

Abstract
The chronic phase of pulmonary arterial hypertension (PAH) is associated with vascular remodeling, especially thickening of the smooth muscle layer of large pulmonary arteries and muscularization of small pulmonary vessels, which normally have no associated smooth muscle. Serotonin (5-hydroxytryptamine, 5-HT) has been shown to induce proliferation and hypertrophy of pulmonary artery smooth muscle cells (PASMC), and may be important for in vivo pulmonary vascular remodeling. Here, we show that 5-HT stimulates migration of pulmonary artery PASMC. Treatment with 5-HT for 16 h increased migration of PASMC up to four-fold as monitored in a modified Boyden chamber assay. Increased migratory responses were associated with cellular morphological changes and reorganization of the actin cytoskeleton. 5-HT-induced alterations in morphology were previously shown in our laboratory to require cAMP [Lee SL, Fanburg BL. Serotonin produces a configurational change of cultured smooth muscle cells that is associated with elevation of intracellular cAMP. J Cell Phys 1992;150(2):396–405], and the 5-HT4 receptor was pharmacologically determined to be the primary activator of cAMP in bovine PASMC [Becker BN, Gettys TW, Middleton JP, Olsen CL, Albers FJ, Lee SL, et al. 8-Hydroxy-2-(di-n-propylamino)tetralin-responsive 5-hydroxytryptamine4-like receptor expressed in bovine pulmonary artery smooth muscle cells. Mol Pharmacol 1992;42(5):817–25]. We examined the role of the 5-HT4 receptor and cAMP in 5-HT-induced bovine PASMC migration. PASMC express 5-HT4 receptor mRNA, and a 5-HT4 receptor antagonist and a cAMP antagonist completely blocked 5-HT-induced cellular migration. Consistent with our previous report that a cAMP-dependent Cl− channel is required for 5-HT-induced morphological changes in PASMC, phenylanthranilic acid, a Cl− channel blocker, inhibited actin cytoskeletal reorganization and migration produced by 5-HT. We conclude that 5-HT stimulates PASMC migration and associated cytoskeletal reorganization through the 5-HT4 receptor and cAMP activation of a chloride channel.
Keywords: 5-HT, Migration, cAMP, Cl− channel, Cytoskeleton, 5-HTT, 5-HT4 receptor, 5-HT1B/1D receptor, MAPK
1. Introduction
Pulmonary arterial hypertension (PAH) is characterized by increased pulmonary vascular resistance, eventually leading to death from right ventricular heart failure [1]. Acutely, PAH occurs as a result of pulmonary artery vasoconstriction; however, more chronically, PAH involves remodeling of the pulmonary arterial wall. Pathology studies of the lungs of patients with PAH reveal increased medial wall thickness of muscular arteries and decreased elasticity of pulmonary blood vessels [2,3]. Additionally, studies show the development of a muscular layer in smaller vessels (arterioles), which normally have no associated smooth muscle [4]. The extension of pulmonary artery smooth muscle cells (PASMC) into nonmuscularized vessels suggests the involvement of factors that stimulate smooth muscle cell migration as well as growth in the pathogenesis of PAH.
An abundance of evidence suggests that serotonin (5-hydroxytryptamine, 5-HT) plays an important role in the pathogenesis of PAH [5,6]. Clinically, the use of serotonergic anorexigens (such as fenfluoramine) is a major risk factor for development of PAH [7,8]. PASMC taken from patients with familial PAH have been demonstrated to have altered expression of the serotonin transporter and receptors, and to proliferate more rapidly in response to 5-HT compared to PASMC from control patients [2,9-12]. In animal models of PAH, 5-HT has been shown to play a role in the development of pulmonary vascular remodeling [13-15]. 5-HT increases PAH in rats subjected to long-term hypoxia, and this effect can be blocked by a 5-HT antagonist [16]. In response to hypoxia, knockout mice for the serotonin transporter (5-HTT) have decreased pulmonary vascular remodeling compared to control mice [16].
In primary cultures of bovine PASMC, 5-HT induces hyperplasia and hypertrophy [17-21]. Experiments to investigate 5-HT signal transduction have shown that PASMC proliferation requires production of reactive oxygen species, activation of the small GTP-binding proteins Ras, Rac 1 and Rho, and activation of the p42/p44 mitogen-activated protein kinase (MAPK) [20-23]. Pharmacological experiments in cell cultures, using receptor and transporter antagonists, have shown that 5-HT-induced hyperplasia and hypertrophy are sensitive to inhibition of the serotonin transporter [17,18,24-26]. Recent cell culture and animal model experiments indicate that proliferation is also mediated by the 5-HT receptor subtype 1B/1D (5-HT1B/1D), which activates Rho/ROCK, suggesting that cross-talk occurs between 5-HT receptors and the 5-HTT for 5-HT-induced growth [22,27].
Our laboratory previously demonstrated that 5-HT stimulates cAMP-associated chloride (Cl−) efflux from PASMC, which is required for a 5-HT-induced change in PASMC morphology [28]. We now show that the change in cellular morphology correlates with alterations of the cellular actin cytoskeleton and migration in PASMC. Migration in PASMC is mediated by the 5-HT4 receptor, a positive regulator of adenylyl cyclase, and a cAMP antagonist (and inhibitor of cAMP-dependent protein kinase (PKA)) inhibits 5-HT-induced migration. Further, a chloride ion channel blocker phenylanthranilic acid inhibits 5-HT-induced migration and actin depolymerization, suggesting the involvement of Cl− transport in the mechanism of 5-HT-induced PASMC migration.
2. Materials and methods
2.1. Reagents
Citalopram, diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS), imipramine, 3-isobutyl-1-methyl-xanthine (IBMX), forskolin, methiothepin, N-phenylanthranilic acid (PAA; also sold as diphenylamine-2-carboxylate or DPC), serotonin (5-HT) and spiperone were purchased from Sigma–Aldrich (St. Louis, MO). RS 39604 hydrochloride (5-HT4 receptor antagonist), RS 23597-190 (a 5-HT4 receptor partial agonist), GR 55562 (5-HT1B/1D receptor antagonist) and SB 258585 (5-HT6 receptor antagonist) were purchased from Tocris (Ellisville, MO). AM8 (8-bromo-adenosine 3′,5′-cyclic monophosphorothioate, Rp-isomer) was purchased from Calbiochem (La Jolla, CA). 5-Nitro-2-(3-phenylpropylamino)-benzoic acid (NPPB) was purchased from Biomol (Plymouth Meeting, PA). Fetal bovine serum (FBS) was from Sigma or from Gemini Bioproducts (Woodland, CA). U0126 was purchased from Cell Signaling (Beverly, MA). Antibodies for phosphorylated and total Erk1/2 MAPK were purchased from Cell Signaling.
2.2. Cell culture
Bovine PASMC were prepared from the pulmonary arteries of fresh cow hearts as previously described [28]. Briefly, pulmonary arteries and aorta were separated from other tissue, immersed in a 0.1% iodide solution and then rinsed in cold PBS, pH 7.4, containing 300 units/mL penicillin, 300 μg/mL streptomycin and 1.25 μg/mL fungizone. Under aseptic conditions, each vessel was cut longitudinally with large iris scissors and unrolled to form a single sheet, which was briefly bathed in iodine solution, then rinsed again in sterile PBS with 100 U/mL penicillin, 100 μg/mL streptomycin and 1.25 μg/mL fungizone. The luminal aspect of the vessel was scraped with a sterile scalpel to remove the endothelium. For the bovine PASMC, 25 mm2 samples of the tunica media were collected using tissue forceps and small iris scissors. Samples were transferred luminal side up to six-well cell culture plates and allowed to adhere to the wells using several drops of medium for 30 min. Two milliliters of RPMI 1640 media with 20% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 2.5 μg/mL Plasmocin (Invivogen, San Diego, CA) and 1.25 μg/mL fungizone was added to each well without dislodging the tissue samples. The plates were incubated (5% CO2, 37 °C, in a humidified incubator) undisturbed for three days, in order to allow the pulmonary artery smooth muscle cells to grow out onto the culture wells. Subsequently, the cells were fed every two days until they reached confluence, when the tissue explants were removed and the cells were passaged. Cells were used from P1–P4.
