

Abstract
Mitochondria often reside in subcellular regions with high metabolic demands. We examined the mechanisms that can govern the relocation of mitochondria to these sites in respiratory neurons. Mitochondria were visualized using tetramethylrhodamineethylester, and their movements were analyzed by applying single-particle tracking. Intracellular ATP ([ATP]i) was assessed by imaging the luminescence of luciferase, the fluorescence of the ATP analog TNP-ATP, and by monitoring the activity of K(ATP) channels. Directed movements of mitochondria were accompanied by transient increases in TNP-ATP fluorescence. Application of glutamate and hypoxia reversibly decreased [ATP]i levels and inhibited the directed transport. Injections of ATP did not rescue the motility of mitochondria after its inhibition by hypoxia. Introduction of ADP suppressed mitochondrial movements and occluded the effects of subsequent hypoxia. Mitochondria decreased their velocity in the proximity of synapses that correlated with local [ATP]i depletions. Using a model of motor-assisted transport and Monte Carlo simulations, we showed that mitochondrial traffic is more sensitive to increases in [ADP]i than to [ATP]i depletions. We propose that consumption of synaptic ATP can produce local increases in [ADP]i and facilitate the targeting of mitochondria to synapses.
INTRODUCTION
Neuronal activity disturbs transmembrane ion gradients, and their restoration requires intracellular ATP, [ATP]i. This can be most prominent at sites of intense activity such as synapses, and relocation of mitochondria to these strategic foci may be advantageous. Indeed, the distribution of mitochondria in fixed preparations correlates with the previously imposed neuronal activity (1,2): mitochondria tend to accumulate in the vicinity of active growth cones of developing neurons (3) and slow down in the vicinity of active synapses (4). In various neuronal preparations (5–8), the movements of mitochondria are sensitive to mitochondrial poisons, indicating that ATP synthesis is necessary for providing a fuel for molecular motors that transport mitochondria. This suggestion, albeit plausible, has been never confirmed by measuring ATP levels in the living neurons. If metabolically active regions such as synapses and growth cones are really exhausted, [ATP]i gradients can act as local chemical signals that would modulate the traffic of mitochondria. However, it is yet unclear whether intracellular [ATP]i gradients exist and how they influence the transport of mitochondria.
To examine these issues, we used respiratory neurons that demonstrate persistent rhythmic activity (4). Similar to other organs capable of sensing enviromental low Po2 (<50 mm Hg) (9), a hypoxic stimulation of the respiratory kernel modifies inspiratory activity of neurons (10–14) and induces specific physiological responses. Mitochondria substantially contribute to the generation of respiratory activity and its modulation by hypoxia, as demonstrated by periodic changes in the oxygen consumption (10), in intracellular [Ca2+] (11), in mitochondrial properties (12), and in the activity of KATP-sensitive channels (13). To measure [ATP]i levels in neurons, we used the expression of luciferase, the fluorescent ATP analog TNP-ATP, and the recordings of activity of the plasmalemmal K(ATP) channels. Spatiotemporal changes in mitochondrial movements correlated well with the measured [ATP]i levels in perisynaptic regions. Hypoxia and glutamate inhibited the directed transport of mitochondria, and the suppression was not reversed after direct ATP injections. On the other hand, the movements of mitochondria became slower after injections of ADP, and subsequent application of hypoxia was ineffective. To explain the data, we developed a model through which, using analysis and Monte Carlo simulations, we confirmed a larger sensitivity of mitochondrial transport to increases in [ADP]i than to [ATP]i depletions. We therefore propose that [ADP]i gradients, which can be established in the proximity of synapses, slow down the movements of mitochondria and facilitate targeting of mitochondria to the “strategic hot spots” in neurons.
