which resulted in approximately 24% apoptotic cells at 24 h, as determined by sub-G1 analysis (Figure 7D). However, analysis at 48 h post-transfection revealed similar levels of apoptosis (data not shown), indicating that HeLa cells are less dependent on Mcl-1 for survival than T98G cells.
Knockdown of Bak, Bax, or Bim protects cells from apoptosis induced by inhibition of translation
Most apoptotic stimuli activate pro-apoptotic BH3-only proteins, which then associate with anti-apoptotic Bcl-2 proteins through interactions that displace and activate Bak and Bax (2). Multiple reports have shown that Mcl-1 interacts with and inhibits the pro-apoptotic BH3-only protein Bim (57-60). In addition, Mcl-1 interacts directly with Bak to sequester Bak in an inactive complex (61, 62). The degradation of Mcl-1 following translation inhibition would thus be predicted to result in the direct activation of both Bak and Bim, with the activation of Bim then leading to subsequent activation of Bax as well as Bak. We therefore tested the roles of these downstream targets of Mcl-1 in apoptosis resulting from inhibition of translation.
RNA interference was used to knockdown Bak, Bax, and Bim in HeLa cells prior to treatment with cycloheximide for 24 h. Transfection with siRNAs resulted in a 5-10 fold knockdown of the respective proteins (Bim is expressed in three isoforms: BimEL, BimL, and BimS [63]) (Figure 8A). Apoptosis following cycloheximide treatment was significantly reduced from approximately 50% to 30-35% apoptotic cells by knockdown of either Bak or Bax (p < 0.001) (Figure 8B), consistent with our earlier observations showing activation of both proteins in response to cycloheximide (see Figure 3). Knockdown of Bim resulted in a similar inhibition of apoptosis following cycloheximide treatment (p < 0.001) (Figure 8C), indicating that Bim, as well as Bak and Bax, is involved in apoptosis induced by inhibition of translation.
Fig 8.
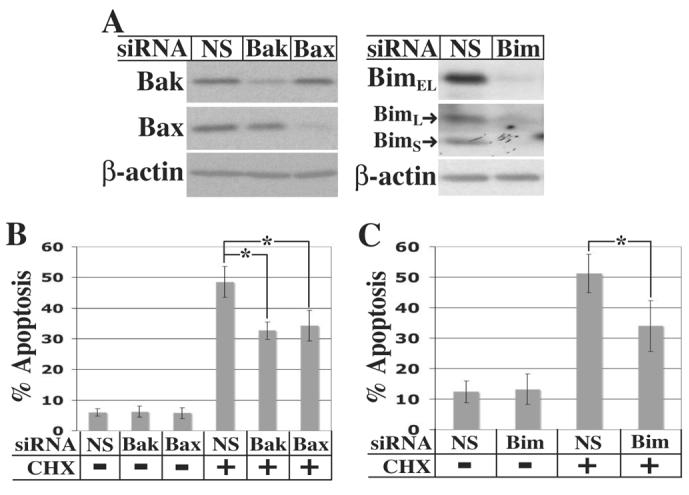
Knockdown of Bak, Bax, or Bim partially blocks cycloheximide-induced apoptosis. A, HeLa cells were transfected for 72 h with nonspecific (NS), Bak, Bax, or Bim siRNA and harvested for immunoblot analysis. B and C, 72 h after transfection with the indicated siRNA, HeLa cells were either left untreated or treated with 10 μg/ml cycloheximide for an additional 24 h. Cells were stained with propidium iodide and the fraction of cells with sub-G1 DNA content determined by flow cytometry. For Bak and Bax knockdowns (panel B), data are averages ± SD of ten independent cultures. Two different siRNAs (ID #'s 120199 and 120201) were used with comparable results. For Bim knockdowns (panel C), data are averages ± SD of seven independent cultures. *A Student's t-test comparing levels of apoptosis following cycloheximide treatment of cells transfected with nonspecific siRNA to cells transfected with Bak, Bax, or Bim siRNA all yielded a p-value < 0.001.