2.3. Migration assay
Migration assays were performed using a standard modified Boyden chamber assay which determines cellular migration through a porous membrane [29]. Briefly, bovine PASMCs (P1–P4) were grown to confluence, trypsinized and washed in serum-free RPMI before plating into a Transwell plate (12 mm diameter, 12 μm pore size, Corning Incorporated Costar) at 4 × 105 cells per 12-well/plate. Cells were then treated with 10 μM 5-HT in the upper and lower chambers with or without 30 min pretreatment with inhibitors (also in upper and lower chambers). After 16–20 h of incubation the media were removed and cells were fixed with 0.75 mL 100% methanol for 15 min and then stained in 0.75 mL 1.88% Crystal Violet (in 20% methanol) for 1 h. The dye was then rinsed in cold water and the top surface of the Transwell membrane was wiped with a cotton swab to remove cells remaining on the upper side of the membrane. The number of cells that had traversed the membrane was counted in bright field microscopy. Each experiment was performed at least three times, with a minimum of n = 3.
2.4. Actin cytoskeletal staining
Bovine PASMCs (P1–P4) were seeded in 35 mm dishes onto glass coverslips coated with 0.015–0.15% gelatin for 8–16 h. Cells were grown to confluence and treated with 5-HT in the presence or absence of other agents as described in individual experiments. Following treatment, cells were washed once with 1 mL warm (37 °C) serum-free RPMI for 3 min. Cells were then washed once with 1 mL warm PBS for 3 min before fixing for 5 min in 1 mL formalin (4% formaldehyde in PBS). Cells were then gently washed three times with 1 mL room temperature PBS for 3 min. Cells were permeabilized for 5 min with Triton buffer (0.1% Triton, 50 mM PIPES, pH 7.0, 90 mM HEPES, pH 7.0, 0.5 mM MgCl2, 75 mM KCl and 0.5 mM EGTA) and again washed three times in PBS. Rhodamine-labeled phalloidin (Molecular Probes, Eugene, OR) (1:50 in 1% BSA in PBS) was added dropwise onto the center of the coverslip and incubated 1 h at ambient temperature. Finally, cells were washed twice with 1 mL PBS at ambient temperature for 3 min. Coverslips were mounted in 9:1 glycerol PBS. Fluorescent proteins were visualized on a Zeiss fluorescent microscope using 100× magnification, and pictures were obtained digitally with Metamorph software, Version 5.0 (Universal Imaging Corporation, Downingtown, PA). Microscopy was performed at the New England Medical Center/Tufts University GRASP Center for Imaging and Cell Analysis.
2.5. Tubulin cytoskeletal staining
Bovine PASMCs were cultured and treated as described above. Following treatment, cells were washed once with 1 mL warm (37 °C) serum-free RPMI for 3 min. Cells were then washed twice with 1 mL warm PBS for 3 min before fixing for 5 min in 1 mL of cold (−20 °C) absolute methanol. Cells were then gently washed three times for 5 min with 1 mL room temperature PBS. PBS was removed by aspiration. Dishes were incubated at ambient temperature for 1 h in 100 μL monoclonal mouse anti-α-tubulin antibody (Sigma–Aldrich, prepared 1:2000 in PBS), added dropwise to the center of the coverslips. Cells were again washed three times with PBS. After the third PBS wash was aspirated, dishes were incubated in 100 μL fluorescein-conjugated goat anti-mouse IgG (Jackson Immunoresearch, West Grove, PA, prepared 1:200 in PBS) for 1 h at ambient temperature. Finally, cells were washed twice with 1 mL room temperature PBS for 3 min. Coverslips were mounted in 9:1 glycerol PBS. Fluorescent proteins were visualized and photographed as above.
2.6. RNA isolation and PCR of 5-HT receptor subtypes and 5-HTT
Total cell RNA was obtained from cultured cells using Trizol (Invitrogen, Carlesbad, CA). RNA concentrations were determined spectrophotometrically at 260 nm. One microgram of RNA from each sample was reverse transcribed for 10 min at ambient temperature, followed by 30 min at 42 °C, in a 20 μL reaction containing 200 U Moloney murine leukemia virus reverse transcriptase (MMLV-RT) in 50 mM Tris–HCl (pH 8.3), 40 mM KCl, 6 mM MgCl2, 1 mM dithiothreitol (DTT), 660 pmol oligo-(dT)16, 0.5 mM each deoxynucleoside triphosphate (dNTP) and 1 U RNase inhibitor. Samples were heated to 95 °C for 5 min to inactivate the MMLV-RT and stored at −20 °C. All RT reagents were purchased from Applied Biosystems, Foster City, CA. As a control for DNA contamination, RNA was treated with DNase in control experiments. Two micrograms of RNA from each sample was incubated with 10 U DNase in supplied buffer (Boehringer Mannheim, Indianapolis, IN) for 15 min at ambient temperature before the addition of EDTA at 2.5 mM (final concentration).
PCR primers for bovine 5-HT receptor subtypes 4, 1B/1D, and 5-HTT were designed based on mRNA sequences in the NIH GenBank [30]. Primers for serotonin receptors and transporters were 100% homologous to the bovine sequences; all primers, including tubulin, spanned at least two exons, and the sizes of products were verified to ensure that products were obtained from cDNA and not the result of contaminating genomic DNA, which would produce larger PCR products. The following primers were used (NIH accession numbers in parentheses): 5-HT1B/1D receptor (AJ491859) forward 5′-TGCTCCTCATCGCCCTCTAT-3′, reverse 5′-TAGCGGCC ATGAGTTTCTTCT-3′ (258 bp product); 5-HT4 receptor (AJ491866) forward 5′-GGACAAACTTGATGCTAATGTGAG-3′, reverse 5′-GAAACCAGCA-GATCCGCAAA-3′ (250 bp product). For 5-HTT (AF119122) primers were: forward 5′-TGGGCC-AAGAAGGTGGATTT-3′, reverse 5′GATGGTGGTGTAGTAGGAGGCGA-3′ (294 bp product).
One microgram cDNA from the RT reaction was used in a PCR reaction with 0.4 μM each forward and reverse primer, 200 μM each dNTP and 1 U/50 μl iTaq DNA polymerase and supplied PCR buffer (BioRad Laboratories Inc., Hercules, CA). Control reactions were performed with RNA with no RT to ensure that the PCR product obtained was from cDNA and not genomic DNA. PCR reactions were optimized for annealing temperatures using a temperature gradient in a BioRad iCycler. For 5-HT receptor subtypes, reactions were carried out for 36 cycles using the following conditions: 30 s, 94 °C denaturation; 1 min, 56–60 °C annealing; 2 min, 72 °C extension. The last cycle extension was for 10 min. Conditions and primers for tubulin PCR were as previously described [31]; using 0.1 μl cDNA, 30 cycles were performed: 30 s, 94 °C denaturation; 2 min, 68 °C combined annealing/extension. The last cycle extension was for 10 min. A deletion mutant internal standard was used to normalize for differences in PCR efficiency for tubulin PCR reactions. PCR reactions were analyzed on a 3% agarose gel in Tris EDTA buffer and bands were visualized using ethidium bromide.