MATERIALS AND METHODS
Preparation and chemicals
Cultured respiratory neurons were obtained from neonatal mice (NMRI, P3–P6) as described previously (4,8). The use and care of animals in this study complied with the guidelines of the European Commission (No. L358, ISSN 0378-6978) and the Committee for Animal Research, Göttingen University. During the experiments, the coverslips with cells were mounted in the recording chamber, which was continuously superfused at 34°C with ACSF (artificial cerebrospinal fluid) containing (in mM) 136 NaCl, 5 KCl, 1.25 CaCl2, 0.8 MgSO4, 0.4 NaH2PO4, 0.3 K2HPO4, 3.3 NaHCO3, 6 glucose, pH 7.4, and was saturated with 95% O2–5% CO2. ATP and ADP were from Sigma (Deisenhofen), and fura-2/AM, tetramethylrhodamineethylester (TMRE), and 2′-o-trinitrophenyl)-adenosine-5′-triphoshate (TNP-ATP) were purchased from Molecular Probes (Eugene, OR).
Each experiment was repeated for at least three different cell preparations, and mean ± SE were compared using Student's t-test, with P < 0.05 being the criterion for statistical significance. For multiple comparisons, the analysis of variance (ANOVA) was used. All shown traces and images are representative of at least five separate experiments.
Imaging
In the experiments, we used both a two-photon scanning microscope (TPSM) and cooled CCD camera (MicroMax, Princeton Instruments, Monmouth, NJ), as described previously in full (4,8,11,12). The experimental setups were routinely tested to exclude mechanical artifacts. The images were background subtracted, 3 × 3 median filtered, and analyzed using Metamorph (Princeton Instruments). TPSM was used in all experiments where only one indicator dye was imaged, and in the experiments with two indicator dyes, a CCD camera was used. In the experiments where fluorescence of two dyes was measured, the recorded signals were efficiently separated using appropriate illumination wavelengths and emission filters (4,8,11,12). The corresponding controls showed that contamination of signals (“bleeding”) measured in two recorded channels was negligible.
To monitor locations of mitochondria and their potential, the cells were stained with TMRE. The dye was added to ASCF at 100 nM, and the cells were allowed to equilibrate for 20 min. TMRE was excited at 515 nm, and the emission was filtered at 560 nm. The trajectories of single mitochondria were obtained using a single-particle tracking (SPT) method as described previously (4), and the traces were subsequently analyzed to characterize the directed transport of mitochondria. The presented data were obtained in dendrites. We did not find significant differences between the behavior of mitochondria that were located close to the soma and those in the distal dendrites.
Synaptic vesicles were visualized using FM 1-43 (15). The neurons were stained with 8 μM FM 1-43 in the presence of 60 mM KCl for 60–120 s and were then washed with fresh ASCF for 15 min. FM 1-43 was excited at 460 nm, and the emission was filtered at 535 nm. To unequivocally identify the active synaptic sites, we induced exocytosis in the end of each experiment by applying 45 mM KCl for 5 s to the whole cell including dendrites. Only those bright spots (>85% of all observed) in which fluorescence signal decreased by >20% (4) were considered as true synaptic endings. In five separate experiments, we imaged neurons that were transfected with GFP-synaptophysin and subsequently stained them with FM 1-43. The distributions of synaptophysin and FM 1-43 in five neurons showed an overlap of 94 ± 6%.
For the measurements of [ATP]i, cells were transfected with pEGFP-Luciferase (Clontech, Heidelberg). The luminescence was recorded with VersArray:512B back-illuminated and liquid-nitrogen-cooled CCD camera (Visitron Systems, Puchheim, Germany). Differences in the intracellular distributions of luciferase were accounted for by normalizing the luminescence by the accompanying fluorescence of EGFP. The luminescence was weak, and the signals were accumulated for 15 to 60 s, and the pixels were cumulated (2 × 2 to 8 × 8 binning was used). To compare luminescence images and fluorescence recordings, the formers were appropriately zoomed by applying corresponding routines of Metamorph.