DISCUSSION
The modulation of global translation plays a role in the pro-survival and pro-apoptotic effects of a wide range of cellular stimuli. For example, growth factors promote cell survival, in part, by stimulating translation through activation of the PI3-kinase/Akt pathway, which activates several components of the translation machinery, including the initiation factors eIF2B and eIF4E (17, 26-30). In contrast, several pro-apoptotic stimuli, including dsRNA (produced during viral infection), interferon, lipopolysaccharide, and TNFα, induce apoptosis via inhibition of the translation initiation factor eIF2α by the protein kinase PKR (13-16).
Despite its pro-apoptotic effects in response to various stimuli, the mechanism by which global inhibition of translation promotes apoptosis has not been established. Potential mechanisms include selective translation of proapoptotic proteins from mRNAs containing IRES or upstream ORFs (10, 38). However, complete inhibition of translation with cycloheximide induces apoptosis in a wide range of cell types (17, 39-42), indicating that translation inhibition can induce apoptosis by a mechanism independent of increased translation of pro-apoptotic proteins. Since endogenous levels of cellular proteins are regulated by their rates of degradation as well as by their rates of translation, selective degradation of anti-apoptotic proteins is another mechanism by which global inhibition of translation could induce apoptosis. In the present study, we have tested this model by examining the induction of apoptosis following inhibition of translation with cycloheximide. Our results indicate that translation inhibition activates the mitochondrial pathway of apoptosis due to degradation of one or more anti-apoptotic proteins via the proteasome, and identify the Bcl-2 family protein Mcl-1 as one such protein whose degradation triggers the apoptotic machinery.
The induction of apoptosis by cycloheximide was blocked by proteasome inhibitors in several human and rat cell lines, indicating that selective degradation of cellular proteins was required for apoptosis resulting from inhibition of translation. Multiple observations demonstrated that apoptosis resulting from this proteasome-mediated degradation of cellular proteins was due to specific activation of the mitochondrial pathway of apoptosis rather than a generalized degradation of cellular constituents. First, the induction of apoptosis by cycloheximide was blocked by inhibition of caspases or by overexpression of the anti-apoptotic Bcl-2 family protein Bcl-xL. In addition, cycloheximide induced rapid activation of the pro-apoptotic Bcl-2 proteins Bak and Bax, consistent with previous findings that cycloheximide induced the release of mitochondrial cytochrome c (17). Inhibition of the proteasome prevented Bak and Bax activation as well as apoptosis, indicating that the degradation of one or more proteins acting upstream of Bak and Bax leads to activation of the mitochondrial pathway of apoptosis.
Bak and Bax are normally antagonized through interactions with anti-apoptotic Bcl-2 proteins, including Bcl-2, Bcl-xL, and Mcl-1 (1). Previous studies have demonstrated that Mcl-1 is targeted for proteasome-mediated degradation by the E3 ubiquitin ligase MULE (51) and is rapidly degraded with an estimated half-life of 30 min to 3 h (48-50), suggesting that degradation of Mcl-1 may activate Bak and/or Bax following translation inhibition. Consistent with these findings, inhibition of translation with cycloheximide resulted in rapid loss of Mcl-1 in U937, T98G, and HeLa cells. The half-life of Mcl-1 following inhibition of translation was approximately 30 min and its degradation was blocked by proteasome inhibition. Loss of Mcl-1 preceded Bak and Bax activation, cleavage of the caspase substrate PARP, and the onset of apoptosis as assessed by DNA fragmentation.