2.7. Western blot analysis
Cells for Western blotting were grown on 35 mm dishes. Cells were washed two times with ice-cold PBS. Cells were lysed in 100 μL buffer containing 50 mM HEPES, pH 7.4, 1% (v/v) Triton X-100, 4 mM EDTA, 1 mM tetrasodiumpyrophosphate, 0.1 mM sodium orthovanadate, 2 mM sodium fluoride, 10 μg/mL aprotinin and 10 μg/mL leupeptin at 4 °C. The insoluble material was removed by centrifugation (14,000 × g, 2 min, 4 °C), and the supernatant was used for analysis. Ten to 15 μg protein of whole cell lysate was used for SDS-PAGE on a 12% gel. After electrophoresis, proteins were transferred to Immobilion-P (Millipore, Bedford, MA). Blots were blocked in 5% BSA in Tris buffered saline (TBS; 20 mM Tris–HCl, pH 7.5, 150 mM NaCl and 0.05% Tween 20) for 1 h at ambient temperature before incubating overnight with 1:1000 dilution of primary antibody in 0.5% BSA protein/TBS at 4 °C. Blots were washed three times in TBS for 1 h total. Secondary antibody was diluted 1:1000 in TBS/0.1% Tween 20 for 2 h; blots were washed as above. ECL (Amersham, Piscataway, NJ) was applied according to the manufacturer's instructions before film exposure. Blots for phosphor-MAPK were stripped and reprobed for total MAPK to provide loading controls. Densitometry was performed on both blots to determine normalized levels of phospho-MAPK.
2.8. Quantification of stress fiber content in phalloidin-stained PASMC
Digital images of cells were individually scored in four categories by two investigators blinded to the treatment groups. The four categories were—1: no stress fibers; 2: 1–5 fibers; 3: 5–10 fibers; 4: >10 fibers. All viable cells in each field were scored, and a minimum of 100 cells were scored per category. No significant differences were found between the scores of the two investigators.
2.9. Statistical analysis
Stress fiber content scores were compared using the Kruskal–Wallis non-parametric test, followed by Dunn's multiple comparison test. Cell counts from migration studies were compared by Student's t-test, or by one-way ANOVA, followed by the Bonferroni test, as appropriate. p-Values < 0.05 were considered statistically significant.
3. Results
3.1. 5-HT induces morphological changes, actin depolymerization and migration of PASMC
Previous studies have shown that short-term treatment of PASMC with 5-HT induces specific morphological alterations [17,18] (Fig. 1A). The normally flat and spindle-shaped cells become rounded in the body with dendrite-like processes extending outward. Changes in cellular morphology, such as those induced by 5-HT, are suggestive of cytoskeletal alterations. Therefore, we investigated the effect of 5-HT on the actin cytoskeleton. Staining of cells with rhodamine–phalloidin showed that control cells contain actin stress fibers, generally organized lengthwise through the bodies of the cells (Fig. 1B). Treatment of cells with 5-HT for 30 min reduced the numbers of actin stress fibers, and caused the appearance of punctate staining throughout the body of the cells, indicating that the actin has undergone reorganization. In some cases, strongly stained cortical actin was also visible. Quantitative analysis of stress fibers per cell before and after treatment with 5-HT is shown in Fig. 1C. In resting conditions, most cells contained >10 fibers/cell, while after treatment with 5-HT, only ∼20% of cells contained >10 fibers, while over half the cells contained 5 fibers/cell or fewer. Staining for tubulin showed that microtubules are present in both control and 5-HT-treated cells, indicating that tubulin is not depolymerized or reorganized (Fig. 1D).
Fig. 1.

5-HT induces migration, morphological changes and actin depolymerization in BPASMC. (A and B) BPASMC were grown to 80% confluence on gelatin-treated coverslips and placed in 1% FBS/RPMI overnight before treatment with 5-HT (10 μM) for 30 min, 37 °C. All fields are representative, 40× magnification. (A) Phase contrast microscopy of cells. (B) Actin cytoskeleton stained with rhodamine-conjugated phalloidin. Increase in cortical actin and membrane ruffles can be observed. (C) Quantification of stress fiber per cell for control and 5-HT-treated PASMC. At least 100 cells were analyzed per condition as described in Section 2. All cells were analyzed in each field using at least six fields per condition from at least three different experiments. Conditions were scored independently by two blinded investigators—1: 0 fibers/cell; 2: 1–5 fibers/cell; 3: 5–10 fibers/cell; 4: >10 fibers/cell. (D) Cells were stained with a primary antibody for tubulin and a FITC-conjugated secondary antibody. (E) Migration of low passage (P1–P4) BPASMC in a modified Boyden chamber assay. Cells were plated into a Transwell plate in the presence or absence of 5-HT (10 μM) in both upper and lower chambers, and allowed to migrate for 16–20 h. Asterisk (*) indicates statistical significance from control levels, with p < 0.01, n = 3.
Actin cytoskeletal alterations are associated with a variety of biological activities, including migration [32]. We therefore tested the ability of 5-HT to increase migration in PASMC using a modified Boyden chamber assay to monitor cell migration through a porous membrane [29]. 5-HT added to the upper and lower chambers increased cell migration up to approximately four-fold over basal levels at 16–20 h (Fig. 1E). The change in cell migration is not due to increased cell growth, as measurable growth in response to 5-HT can be detected only at 48–72 h [33].
3.2. Effects of adenylyl cyclase-activating 5-HT receptors and cAMP on 5-HT-induced migration
Our laboratory has previously shown that cyclic AMP is required for 5-HT-induced morphological changes in PASMC [28], and that 5-HT induces an increase in cAMP in PASMC within 10–20 min [28,34]. Pharmacological experiments by Becker et al. identified 5-HT4 receptor in bovine PASMC as the primary receptor subtype responsible for stimulation of adenylyl cyclase by 5-HT [34]; however, at that time specific antagonists were not available for other receptor subtypes which may positively regulate cAMP. To confirm the presence of the 5-HT4 receptor, we examined the expression of mRNA in bovine PASMC using RT-PCR (Fig. 2A). The results show a PCR product of ∼220 bp, consistent with the predicted size for 5-HT4 receptor mRNA, but not the genomic DNA [30]. The control experiment without RT lacks this PCR product.
Fig. 2.

Effects of 5-HT4 receptor and cAMP antagonists on 5-HT-induced migration. (A) Total RNA was purified from bovine PASMC from bovine pulmonary artery tissue. RT-PCR was performed to determine the mRNA expression of 5-HT4. Results are representative for passages 0–4, from four different cell culture explants prepared from the pulmonary arteries of two animals. RT: reverse transcriptase. No RT control indicates that the reverse transcription was not performed. Tubulin was performed as housekeeping gene control (IS: internal standard). (B and C) Migration of low passage (P1–P4) BPASMC in a modified Boyden chamber assay. Cells were plated into migration chambers as described in serum free RPMI. (B) Cells were pretreated for 30 min ± 5-HT receptor antagonist RS 39604 (10–0.1 μM) in the upper and lower chamber before treatment with or without 5-HT (10 μM) for 16–20 h. Asterisk (*) indicates statistical significance from control levels of migration, p < 0.05; symbol (†) indicates p < 0.01 difference from 5-HT treatment alone, n ≥ 3. (C) Western blots of 5-HT-induce p42/p44 MAPK phosphorylation and total MAPK following 5-HT treatment (10 μM) alone for the indicated number of minutes, or pretreated with RS 39504 (1.0 or 0.1 μM) for 30 min. (D) Cells were pretreated for 30 min ± 5-HT receptor antagonist SB 258585 (10, 1 or 0.1 μM) in the upper and lower chamber before treatment with or without 5-HT (10 μM) for 16–20 h. Asterisk (*) indicates statistical significance from control levels of migration, p < 0.05; symbol (†) indicates p < 0.01 difference from 5-HT treatment alone, n = 3. (E) Western blots of 5-HT-induce p42/p44 MAPK phosphorylation and total MAPK following 5-HT treatment (10 μM) alone for the indicated number of minutes, or pretreated with SB 258585 (1.0 or 0.1 μM) for 30 min.