To image intracellular ATP-binding sites, we used the fluorescent analog TNP-ATP. Its fluorescence increases severalfold after binding to proteins and has been used to study ATP binding sites in various proteins including molecular motors (16). The neurons were loaded with TNP-ATP using a weak-base peptide (Endo-Porter, Gene Tools, Philomath, OR) according to the prescriptions of the manufacturer. The cells were incubated with 0.1 mM TNP-ATP + peptide on ice for 4 h. TNP-ATP was excited at 460 nm, and the emission was collected at 560 nm. The excitation spectrum of TNP-ATP in cells was identical to that recorded in drops that contained 0.1 mM dye in ACSF. In the TNP-ATP-loaded neurons, the movements of mitochondria were not modified, indicating that this background metabolite did not disturb ATP homeostasis.
For measuring free intracellular Ca2+, [Ca2+]i, cells were loaded with fura-2-AM (3 μM) for 30 min and then washed with fresh ACSF for 30 min before experiments began. Fura-2 was excited at 385 and 360 nm, and, after subtraction of background fluorescence, the fluorescence ratios (R) were formed and converted into [Ca2+]i values as described previously (19).
Electrophysiology
ATP sensitivity of KATP channels is similar to that of molecular motors (14), and we used their activity in cell-attached patches as another measure of intracellular ATP. Patch electrodes from borosilicate glass (Clark Instruments, Pangbourne, UK) had tip openings of 1.5–2 μm and resistances of 1.5–2.5 MΩ. The single-channel currents were measured using the amplifier EPC-7 (ESF, Friedland, Germany) as described previously (12–14). Membrane currents were filtered at 3 kHz (−3 dB), digitized at 5 kHz, and stored for off-line analysis. The pipette solution contained (in mM): 125 K+-gluconate, 10 NaCl, 2 MgCl2, 10 HEPES, 0.5 Na2ATP, pH adjusted to 7.4 with KOH (KATP channels). The open probability Popen was obtained by dividing the mean current by the unitary current. In measuring synaptic currents, the pipette solution was supplied with nystatin (100 μg/ml), and the patch was perforated as described previously (11,18). Synaptic activity was measured at the holding potential of −40 mV, which allowed us to measure synaptic currents.
The gases dissolved in ACSF can be lost during transportation of solutions and in the open experimental chamber. Therefore, we measured oxygen pressure in situ using oxygen-sensitive electrodes (tip diameter 20 μm) and an amplifier (Diamond Electro-Tech, Ann Arbor, MI) as described previously (10,14). After calibration of the O2-sensitive electrode in a beaker saturated with 95% O2 or N2 solutions at 34°C, the electrode was placed into the chamber in the center of the image field, 20 μm above the coverslip surface. The measured Po2 was 178 ± 21 mm Hg in control and was stabilized at 16 ± 5 mm Hg 20 s after starting N2–perfusion (n = 21). Application of brief hypoxia did not damage the neurons as most cells preserved a healthy appearance and showed no deterioration of electrical excitability.
[ATP] and [ADP] gradients in dendrites
We described the spatial profile of [ATP]i in the dendrite containing an active synapse using a 1D-reaction-diffusion equation
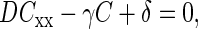 |
(1) |
which takes into account the processes of diffusion, consumption, and production of ATP in the cytoplasm. Here C = [ATP], Cxx is the second spatial derivative, x is the axial coordinate, D is the diffusion coefficient, and γ and δ are the rate constants of ATP consumption and production, respectively. When ATP concentration at synapse (x = 0) is maintained at C1, the solution of Eq. 1 is
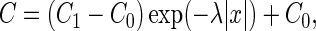 |
(2) |