Overexpression of Mcl-1 inhibited apoptosis in response to cycloheximide, similar to the effect of Bcl-xL overexpression, consistent with the loss of Mcl-1 contributing to apoptosis following translation inhibition. Further support for the role of Mcl-1 was obtained using RNA interference to knockdown Mcl-1 levels. Near complete knockdown of Mcl-1 in T98G cells efficiently induced apoptosis, with 35% and 58% apoptotic cells at 24 and 48 h, respectively. Strikingly, only a 2- to 3-fold knockdown of Mcl-1 in these cells also induced apoptosis, though at more modest levels of 15% and 36% at 24 and 48 h, respectively. These results suggest a critical role for the maintenance of a threshold level of Mcl-1 for cell survival. However, knockdown of Mcl-1 in HeLa cells had more modest effects, with near complete knockdown resulting in approximately 24% apoptotic cells at 24 h without any further increase at 48 h. Previous reports have shown that knockdown of Mcl-1 sensitized HeLa cells to apoptosis, but did not significantly induce apoptosis by itself (48, 61), suggesting that their survival is less dependent on Mcl-1 than that of T98G cells or other cells, including several hematopoietic cell lines (55, 64-66), breast carcinoma cells (67), lung cancer cells (68), and B and T lymphocytes (60) in which knockdown or elimination of Mcl-1 has also been shown to induce apoptosis. The explanation for the differential effects of Mcl-1 knockdown on survival of different cells is not entirely clear, but might reflect different expression levels of other Bcl-2 family proteins relative to Mcl-1.
Further evidence implicating Mcl-1 degradation in apoptosis induced by cycloheximide was obtained by analysis of its downstream targets. Most stimuli that induce apoptosis lead to activation of pro-apoptotic BH3-only proteins, which then activate Bak and/or Bax (1). Previous studies have shown that Mcl-1 specifically interacts with the BH3-only protein Bim and inhibits apoptosis, at least in part, by sequestering Bim in an inactive state (57-60). In addition, Mcl-1 directly sequesters Bak in an inactive complex (61, 62). We therefore tested the effects of RNAi against Bim, Bak, and Bax and found that knockdown of each significantly inhibited apoptosis following cycloheximide treatment. Collectively, these results suggest that degradation of Mcl-1 upon translation inhibition induces apoptosis through the release of both Bim and Bak, with release of Bim then activating Bak and Bax through their displacement from anti-apoptotic Bcl-2 family proteins other than Mcl-1.
We note that this model may account for the differential effects of Bcl-2 and Bcl-xL overexpression on cycloheximide-induced apoptosis. Bcl-xL, like Mcl-1, largely blocked apoptosis resulting from cycloheximide treatment, while overexpression of Bcl-2 only inhibited apoptosis by about 50% (see Fig. 6). Bim can interact with Mcl-1, Bcl-xL and Bcl-2 (69). However, Willis et al (62) recently demonstrated that while Mcl-1 and Bcl-xL sequester Bak in inactive complexes, Bcl-2 does not. Therefore, overexpression of Bcl-2 may yield incomplete protection due to an inability to sequester Bak released from Mcl-1/Bak complexes.
Loss of Mcl-1 has been shown to play a key role in apoptosis induced by adenovirus E1A expression, UV irradiation, and the protein kinase inhibitor BAY 43-9006 (48, 49, 61). The present findings indicate a general role for loss of Mcl-1 in apoptosis resulting from global inhibition of protein synthesis. It is noteworthy that many apoptotic stimuli, including UV irradiation, inhibit global translation as well as activate one or more pro-apoptotic BH3-only proteins. Due to its rapid degradation, intracellular levels of Mcl-1 are critically affected by translation inhibition, suggesting that Mcl-1 may serve as a convergence point for distinct apoptotic stimuli, directly coupling global translation to cell survival and apoptosis.
The abbreviations used are
- 4E-BP1
eIF4E-binding protein
- IRES
internal ribosome entry site
- DMEM
Dulbecco's modified Eagle's medium
- MEM
minimum essential medium
- PBS
phosphate-buffered saline
- BMH
bismaleimidohexane
- PSI
proteasome inhibitor I
- MOMP
mitochondrial outer membrane permeabilization
- CHX
cycloheximide
Footnotes
This research was supported by grant RO1 CA18689 from the National Institutes of Health.