A partial agonist of 5-HT4, RS 23597-190 (30 μM), was able to partially induce actin rearrangements in PASMC (data not shown). We therefore investigated the effects of a 5-HT4 antagonist in the modified Boyden chamber assay for cell migration [29]. A dose–response of the 5-HT4 antagonist RS 39604 is shown in Fig. 2B. Concentrations of RS 39604 from 10 to 1 μM blocked 5-HT-induced migration. At 0.1 μM we observed partial inhibition, as expected for a competitive inhibitor. RS 39604 is specific for 5-HT4 at ∼1 μM (manufacturer's information), and our results therefore indicate that 5-HT4 activation is required for migration. Investigation of the effect of RS 39604 on 5-HT-induced p42/p44 MAPK activation showed no inhibition, indicating that the effect on migration is independent of MAPK activation (Fig. 2C).
The 5-HT6 receptor has also been shown to activate cAMP [35], however the bovine sequence for this receptor subtype is unknown and we were unable to determine its expression by RT-PCR. The 5-HT6 receptor antagonist SB 258585 blocked migration at 10 μM, but lower concentrations of this inhibitor had no significant inhibition of 5-HT-induced migration (Fig. 2D). SB 258585 binding to 5-HT6 is only specific below 0.4 μM (manufacturer's information), so our findings suggest that inhibition at 10 μM is a non-specific effect, and that 5-HT6 is not likely to be involved in 5-HT-induced migration. Investigation of the effect of SB 258585 on 5-HT-induced p42/p44 MAPK activation showed no inhibition (Fig. 2E).
Next we investigated the requirement of cAMP for 5-HT-induced PASMC migration. The cAMP antagonist, AM8, was able to partially block 5-HT-induced actin changes (data not shown). Pretreatment of cells with AM8 (50 μM) for 30 min before the addition of 5-HT inhibited the induced migration to basal levels (Fig. 3A).
Fig. 3.

Effect of cAMP on 5-HT-induced cytoskeletal changes and migration. (A) Cell migration assays in a modified Boyden chamber. PASMC were plated into migration chambers as described in the Methods in serum free RPMI. Cells were pretreated with 50 μM 8-bromo-adenosine 3′,5′-cyclic monophosphorothioate, Rp-isomer (AM8), upper and lower chambers, before addition of 5-HT. For this experiment, the mean percent of control is shown ± S.D. (B) Bovine PASMC were grown to 80% confluence and placed in 1% FBS/RPMI overnight. Cells were then treated ± forskolin (10 μM) for 30 min. Actin cytoskeleton stained with rhodamine-conjugated phalloidin. (C) Cell migration assays in a modified Boyden chamber. PASMC were plated into migration chambers as described in Section 2 in serum free RPMI. Cells were treatment with either 5-HT (10 μM) or forskolin (1 or 0.1 μM) for 16–20 h. Asterisks (**) indicate statistical significance from control levels, p < 0.01; asterisk (*) indicates statistical significance from control levels, p < 0.05; symbol (†) indicates p < 0.01 difference from 5-HT treatment alone, n = 3.
The direct effect of cAMP on actin depolymerization in PASMC was determined by treatment of cells for 30 min with forskolin, which directly activates adenylyl cyclase and increases cellular cAMP. Forskolin caused actin stress fiber depolymerization and the appearance of punctate actin staining (Fig. 3B) similar to that observed in response to 5-HT. Thus, activation of cAMP alone is sufficient to induce cytoskeletal rearrangement in PASMC. However, the addition of forskolin (1 or 0.1 μM) alone inhibited migration in PASMC (Fig. 3C). Higher doses of forskolin, or the inclusion of IBMX with 5-HT, also inhibited migration (data not shown).
To summarize this section, we have shown that 5-HT-induced PASMC migration is mediated by the 5-HT4 receptor, an activator of adenylate cyclase; that a cAMP antagonist inhibits 5-HT-induced migration; and that directly increasing cAMP levels causes actin depolymerization but inhibits 5-HT-induced migration. Taken together, these results suggest that cAMP is necessary but not a sufficient second messenger in 5-HT-induced PASMC migration.
3.3. Inhibition of 5-HT-induced actin depolymerization by the Cl− channel blocker phenylanthranilic acid
We previously found that 5-HT-induced changes in morphology involve the activation of a cAMP-dependent, outward rectifying Cl− channel [28]. Cyclic AMP-responsive chloride channels have been demonstrated in PASMC and other vascular smooth muscle cells [36,37]. 5-HT induced a Cl− efflux in PASMC, and changes in cell shape induced by 5-HT were inhibited by phenylanthranilic acid, a Cl− channel blocker. We tested the ability of PAA to inhibit the actin depolymerization observed with 5-HT. PAA potently inhibited both dendrite formation and actin depolymerization induced by 5-HT or forskolin (Fig. 4A). Results using the modified Boyden chamber assay show that PAA also inhibits migration. A dose–response of PAA inhibition of migration is shown in Fig. 4B. At high concentrations (1 mM), PAA blocks basal and 5-HT-induced migration. At 0.1 mM, PAA inhibits 5-HT-induced but not basal levels of migration, and at 0.01 mM PAA, no significant inhibition is observed. Taken together these results suggest that Cl− channel activation is necessary for 5-HT-induced migration and that the activation of the Cl− channel might be downstream of cAMP.
Fig. 4.
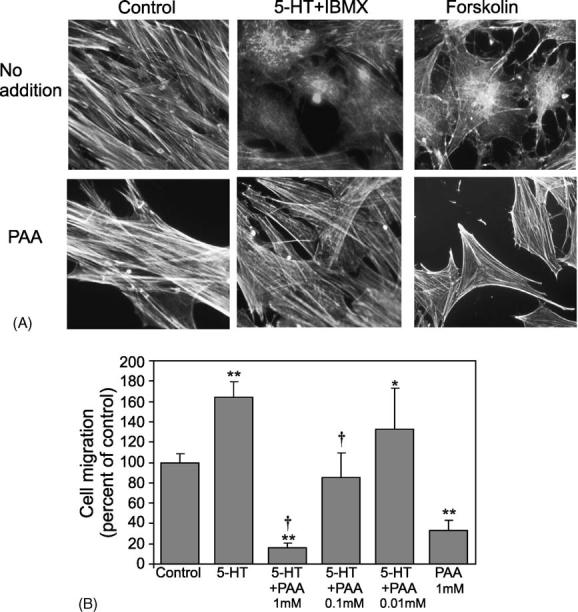
5-HT-induced cytoskeletal changes and motility are inhibited by the Cl− channel blocker phenylanthranilic acid. (A) Bovine PASMC (P1–P4) were grown to 80% confluence on coverslips and placed in 1% FBS/RPMI overnight. Cells were pretreated ± PAA, 1 mM, for 30 min, and were then treated with 5-HT (10 μM) + IBMX (0.1 mM) or forskolin (10 μM) for 30 min, 37 °C, as indicated. Cells were stained to visualize the actin cytoskeleton using rhodamine-conjugated phalloidin as described in Section 2. All fields shown are representative; high magnification is shown to so that cytoskeletal differences can be observed. (B) Cell migration assays. PASMC were plated into migration chambers as described in serum free RPMI. Cells were pretreated ± phenylanthranilic acid (PAA) (1, 0.1 and 0.01 mM) in the upper and lower chambers for 30 min before treatment with 5-HT (10 μM) for 16–20 h. Asterisks (**) indicate statistical significance from control levels, p < 0.01; asterisk (*) indicates statistical significance from control levels, p < 0.05; symbol (†) indicates p < 0.01 difference from 5-HT treatment alone, n = 3.