where 1/λ = √D/γ is the space constant for a substance diffusing from a point source (17) and C0 = δ/γ = 1 mM is the resting concentration of ATP. The value of γ was obtained from the rate constant of ATP-dependent clearance of [Ca2+]i in dendrites (≈2 s−1 (18,19)). Removal of excess [Na+]i and repletion of [K+]i via Na+/K+-ATPase is characterized by a similar rate constant that gives γ = 4 s−1. For D = 140 μm2s−1 (20), the space constant 1/λ is 6 μm. The local ATP sink acts as a source of ADP, and its concentration profile can be written as
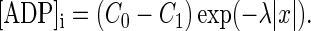 |
(3) |
Motor-assisted transport of mitochondria
Many motors, regardless of their particular family of origin, operate similarly to the processive movement of kinesin on microtubules (21–23). For the directed transport of mitochondria, we used a model
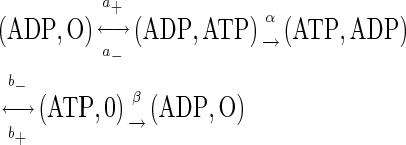 |
(4) |
that contains all essential steps of motor operation. Here, the two symbols in brackets indicate the occupancy of the ATP-binding sites of the motor, the terms a+ (a−) and b+ (b−) are the on-(off)-rate constants for the binding of nucleotides, and α and β represent irreversible translocation steps. The full motor cycle consists of the binding of ATP to the empty head of the motor, its rotation and the motor advance (the first half-step of kinesin, l/2 = 4 nm), dissociation of ADP from the motor, ATP hydrolysis, and the motor advance for another half-step (note that the first and the final states of the motor have identical configuration and only the position of the motor is changed). The mean turnover time (τo) of the motor is given by the steady-state solution of Eq. 4 as
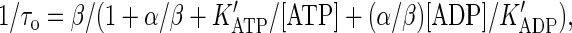 |
(5) |
where the apparent dissociation constants are 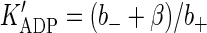 and
and 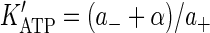 .
.
According to the data obtained in vitro for kinesin (22,23) and myosin V (21), we set the rate constants to α = β = 200 s−1, b+ = a+ = 10 μM−1s−1 and b− = a− = k = 100 s−1. Under these assumptions, Eq. 5 simplifies to
 |
(6) |
where 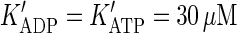 . At resting values of [ATP] = 1 mM and [ADP] = 0.1 mM, the model predicts the motor velocity l/τo = 8 nm × 50 s−1 = 0.4 μm/s, i.e., two times lower than the maximal velocity.
. At resting values of [ATP] = 1 mM and [ADP] = 0.1 mM, the model predicts the motor velocity l/τo = 8 nm × 50 s−1 = 0.4 μm/s, i.e., two times lower than the maximal velocity.
A constant turnover of motors does not allow us to consider them as fully saturated with ATP even under physiological conditions. The situation is dynamic, and it would be more correct to speak about the occupancy of corresponding states. For the resting conditions, the occupancies of the states (ADP,O), (ADP,ATP), (ATP,ADP), and (ATP,0) are 0, 20, 60, and 20%, respectively.
Describing the motor behavior in terms of the Michaelis-Menten kinetics, we can consider the “enzyme” with a single ATP/ADP binding site, which corresponds to a scheme
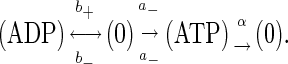 |
(7) |
The enzyme “production” rate is
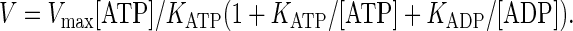 |
(8) |
At [ATP] = 1 mM, the binding site is always occupied by ATP. V is close to Vmax and only weakly depends on [ADP] under most physiological conditions.
The simplest scheme of a motor that involves ATP hydrolysis for movement is
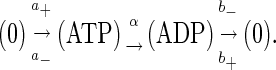 |
(9) |
It has a steady-state solution
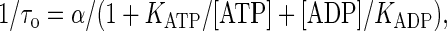 |
(10) |
the form of which is similar to that of Eq. 5, indicating that the order of states in (7) versus (9) makes the difference. The simplest scheme (9) is, however, unrealistic as it corresponds to a motor with one head, which does not exist.