REFERENCES
- 1.Danial NN, Korsmeyer SJ. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 2.Willis SN, Adams JM. Curr. Opin. Cell Biol. 2005;17:617–625. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakano K, Vousden KH. Mol. Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 4.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. Mol. Cell. 2001;7:673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- 5.Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 6.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 7.del Peso L, González-García M, Page C, Herrera R, Nuñez G. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 8.Dijkers PF, Medema RH, Lammers J-WJ, Koenderman L, Coffer PJ. Curr. Biol. 2000;10:1201–1204. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 9.Dever TE. Cell. 2002;108:545–556. doi: 10.1016/s0092-8674(02)00642-6. [DOI] [PubMed] [Google Scholar]
- 10.Holcik M, Sonenberg N. Nat. Rev. Mol. Cell. Biol. 2005;6:318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- 11.Morley SJ, Coldwell MJ, Clemens MJ. Cell Death Diff. 2005;12:571–584. doi: 10.1038/sj.cdd.4401591. [DOI] [PubMed] [Google Scholar]
- 12.Barber GN. Cell Death Diff. 2005;12:563–570. doi: 10.1038/sj.cdd.4401643. [DOI] [PubMed] [Google Scholar]
- 13.Gil J, Alcamí J, Esteban M. Mol. Cell Biol. 1999;19:4653–4663. doi: 10.1128/mcb.19.7.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu L-C, Park JM, Zhang K, Luo J-L, Maeda S, Kaufman RJ, Eckmann L, Guiney DG, Karin M. Nature. 2004;428:341–345. doi: 10.1038/nature02405. [DOI] [PubMed] [Google Scholar]
- 15.Scheuner D, Patel R, Wang F, Lee K, Kumar K, Wu J, Nilsson A, Karin M, Kaufman RJ. J. Biol. Chem. 2006;281:21458–21468. doi: 10.1074/jbc.M603784200. [DOI] [PubMed] [Google Scholar]
- 16.Srivastava SP, Kumar KU, Kaufman RJ. J. Biol. Chem. 1998;273:2416–2423. doi: 10.1074/jbc.273.4.2416. [DOI] [PubMed] [Google Scholar]
- 17.Pap M, Cooper GM. Mol. Cell. Biol. 2002;22:578–586. doi: 10.1128/MCB.22.2.578-586.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cross DAE, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 19.Frame S, Cohen P. Biochem. J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jope RS, Johnson GVW. Trends Biochem. Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 21.Pap M, Cooper GM. J. Biol. Chem. 1998;272:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- 22.Pavitt GD. Biochem. Soc. Trans. 2005;33:1487–1492. doi: 10.1042/BST0331487. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Paulin FEM, Campbell LE, Gomez E, O'Brien K, Morrice N, Proud CG. EMBO J. 2001;20:4349–4359. doi: 10.1093/emboj/20.16.4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Welsh GI, Miller CM, Loughlin AJ, Price NT, Proud CG. FEBS Lett. 1998;421:125–130. doi: 10.1016/s0014-5793(97)01548-2. [DOI] [PubMed] [Google Scholar]
- 25.Sarbassov DD, Ali SM, Sabatini DM. Curr. Opin. Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 26.Li S, Takasu T, Perlman DM, Peterson MS, Burrichter D, Avdulov S, Bitterman PB, Polunovsky VA. J. Biol. Chem. 2003;278:3015–3022. doi: 10.1074/jbc.M208821200. [DOI] [PubMed] [Google Scholar]