PAA blocks some outward rectifying chloride channels (ORCC) and the cystic fibrosis transmembrane conductance regulator (CFTR). Interestingly, diisothiocyanatostilbene-2,2′-disulfonic acid and 5-nitro-2-(3-phenylpropylamino)-benzoic acid, which block Ca2+-regulated and other ORCC Cl− channels, had no effect on 5-HT-induced cytoskeletal changes (data not shown). This is in agreement of our previous findings showing that these other Cl− channel inhibitors had no effect on 5-HT-induced changes in PASMC morphology [28].
3.4. p42/p44 MAPK is required for 5-HT-induced SMC migration
We identified cAMP as a second messenger required for 5-HT-induced migration; however, our results with forskolin-treated PASMC showed that elevation of cAMP alone is insufficient to induce migration. The p42/p44 mitogen-activated protein kinase pathway is associated with cellular migration induced by multiple factors [29,38]. 5-HTT has been shown in fibroblasts to be required for MAPK activation by 5-HT [20,21]; recently, we showed that activation of 5-HT1B/1D is necessary for Rho/ROCK activation and the nuclear translocation of MAPK by 5-HT [20-22]. We investigated the roles of 5-HTT, 5-HT1B/1D, and MAPK in PASMC migration. Although the presence of 5-HT1B/1D and 5-HTT have been shown pharmacologically in these cells, we used RT-PCR to confirm the expression of 5-HT1B/1D and 5-HTT in low passage bovine PASMC (Fig. 5A). RT-PCR results show the generation of a ∼250 bp product for 5-HT1B/1D and a ∼295 bp product for 5-HTT; these sizes are consistent with the predicted size for the respective mRNAs but not products resulting from the genomic DNA [30]. Control experiments without RT lack the PCR product.
Fig. 5.

Expression of 5-HT1B/1D and 5-HTT in BPASMC and effects of MAPK inhibition on 5-HT-induced migration. (A) Total RNA was purified from bovine PASMC from bovine pulmonary artery tissue. RT-PCR was performed to determine the mRNA expression of 5-HT1B/1D and 5-HTT. Results are representative for passages 0–4, from four different cell culture explants prepared from the pulmonary arteries of two animals. RT: reverse transcriptase. No RT control indicates that the reverse transcription was not performed. Tubulin control is shown in Fig. 2A. (B) PASMC were plated into migration chambers as described in serum free RPMI. Cells were pretreated in upper and lower chambers for 30 min ± citalopram (1 × 10−5 M) or GR55562 (5 × 10−6 M), before the addition of 5-HT (10 μM) for 16–10 h; n = 3. (C) PASMC were plated into migration chambers in serum free RPMI. Cells were pretreated in upper and lower chambers for 30 min ± U0126 MAPK inhibitor (10 mM), before the addition of 5-HT (10 μM) for 16–20 h. Asterisks (***) indicate statistical significance from control levels, p < 0.001; symbol (†) indicates p < 0.01 difference from 5-HT treatment alone, n = 3. Experiments were performed at least three times. (D) Western blots of 5-HT-induce p42/p44 MAPK phosphorylation and total MAPK following 5-HT treatment (10 μM) alone for the indicated number of minutes, or pretreated with phenylanthranilic acid (PAA; 1 mM) for 30 min. Lower panel shows densitometry of phosphorylated p42/p44 MAPK protein normalized to total p42/p44 MAPK.
5-HT-induced changes in PASMC actin cytoskeleton were not inhibited by receptor antagonists for 5-HT1B/1D (GR 55562, 5 μM) or a 5-HTT inhibitor (imipramine, 100 μM) (data not shown). Neither citalopram (10 μM), a 5-HTT antagonist, nor the 5-HT1B/1D antagonist GR 55562 (5 μM) resulted in statistically significant reductions of migration (Fig. 5B).
To directly determine the effect of MAPK inhibition on 5-HT-induced migration, cells were pretreated with the MAPK inhibitor (U0126, 10 μM) for 30 min before the addition of 5-HT (Fig. 5C). Results show that MAPK inhibition significantly but partially blocks migration in response to 5-HT. Although MAPK inhibition blocks migration of SMC, 5-HT-induced actin depolymerization was not blocked (data not shown), suggesting that the role of MAPK in 5-HT-induced migration does not involve actin depolymerization. We next tested the effect of PAA on MAPK activation by 5-HT (Fig. 5D). Results show that PAA does not block 5-HT activation of MAPK, suggesting that the MAPK pathway activation is independent of Cl− channel activation.
4. Discussion
The major finding of this study is that 5-HT-induced migration of PASMC requires the activation of cAMP and a chloride channel through activation of the 5-HT4 receptor. Although 5-HT has previously been shown to function as a chemotactic factor for neurons, eosinophils and some smooth muscle cells [39-42], mechanisms of 5-HT-induced migration have not been defined. Our work is the first demonstration of the involvement of 5-HT4 receptor and a Cl− channel in migration.
5-HT-induced migration in PASMC is accompanied by actin rearrangement, but not by changes in microtubules. Dynamic regulation of the actin cytoskeleton is critical for cell motility and invasion by a variety of factors [32,43-45]; in this process, actin stress fibers are first depolymerized, and cortical actin is repolymerized at the leading edge of the cell [32]. Disruption of the actin cytoskeleton by pharmacological agents, such as cytochalasin D, inhibits cell motility [46]. A variety of mechanisms for actin depolymerization in motility have been described, including cAMP- and PKA-dependent [47], cAMP-dependent and PKA-independent [48], and MAPK-dependent events [29]. Our data show that cAMP is necessary but not a sufficient second messenger in 5-HT-induced migration in PASMC. In several cell types pharmacologically induced increases in cellular cAMP have been demonstrated to induce prolonged actin depolymerization and to inhibit cellular motility [48-52]. Ydrenius et al. describe findings similar to ours for fMLP-induced migration in granulocytes, where either blockade of cAMP by an antagonist or induction of high levels of cAMP by IBMX or forskolin were inhibitory [50]. We postulate that the biphasic effects of cAMP may be due to its function in regulating actin polymerization: an increase in cAMP is necessary for actin depolymerization, but cellular cAMP must then decrease in order to allow for actin repolymerization at the leading edge of the migrating cell.
Interestingly, in some systems, the effects of cAMP on the actin cytoskeleton have been shown to be counteracted by activation of RhoA and Rho kinase [49]. We have demonstrated here that an antagonist of 5-HT1B/1D, the receptor required for RhoA and Rho kinase activation in PASMC [22], did not significantly inhibit migration. Rho may be activated at later time points by other mechanisms, such as integrins. Further work is required to determine whether Rho and Rho kinase are indirectly involved in 5-HT-induced motility.