In the Monte Carlo simulations, we applied the algorithms designed to describe a probabilistic behavior of single ion channels (24). At each step of the motor, the turnover time was calculated from Eq. 6 at presumed local values of [ATP]i and [ADP]i. In addition, we modeled the dynamics of small mitochondrial ensembles. We also took into account a constant switching of mitochondria between the Brownian (or stochastic) motion (mode B) and the directed antero- or retrograde transport (mode A and mode R, respectively), which was described by the sequential scheme: A ↔ B ↔ R (4). After a single run, the mode was changed (from A and R to B, and from B to A or to R with equal probability). The run times of particles in a given mode were determined as T = −To ln(R), where To = 5 s in all three modes, and 0 < R < 1 is a random number. The run lengths were calculated as L = −Lo ln(r) where Lo = 3 μm (mode A), −4 μm (mode R), and 0 (mode B) (4) and 0 < r < 1 is another random number. The run times and run lengths were scaled according to the dependence of the motor processivity on the local levels of [ATP] and [ADP].
Qualitative injection of adenine nucleotides
Intracellular injections of ATP and ADP were made using a Femtojet (Eppendorf, Hamburg). Injection pipettes (tip diameter <0.3 μm, resistance >50 MΩ) were manufactured from borosilicate glass with filament (GC150F, Clark Instruments, Salisbury, UK). Injection volumes were calibrated by measuring the diameter of the bubble injected into air before the trial. Intracellular concentrations were obtained by dividing the injected volume by the volume of the cell soma calculated from its area and thickness. In four neurons transfected with luciferase, the luminescence increased by 35 ± 7% after injection of 0.4 mM ATP. Such changes agree with a presumed resting concentration of [ATP]i = 1 mM. Mitochondrial movements were not modified after ATP injections, which indicated that the motors apparently had no ATP deficit (see also Discussion).
The results of experiments with quantitative injection of nucleotides could potentially be distorted by the intrinsic activity of adenylate kinase (AK), which disproportionates ADP according to the reaction
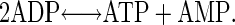 |
(11) |
This enzyme is expressed in various cells (25–27) including neurons and can influence the homeostasis of adenine nucleotides. In biochemistry, the reaction is assumed to be fast and reversible (27) and has an equilibrium constant
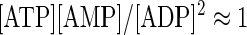 |
(12) |
The reported free concentrations of adenine nucleotides vary, depending on the tissue, the methods of measurements, and other experimental conditions. In the analysis of AK effects, we assumed that at rest [ATP] = 1 mM, [ADP] = 100 μM, and [AMP] = 30 μM: the first two values correspond to the data obtained in neocortical neurons (28), and the value of [AMP] was chosen to satisfy Eq. 12.
During hypoxia, ATP hydrolyzes to ADP. When the total concentration of nucleotides is constant (= N) and AK decreases newly formed ADP by x, Eq. 12 can be put as (N − D + x/2)(x/2)/(D − x)2 = 1, where D is initial ADP concentration. For [ATP] = 0.6 mM (as measured during hypoxia, see below) and D = 0.4 mM, the solution of the above equation corresponds to [ADP] = D − x ≈ 0.2 mM. When ATP is injected at rest, new ADP cannot be produced by AK because [AMP] is too small. When ATP is injected during hypoxia, the equilibrium maintained by AK satisfies the equation (T − x)(M − x)/(D + 2x)2 = 1, where T = 1 mM (the measured [ATP] plus the injected quantity), and D = 0.2 mM and M = [AMP] = 0.2 mM are the “equilibrium” concentrations of ATP, ADP, and AMP during hypoxia as estimated above. The solution of this quadratic equation gives [ATP] = 0.93, [ADP] = 0.44, and [AMP] = 0.13 mM. When D equivalents of ADP are injected at normal conditions, the equilibrium corresponds to the equation (T + x/2)(x/2)/(D − x)2 = 1. The resulting [ADP] = 0.23 mM, which is close to the maximal [ADP] levels that can be established during hypoxia in the presence of AK.
RESULTS
In neurons transfected with ECFP-mito (Clontech, Heidelberg) and subsequently stained with TMRE, the fluorescence images were almost identical (95 ± 4% overlap in seven neurons), indicating that TMRE stained only mitochondria but not other organelles. In the images of luciferase luminescence (leftmost panel in Fig. 1 A), the local decreases often coincided with the positions of mitochondria, indicating their preferential location in the regions of low [ATP]i. The mean widths of peaks that represented mitochondria (TMRE), synapses (FM 1-43), and ATP-binding sites (TNP-ATP) were equal to 0.72 ± 0.12 (n = 23), 0.95 ± 0.17 (n = 21), and 0.86 ± 0.15 μm (n = 22), respectively. Distributions of luciferase luminescence showed much shallower troughs. This speaks against possible effects of exclusion of cytosolic volume by intracellular organelles, which might contribute to the observed correlation between the local decreases in luminescence and TMRE and FM 1-43 fluorescence spots.