- 27.Polunovsky VA, Rosenwald IB, Tan AT, White J, Chiang L, Sonenberg N, Bitterman PB. Mol. Cell Biol. 1996;16:6573–6581. doi: 10.1128/mcb.16.11.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan A, Bitterman P, Sonenberg N, Peterson M, Polunovsky V. Oncogene. 2000;19:1437–1447. doi: 10.1038/sj.onc.1203446. [DOI] [PubMed] [Google Scholar]
- 29.Li S, Sonenberg N, Gingras A-C, Peterson M, Avdulov S, Polunovsky VA, Bitterman PB. Mol. Cell Biol. 2002;22:2853–2861. doi: 10.1128/MCB.22.8.2853-2861.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Polunovsky VA, Gingras A-C, Sonenberg N, Peterson M, Tan A, Rubins JB, Manivel JC, Bitterman PB. J. Biol. Chem. 2000;275:24776–24780. doi: 10.1074/jbc.M001938200. [DOI] [PubMed] [Google Scholar]
- 31.Huang S, Houghton PJ. Curr. Opin. Pharmacol. 2003;3:371–377. doi: 10.1016/s1471-4892(03)00071-7. [DOI] [PubMed] [Google Scholar]
- 32.Sarbassov DD, Ali SM, Sengupta S, Sheen J-H, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Mol. Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 33.Browne GJ, Proud CG. Mol. Cell Biol. 2004;24:2986–2997. doi: 10.1128/MCB.24.7.2986-2997.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spriggs KA, Bushell M, Mitchell SA, Willis AE. Cell Death Diff. 2005;12:585–591. doi: 10.1038/sj.cdd.4401642. [DOI] [PubMed] [Google Scholar]
- 35.Holcik M, Lefebvre C, Yeh C, Chow T, Korneluk RG. Nat. Cell Biol. 1999;1:190–192. doi: 10.1038/11109. [DOI] [PubMed] [Google Scholar]
- 36.Warnakulasuriyarachchi D, Cerquozzi S, Cheung HH, Holcík M. J. Biol. Chem. 2004;279:17148–17157. doi: 10.1074/jbc.M308737200. [DOI] [PubMed] [Google Scholar]
- 37.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. Mol. Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 38.Balachandran S, Kim CN, Yeh W-C, Mak TW, Bhalla K, Barber GN. EMBO J. 1998;17:6888–6902. doi: 10.1093/emboj/17.23.6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alessenko AV, Boikov PY, Filippova GN, Khrenov AV, Loginov AS, Makarieva ED. FEBS Lett. 1997;416:113–116. doi: 10.1016/s0014-5793(97)01161-7. [DOI] [PubMed] [Google Scholar]
- 40.Blom WM, de Bont HJGM, Meijerman I, Mulder GJ, Nagelkerke JF. Biochem. Pharmacol. 1999;58:1891–1898. doi: 10.1016/s0006-2952(99)00268-3. [DOI] [PubMed] [Google Scholar]
- 41.Martin SJ, Lennon SV, Bonham AM, Cotter TG. J. Immunol. 1990;145:1859–1867. [PubMed] [Google Scholar]
- 42.Tang D, Lahti JM, Grenet J, Kidd VJ. J. Biol. Chem. 1999;274:7245–7252. doi: 10.1074/jbc.274.11.7245. [DOI] [PubMed] [Google Scholar]
- 43.Hockenbery D, Nuñez G, Milliman C, Schreiber RD, Korsmeyer SJ. Nature. 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 44.Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, Korsmeyer SJ. Genes & Dev. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- 45.Tullai JW, Schaffer ME, Mullenbrock S, Kasif S, Cooper GM. J. Biol. Chem. 2004;279:20167–20177. doi: 10.1074/jbc.M309260200. [DOI] [PubMed] [Google Scholar]
- 46.Yao R, Cooper GM. Oncogene. 1996;13:343–351. [PubMed] [Google Scholar]
- 47.Erhardt P, Cooper GM. J. Biol. Chem. 1996;271:17601–17604. doi: 10.1074/jbc.271.30.17601. [DOI] [PubMed] [Google Scholar]