5-HT-induced changes in morphology, actin depolymerization and motility were also inhibited by the chloride channel inhibitor PAA, but not by DIDS or NPPB, which inhibit different Cl− channel subtypes from PAA. Previous work showed that the treatment of PASMC with 5-HT caused a cAMP-dependent chloride efflux, but not changes in calcium currents [28]. Cyclic AMP-responsive chloride channels have been demonstrated in intestine, aorta and pulmonary artery SMC, and recent studies have shown the activation of Cl− channels in intact tissues in response to 5-HT downstream of 5-HT4 signaling [36,53-55]. Also, expression of the cAMP-dependent cystic fibrosis transmembrane regulator has been demonstrated in vascular smooth muscle cells [37]. The primary outward rectifying chloride channel expressed by PASMC has been shown by immunohistochemical techniques to be identical to ClC-3, a chloride channel which is also up-regulated in smooth muscle cells in pulmonary artery hypertrophy [56,57]. The mechanism by which chloride channel activation might mediate actin depolymerization is not known. Over 30 genes for putative chloride channels have been found in the human genome [58], and although PAA is known to block the ORCC and CFTR chloride channel types, the channel involved in 5-HT-induced changes in cellular morphology and actin rearrangement has not yet been identified.
p42/p44 MAPK activation is required for cellular migration by a variety of factors including platelet derived growth factor and hepatocyte growth factor [29,59]. Here, we show that the full migratory response of PASMC to 5-HT also requires p42/p44 MAPK activation. We did not, however, observe significant inhibition of 5-HT-induced migration by antagonists for 5-HTT or 5-HT1B/1D receptor, suggesting that another receptor may also be involved in PASMC for activation of MAPK [22]. Interestingly, 5-HT activation of MAPK was not affected by either inhibition of the 5-HT4 receptor (by RS 39604) or inhibition of the cAMP-dependent chloride channel (by PAA); this suggests that mechanism(s) of motility regulation by MAPK may occur in parallel to actin rearrangement. Taken together, our data support a model of 5-HT-induced PASMC migration in which cAMP and a Cl− channel regulate actin depolymerization, while MAPK may be required for parallel or downstream events in motility.
The results of our present studies are of further interest as they complement our previous work demonstrating the specificity of multiple 5-HT cell membrane targets (receptors and 5-HT transporter) for cell signaling pathways and cell function in the same cell type. Thus, as demonstrated in this study, the 5-HT4 receptor participates in PASMC mobility, and we also recently showed that the 5-HT1B/1D receptor regulates Rho/ROCK activation, translocation of MAPK to the nucleus and PASMC proliferation [22]. We have recently found that the 5-HT2A receptor activates the phosphatidylinositol 3-kinase/Akt/mTOR/S6K1 pathway and contributes to cell proliferation [60], while 5-HTT regulates ROS formation, MAPK activation and cell proliferation [17,18,20,21]. These findings suggest that 5-HT signaling through the multiple receptors and 5-HTT is not redundant, but that independent signaling pathways are induced to result in cellular hyper-trophy, hyperplasia and motility in PASMC.
Acknowledgements
This work was supported by National Heart, Lung and Blood Institute Grant HL-32723 (B.L. Fanburg) and HL-073929 (R.M. Day), by an American Lung Association Research Grant (R.M. Day), and an American Heart Association Scientist Development Grant (R.M. Day). M.J. Segel is the recipient of a Ruth L. Kirschstein National Research Service Award, Individual Fellowship (HL-76082-2). Thanks to Kathleen Riley for phase and fluorescent microscopy, and to Young H. Lee for technical assistance. The content of this manuscript is the opinion of the authors and does not reflect the views of The Uniformed Services University of the Health Sciences, the US Department of Defense or the Federal Government.
REFERENCES
- 1.Rubin LJ. Primary pulmonary hypertension. N Engl J Med. 1997;336:111–7. doi: 10.1056/NEJM199701093360207. [DOI] [PubMed] [Google Scholar]
- 2.Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M, Capron F, et al. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest. 2001;108:1141–50. doi: 10.1172/JCI12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eddahibi S, Adnot S. Anorexigen-induced pulmonary hypertension and the serotonin (5-HT) hypothesis: lessons for the future in pathogenesis. Respir Res. 2002;3:9. doi: 10.1186/rr181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pietra G, Capron F, Stewart S, Leone O, Humbert M, Robbins I, et al. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol. 2004;43:25S–32S. doi: 10.1016/j.jacc.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 5.MacLean MR. Pulmonary hypertension, anorexigens and 5-HT: pharmacological synergism in action? Trends Pharmacol Sci. 1999;20:490–5. doi: 10.1016/s0165-6147(99)01389-9. [DOI] [PubMed] [Google Scholar]
- 6.Michelakis ED, Weir EK. Anorectic drugs and pulmonary hypertension from the bedside to the bench. Am J Med Sci. 2001;321:292–9. doi: 10.1097/00000441-200104000-00009. [DOI] [PubMed] [Google Scholar]
- 7.Simonneau G, Fartoukh M, Sitbon O, Humbert M, Jagot JL, Herve P. Primary pulmonary hypertension associated with the use of fenfluramine derivatives. Chest. 1998;114:195S–9S. doi: 10.1378/chest.114.3_supplement.195s. [DOI] [PubMed] [Google Scholar]
- 8.Johnson GJ, Leis LA, Dunlop PC, Weir EK. The effect of the anorectic agent, d-fenfluramine, and its primary metabolite, d-norfenfluramine, on intact human platelet serotonin uptake and efflux. J Thromb Haemost. 2003;1:2663–8. doi: 10.1046/j.1538-7836.2003.00474.x. [DOI] [PubMed] [Google Scholar]
- 9.Herve P, Launay JM, Scrobohaci ML, Brenot F, Simonneau G, Petitpretz P, et al. Increased plasma serotonin in primary pulmonary hypertension. Am J Med. 1995;99:249–54. doi: 10.1016/s0002-9343(99)80156-9. [DOI] [PubMed] [Google Scholar]
- 10.Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M, Capron F, et al. Hyperplasia of pulmonary artery smooth muscle cells is causally related to overexpression of the serotonin transporter in primary pulmonary hypertension. Chest. 2002;121:97S–8S. [PubMed] [Google Scholar]
- 11.Marcos E, Fadel E, Sanchez O, Humbert M, Dartevelle P, Simonneau G, et al. Serotonin-induced smooth muscle hyperplasia in various forms of human pulmonary hypertension. Circ Res. 2004;94:1263–70. doi: 10.1161/01.RES.0000126847.27660.69. [DOI] [PubMed] [Google Scholar]