FIGURE 1.

Correlation among the positions of mitochondria, synaptic vesicles, and ATP. (A) Merged images of TMRE, FM 1-43, and TNP-ATP fluorescence (green) and the luminescence of luciferase (red). The arrows indicate the positions of enlarged (×8) areas shown in insets. (B) Curvilinear line scans of fluorescence (green) and luminescence (red) in dendrites are indicated by the white lines in A. The data were normalized to the maximal luminescence (fluorescence) in the respective frames. Note the negative (TMRE, FM 1-43) and the positive (TNP-ATP) correlations with the luminescence of luciferase in the corresponding line scans. (C) Spatial correlation between the luminescence and fluorescence signals. The black and blue dots indicate the amplitude of respective pixels in the soma and in dendrites. Linear regression gave the slopes of straight lines 1.05 (A), 0.97 (B), and 0.92 (C), respectively, and all correlation coefficients are >0.95. The differences between the overlap in the soma and those in dendrites were insignificant.
After obtaining luminescence images, we switched into the fluorescence mode and recorded the movements of mitochondria in dendrites. The particles, which initially resided in the regions of low luminescence (<25% of the mean in dendrites), moved with an absolute mean velocity of 0.12 ± 0.03 μm/s (n = 29), and the organelles, which were present in the regions of high luminescence (>75% of the mean in dendrites), moved with an absolute mean velocity of 0.41 ± 0.13 μm/s (n = 23).
Mitochondria were often found in the vicinity of synapses visualized by staining synaptic vesicles with FM 1-43 (Fig. 1 B, see also Mironov (4)). Luciferase signals in the perisynaptic regions were smaller (25 ± 6% of the average luminescence, mean for 16 areas analyzed in six cells). The synaptic origin of the observed FM 1-43 signals was confirmed at the end of each experiment by applying 45 mM KCl to induce exocytosis. The positions of the spots of FM 1-43 fluorescence also coincided with the distribution of synaptophysin (five neurons examined). In TMRE- and FM 1-43-stained neurons, mitochondria moved at a mean speed 0.14 ± 0.07 μm/s at distances <2 μm from the synapse. Outside these regions, they moved at 0.49 ± 0.11 μm/s (17 mitochondria examined in seven cells, P < 0.05, see also Mironov (4)).
Because the spatiotemporal resolution of luciferase imaging was clearly insufficient to reveal the fine and fast processes that occur during the trafficking of mitochondria, we used also TNP-ATP, which fluorescence highlights ATP-binding sites (16). In cells loaded with TNP-ATP, the cytoplasmic fluorescence was diffuse and showed bright spots that coincided with peaks of luciferase luminescence (Fig. 1 C).
The respiratory neurons in vivo (11–13) and in vitro (4) demonstrate spontaneous synaptic currents assembled into rhythmic synaptic bursts or drives that are accompanied by [Ca2+]i transients. During hypoxia, the amplitude and frequency of synaptic currents first increased and then decreased and slowly restored on reogygenation (Fig. 2 A). This sequence of events represents a typical respiratory response to hypoxia (11–14).
FIGURE 2.