- 48.Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. Genes & Dev. 2003;17:1475–1486. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rahmani M, Davis EM, Bauer C, Dent P, Grant S. J. Biol. Chem. 2005;280:35217–35227. doi: 10.1074/jbc.M506551200. [DOI] [PubMed] [Google Scholar]
- 50.Yang T, Kozopas KM, Craig RW. J. Cell Biol. 1995;128:1173–1184. doi: 10.1083/jcb.128.6.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhong Q, Gao W, Du F, Wang X. Cell. 2005;121:1085–1095. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 52.Clohessy JG, Zhuang J, Brady HJM. Brit. J. Haematol. 2004;125:655–665. doi: 10.1111/j.1365-2141.2004.04949.x. [DOI] [PubMed] [Google Scholar]
- 53.Gomez-Bougie P, Oliver L, Gouill SL, Bataille R, Amiot M. Oncogene. 2005;24:8076–8079. doi: 10.1038/sj.onc.1208949. [DOI] [PubMed] [Google Scholar]
- 54.Herrant M, Jacquel A, Marchetti S, Belhacène N, Colosetti P, Luciano F, Auberger P. Oncogene. 2004;23:7863–7873. doi: 10.1038/sj.onc.1208069. [DOI] [PubMed] [Google Scholar]
- 55.Michels J, O'Neill JW, Dallman CL, Mouzakiti A, Habens F, Brimmell M, Zhang KYJ, Craig RW, Marcusson EG, Johnson PWM, Packham G. Oncogene. 2004;23:4818–4827. doi: 10.1038/sj.onc.1207648. [DOI] [PubMed] [Google Scholar]
- 56.Weng C, Li Y, Xu D, Shi Y, Tang H. J. Biol. Chem. 2005;280:10491–10500. doi: 10.1074/jbc.M412819200. [DOI] [PubMed] [Google Scholar]
- 57.Gomez-Bougie P, Bataille R, Amiot M. Eur. J. Immunol. 2004;34:3156–3164. doi: 10.1002/eji.200424981. [DOI] [PubMed] [Google Scholar]
- 58.Han J, Goldstein LA, Gastman BR, Froelich CJ, Yin X-M, Rabinowich H. J. Biol. Chem. 2004;279:22020–22029. doi: 10.1074/jbc.M313234200. [DOI] [PubMed] [Google Scholar]
- 59.Han J, Goldstein LA, Gastman BR, Rabinowich H. J. Biol. Chem. 2006;281:10153–10163. doi: 10.1074/jbc.M510349200. [DOI] [PubMed] [Google Scholar]
- 60.Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Nature. 2003;426:671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 61.Cuconati A, Mukherjee C, Perez D, White E. Genes & Dev. 2003;17:2922–2932. doi: 10.1101/gad.1156903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DCS. Genes & Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O'Connor L, Strasser A, O'Reilly LA, Hausmann G, Adams JM, Cory S, Huang DCS. EMBO J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aichberger KJ, Mayerhofer M, Krauth M-T, Skvara H, Florian S, Sonneck K, Akgul C, Derdak S, Pickl WF, Wacheck V, Selzer E, Monia BP, Moriggl R, Valent P, Sillaber C. Blood. 2005;105:3303–3311. doi: 10.1182/blood-2004-02-0749. [DOI] [PubMed] [Google Scholar]
- 65.Derenne S, Monia B, Dean NM, Taylor JK, Rapp M-J, Harousseau J-L, Bataille R, Amiot M. Blood. 2002;100:194–199. doi: 10.1182/blood.v100.1.194. [DOI] [PubMed] [Google Scholar]
- 66.Liu H, Perlman H, Pagliari LJ, Pope RM. J. Exp. Med. 2001;194:113–125. doi: 10.1084/jem.194.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Han J, Goldstein LA, Gastman BR, Rabinovitz A, Rabinowich H. J. Biol. Chem. 2005;280:16383–16392. doi: 10.1074/jbc.M411377200. [DOI] [PubMed] [Google Scholar]
- 68.Song L, Coppola D, Livingston S, Cress D, Haura EB. Cancer Biol. Ther. 2005;4:267–276. doi: 10.4161/cbt.4.3.1496. [DOI] [PubMed] [Google Scholar]
- 69.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DCS. Mol. Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]