- 12.Deraet M, Manivet P, Janoshazi A, Callebert J, Guenther S, Drouet L, et al. The natural mutation encoding a C terminus-truncated 5-hydroxytryptamine 2B receptor is a gain of proliferative functions. Mol Pharmacol. 2005;67:983–91. doi: 10.1124/mol.104.008268. [DOI] [PubMed] [Google Scholar]
- 13.Eddahibi S, Raffestin B, Pham I, Launay JM, Aegerter P, Sitbon M, et al. Treatment with 5-HT potentiates development of pulmonary hypertension in chronically hypoxic rats. Am J Physiol. 1997;272:H1173–81. doi: 10.1152/ajpheart.1997.272.3.H1173. [DOI] [PubMed] [Google Scholar]
- 14.Eddahibi S, Adnot S, Frisdal E, Levame M, Hamon M, Raffestin B. Dexfenfluramine-associated changes in 5-hydroxytryptamine transporter expression and development of hypoxic pulmonary hypertension in rats. J Pharmacol Exp Ther. 2001;297:148–54. [PubMed] [Google Scholar]
- 15.Kanai Y, Hori S, Tanaka T, Yasuoka M, Watanabe K, Aikawa N, et al. Role of 5-hydroxytryptamine in the progression of monocrotaline induced pulmonary hypertension in rats. Cardiovasc Res. 1993;27:1619–23. doi: 10.1093/cvr/27.9.1619. [DOI] [PubMed] [Google Scholar]
- 16.Eddahibi S, Raffestin B, Hamon M, Adnot S. Is the serotonin transporter involved in the pathogenesis of pulmonary hypertension? J Lab Clin Med. 2002;139:194–201. doi: 10.1067/mlc.2002.122181. [DOI] [PubMed] [Google Scholar]
- 17.Lee SL, Wang WW, Lanzillo JJ, Fanburg BL. Regulation of serotonin-induced DNA synthesis of bovine pulmonary artery smooth muscle cells. Am J Physiol. 1994;266:L53–60. doi: 10.1152/ajplung.1994.266.1.L53. [DOI] [PubMed] [Google Scholar]
- 18.Lee SL, Wang WW, Lanzillo JJ, Fanburg BL. Serotonin produces both hyperplasia and hypertrophy of bovine pulmonary artery smooth muscle cells in culture. Am J Physiol. 1994;266:L46–52. doi: 10.1152/ajplung.1994.266.1.L46. [DOI] [PubMed] [Google Scholar]
- 19.Lee SL, Wang WW, Fanburg BL. Dexfenfluramine as a mitogen signal via the formation of superoxide anion. FASEB J. 2001;15:1324–5. doi: 10.1096/fj.00-0431fje. [DOI] [PubMed] [Google Scholar]
- 20.Lee SL, Wang WW, Finlay GA, Fanburg BL. Serotonin stimulates mitogen-activated protein kinase activity through the formation of superoxide anion. Am J Physiol. 1999;277:L282–91. doi: 10.1152/ajplung.1999.277.2.L282. [DOI] [PubMed] [Google Scholar]
- 21.Lee SL, Simon AR, Wang WW, Fanburg BL. H(2)O(2) signals 5-HT-induced ERK MAP kinase activation and mitogenesis of smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:L646–52. doi: 10.1152/ajplung.2001.281.3.L646. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, Suzuki YJ, Day RM, Fanburg BL. Rho kinase-induced nuclear translocation of ERK1/ERK2 in smooth muscle cell mitogenesis caused by serotonin. Circ Res. 2004;95:579–86. doi: 10.1161/01.RES.0000141428.53262.a4. [DOI] [PubMed] [Google Scholar]
- 23.Simon AR, Severgnini M, Takahashi S, Rozo L, Andrahbi B, Agyeman A, et al. 5-HT induction of c-Fos gene expression requires reactive oxygen species and Rac1 and Ras GTPases. Cell Biochem Biophys. 2005;42:263–76. doi: 10.1385/CBB:42:3:263. [DOI] [PubMed] [Google Scholar]
- 24.Kavanaugh WM, Williams LT, Ives HE, Coughlin SR. Serotonin-induced deoxyribonucleic acid synthesis in vascular smooth muscle cells involves a novel, pertussis toxin-sensitive pathway. Mol Endocrinol. 1988;2:599–605. doi: 10.1210/mend-2-7-599. [DOI] [PubMed] [Google Scholar]
- 25.Eddahibi S, Hanoun N, Lanfumey L, Lesch KP, Raffestin B, Hamon M, et al. Attenuated hypoxic pulmonary hypertension in mice lacking the 5-hydroxytryptamine transporter gene. J Clin Invest. 2000;105:1555–62. doi: 10.1172/JCI8678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcos E, Adnot S, Pham MH, Nosjean A, Raffestin B, Hamon M, et al. Serotonin transporter inhibitors protect against hypoxic pulmonary hypertension. Am J Respir Crit Care Med. 2003;168:487–93. doi: 10.1164/rccm.200210-1212OC. [DOI] [PubMed] [Google Scholar]
- 27.Morecroft I, Loughlin L, Nilsen M, Colston J, Dempsie Y, Sheward J, et al. Functional interactions between 5-hydroxytryptamine receptors and the serotonin transporter in pulmonary arteries. J Pharmacol Exp Ther. 2005;313:539–48. doi: 10.1124/jpet.104.081182. [DOI] [PubMed] [Google Scholar]
- 28.Lee SL, Fanburg BL. Serotonin produces a configurational change of cultured smooth muscle cells that is associated with elevation of intracellular cAMP. J Cell Physiol. 1992;150:396–405. doi: 10.1002/jcp.1041500224. [DOI] [PubMed] [Google Scholar]
- 29.Day RM, Cioce V, Breckenridge D, Castagnino P, Bottaro DP. Differential signaling by alternative HGF isoforms through c-Met: activation of both MAP kinase and PI 3-kinase pathways is insufficient for mitogenesis. Oncogene. 1999;18:3399–406. doi: 10.1038/sj.onc.1202683. [DOI] [PubMed] [Google Scholar]
- 30.Reist M, Pfaffl WW, Morel C, Meylan M, Hirsbrunner G, Blum JW, et al. Quantitative mRNA analysis of eight bovine 5-HT receptor subtypes in brain, abomasum, and intestine by real-time RT-PCR. J Recept Signal Transduct Res. 2003;23:271–87. doi: 10.1081/rrs-120026971. [DOI] [PubMed] [Google Scholar]
- 31.Lanzillo JJ, Yu FS, Stevens J, Hassoun PM. Determination of xanthine dehydrogenase mRNA by a reverse transcription-coupled competitive quantitative polymerase chain reaction assay: regulation in rat endothelial cells by hypoxia and hyperoxia. Arch Biochem Biophys. 1996;335:377–80. doi: 10.1006/abbi.1996.0519. [DOI] [PubMed] [Google Scholar]
- 32.Degryse B, Resnati M, Rabbani SA, Villa A, Fazioli R, Blasi F. Src-dependence and pertussis-toxin sensitivity of urokinase receptor-dependent chemotaxis and cytoskeletal reorganization in rat smooth muscle cells. Blood. 1999;94:649–62. [PubMed] [Google Scholar]
- 33.Lee SL, Wang WW, Moore BJ, Fanburg BL. Dual effect of serotonin on growth of bovine pulmonary artery smooth muscle cells in culture. Circ Res. 1991;68:1362–8. doi: 10.1161/01.res.68.5.1362. [DOI] [PubMed] [Google Scholar]
- 34.Becker BN, Gettys TW, Middleton JP, Olsen CL, Albers FJ, Lee SL, et al. 8-hydroxy-2-(di-n-propylamino)tetralin-responsive 5-hydroxytryptamine4-like receptor expressed in bovine pulmonary artery smooth muscle cells. Mol Pharmacol. 1992;42:817–25. [PubMed] [Google Scholar]
- 35.Raymond J, Mukhin Y, Gelasco A, Turner J, Collinsworth G, Gettys T, et al. Multiplicity of mechanisms of serotonin receptor signal transduction. Pharmacol Ther. 2001;92:179–212. doi: 10.1016/s0163-7258(01)00169-3. [DOI] [PubMed] [Google Scholar]
- 36.Zhao YJ, Wang J, Rubin LJ, Yuan XJ. Roles of K+ and Cl− channels in cAMP-induced pulmonary vasodilation. Exp Lung Res. 1998;24:71–83. doi: 10.3109/01902149809046055. [DOI] [PubMed] [Google Scholar]
- 37.Robert R, Thoreau V, Norez C, Cantereau A, Kitzis A, Mettey Y, et al. Regulation of the cystic fibrosis transmembrane conductance regulator channel by beta-adrenergic agonists and vasoactive intestinal peptide in rat smooth muscle cells and its role in vasorelaxation. J Biol Chem. 2004;279:21160–8. doi: 10.1074/jbc.M312199200. [DOI] [PubMed] [Google Scholar]
- 38.Yao JS, Chen Y, Zhai W, Xu K, Young WL, Yang GY. Minocycline exerts multiple inhibitory effects on vascular endothelial growth factor-induced smooth muscle cell migration: the role of ERK1/2, PI3K, and matrix metalloproteinases. Circ Res. 2004;95:364–71. doi: 10.1161/01.RES.0000138581.04174.2f. [DOI] [PubMed] [Google Scholar]
- 39.Boehme S, Lio F, Sikora L, Pandit T, Lavrador K, Rao S, et al. Cutting edge: serotonin is a chemotactic factor for eosinophils and functions additively with eotaxin. J Immunol. 2004;173:3599–603. doi: 10.4049/jimmunol.173.6.3599. [DOI] [PubMed] [Google Scholar]
- 40.Moiseiwitsch J, Lauder J. Serotonin regulates mouse cranial neural crest migration. Proc Natl Acad Sci USA. 1995;92:7182–6. doi: 10.1073/pnas.92.16.7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tamura K, Kanzaki T, Saito Y, Otabe M, Saito Y, Morisaki N. Serotonin (5-hydroxytryptamine, 5-HT) enhances migration of rat aortic smooth muscle cells through 5-HT2 receptors. Atherosclerosis. 1997;132:139–43. doi: 10.1016/s0021-9150(97)00077-4. [DOI] [PubMed] [Google Scholar]
- 42.Bottaro D, Shepro D, Peterson S, Hechtman HB. Serotonin, histamine, and noreinephrine mediation of endothelial and vascular smooth muscle cell movement. Am J Physiol. 1985;248:C252–7. doi: 10.1152/ajpcell.1985.248.3.C252. [DOI] [PubMed] [Google Scholar]
- 43.Sotiropoulos A, Gineitis D, Copeland J, Treisman R. Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell. 1999;98:159–69. doi: 10.1016/s0092-8674(00)81011-9. [DOI] [PubMed] [Google Scholar]
- 44.Hall A. Small GTP-binding proteins and regulation of the actin cytoskeleton. Annu Rev Cell Biol. 1994;10:31–54. doi: 10.1146/annurev.cb.10.110194.000335. [DOI] [PubMed] [Google Scholar]
- 45.Tapon N, Hall A. Rho, Rac Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr Opin Cell Biol. 1997;9:86–92. doi: 10.1016/s0955-0674(97)80156-1. [DOI] [PubMed] [Google Scholar]
- 46.Chen BH, Tzen JT, Bresnick AR, Chen HC. Roles of Rho-associated kinase and myosin light chain kinase in morphological and migratory defects of focal adhesion kinase-null cells. J Biol Chem. 2002;277:33857–63. doi: 10.1074/jbc.M204429200. [DOI] [PubMed] [Google Scholar]
- 47.Lamb NJ, Fernandez A, Conti MA, Adelstein R, Glass DB, Welch WJ, et al. Regulation of actin microfilament integrity in living nonmuscle cells by the cAMP-dependent protein kinas and the myosin light chain kinase. J Cell Biol. 1988;106:1955–71. doi: 10.1083/jcb.106.6.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hirshman CA, Zhu D, Panettieri RA, Emala CW. Actin depolymerization via the beta-adrenoceptor in airway smooth muscle cells: a novel PKA-independent pathway. Am J Physiol Cell Physiol. 2001;281:C1468–76. doi: 10.1152/ajpcell.2001.281.5.C1468. [DOI] [PubMed] [Google Scholar]
- 49.Dong JM, Leung T, Manser E, Lim L. cAMP-induced morphological changes are counteracted by the activated RhoA small GTPase and the Rho kinase ROKalpha. J Biol Chem. 1998;273:22554–62. doi: 10.1074/jbc.273.35.22554. [DOI] [PubMed] [Google Scholar]
- 50.Ydrenius L, Molony L, Ng-Sikorski J, Andersson T. Dual action of cAMP-dependent protein kinase on granulocyte movement. Biochem Biophys Res Commun. 1997;235:445–50. doi: 10.1006/bbrc.1997.6822. [DOI] [PubMed] [Google Scholar]
- 51.Blindt R, Bosserhoff AK, vom Dahl J, Hanrath P, Schror K, Hohlfeld T, et al. Activation of IP and EP(3) receptors alters cAMP-dependent cell migration. Eur J Pharmacol. 2002;444:31–7. doi: 10.1016/s0014-2999(02)01607-2. [DOI] [PubMed] [Google Scholar]
- 52.Goncharova EA, Billington CA, Irani C, Vorotnikov AV, Tkachuk VA, Penn RB, et al. Cyclic AMP-mobilizing agents and glucocoricoids modulate human smooth muscle cell migration. Am J Respir Cell Mol Biol. 2003;29:19–27. doi: 10.1165/rcmb.2002-0254OC. [DOI] [PubMed] [Google Scholar]
- 53.Kellum JM, Albuquerque FC, Stoner MC, Harris RP. Stroking human jejunal mucosa induces 5-HT release and Cl− secretion via afferent neurons and 5-HT4 receptors. Am J Physiol Gastrointest Liver Physiol. 1999;277:G515–20. doi: 10.1152/ajpgi.1999.277.3.G515. [DOI] [PubMed] [Google Scholar]
- 54.Kellum JM, Budhoo MR, Siriwardena AK, Smith EP, Jebraili SA. Serotonin induces Cl− secretion in human jejunal mucosa in vitro via a nonneural pathway at a 5-HT4 receptor. Am J Physiol. 1994;267:G357–63. doi: 10.1152/ajpgi.1994.267.3.G357. [DOI] [PubMed] [Google Scholar]
- 55.Robert R, Norez C, Becq F. Disruption of CFTR chloride channel alters mechanical properties and cAMP-dependent Cl− transport of aortic smooth muscle cells. J Physiol. 2005 October 15;568(Pt 2):438–95. doi: 10.1113/jphysiol.2005.085019. [Epub 2005 August 4] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duan D, Zhong J, Hermoso M, Satterwhite CM, Rossow CF, Hatton WJ, et al. Functional inhibition of native volume-sensitive outwardly rectifying anion channels in muscle cells and Xenopus oocytes by anti-ClC-3 antibody. J Physiol. 2001;531:437–44. doi: 10.1111/j.1469-7793.2001.0437i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dai YP, Bongalon S, Hatton WJ, Hume JR, Yamboliev IA. ClC-3 chloride channel is upregulated by hypertrophy and inflammation in rat and canine pulmonary artery. Br J Pharmacol. 2005;145:5–14. doi: 10.1038/sj.bjp.0706135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kozlowski RZ. Chloride channels: potential therapeutic targets. In: Kozlowski RZ, editor. Chloride channels. ISIS Medical Media Ltd.; Oxford: 1999. pp. 177–86. [Google Scholar]
- 59.Campbell M, Trimble ER. Modification of PI3K- and MAPK-dependent chemotaxis in aortic vascular smooth muscle cells by protein kinase C{beta}II. Circ Res. 2004;96:197–206. doi: 10.1161/01.RES.0000152966.88353.9d. [DOI] [PubMed] [Google Scholar]
- 60.Liu Y, Fanburg BL. Serotonin-induced growth of pulmonary artery smooth muscle requires activation of PI3K/Akt/S6K1. Am J Respir Cell Mol Biol. 2005 Sept 29; doi: 10.1165/rcmb.2005-0163OC. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]