Suppression of mitochondrial mobility by hypoxia and glutamate. (A) Rhythmic [Ca2+]i transients (upper trace) and electrical activity (lower trace) are modulated by hypoxia. The measurements were made in fura-2-loaded respiratory neurons using perforated whole-cell patch-clamp recordings as described in Methods. (B) Mitochondrial movements during hypoxia and their correlation with local changes in TNP-ATP fluorescence. The two traces show the changes in instantaneous mitochondrial velocity (dx/dt) and in the TNP-ATP fluorescence (ΔFTNP). The time-dependent changes were measured in moving regions that encircled single mitochondria in dendrites. The positive and negative step-like changes in velocity correspond to the antero- and retrograde transport, respectively. (C) The run times and run lengths of mitochondria measured 3 min before (open bars) and 3 min after applying hypoxia (solid bars). (D) Inhibition of mitochondrial mobility by glutamate (1 mM) in Ca2+-containing and Ca2+-free solutions. Note that both fluorescence transients and directed movements were reversibly inhibited during the applications of hypoxia and glutamate.
The fluorescence of TNP-ATP showed transient increases during the directed transport of mitochondria. Hypoxia inhibited both fluorescence changes and the directed transport of mitochondria (Fig. 2 B). Using SPT, we observed a decrease in the mean occurrence of transport events from 1/(8 ± 2 s) to 1/(18 ± 4 s) during hypoxia (32 mitochondria in five cells). The run times (Fig. 2 C) were not changed (8.5 ± 1.1 vs. 8.7 ± 1.0 s), and the run lengths decreased from 5.0 ± 1.2 to 2.4 ± 0.5 μm (anterograde transport) and from 4.2 ± 1.1 to 2.2 ± 0.4 μm (retrograde transport). Thus, mitochondria during hypoxia traveled along smaller distances within the same run time at about half velocity. The data also show that during hypoxia mitochondria were picked up by the motors less frequently, or, alternatively, they were docked more frequently. At present, we cannot distinguish between these two possibilities.
The directed transport of mitochondria and the accompanying TNP-ATP transients were reversibly inhibited after the applications of 1 mM glutamate (Fig. 2 D, representative trace of 15 dendritic mitochondria tracked in six cells). The effect might be caused by glutamate-evoked [Ca2+]i increases, which inhibit the mobility of mitochondria (29,30). However, the applications of glutamate (the second experiment in Fig. 2 D) and hypoxia (n = 5, data not shown) in [Ca2+]-free solutions also inhibited the mitochondrial movements, which speaks against the involvement of intracellular Ca2+.
In luciferase-expressing neurons, applications of hypoxia for 3 min decreased the mean luminescence by 37 ± 5% (n = 5), and 1 mM glutamate decreased it by 27 ± 5% (n = 4, P < 0.05). Because luciferase generates no luminescence without ATP (31), the changes in [ATP]i are determined by relative decreases in luminescence. For the resting [ATP]i = 1 mM, this would correspond to an [ATP]i decrease during hypoxia to ∼0.6 mM, which agrees with the measurements of ATP content in neocortical neurons (28).
We attempted to reverse the observed hypoxic suppression of the mitochondrial mobility by injecting 0.4 mM ATP. This was without effect (Fig. 3 A), which might be caused by fast ATP consumption during hypoxia. We therefore repeated the experiments by measuring simultaneously the activity of KATP channels. ATP injection fully reversed the activation of channels by hypoxia (Fig. 3 B). The effect lasted for minutes, which speaks against fast ATP consumption during hypoxia.
FIGURE 3.

Movements of mitochondria are differentially influenced by ATP and ADP. (A) Hypoxia and ATP. The trajectories of mitochondria in dendrites were repositioned to begin at the same point for comparison. Note that inhibition of mobility by hypoxia was not reversed after injection of 0.4 mM ATP. (B) ATP (0.4 mM) reverses hypoxic activation of ATP-sensitive K+ (KATP) channels. Mean open probabilities of the channels (Popen) are indicated near each trace. (C) ADP and hypoxia. Note that after suppression of mitochondrial movements by injection of 0.4 mM ADP, hypoxia produced no effect. All presented trajectories were obtained by applying SPT to the 3-min-long recordings of mitochondrial movements in the control and after the treatments. Note decreases in the lengths of continuous directed movements of mitochondria during hypoxia and at elevated [ADP]i levels.
Injections of 0.4 mM ADP promptly and potently suppressed the movements of mitochondria (Fig. 3 C). The mean velocities during anterograde and retrograde movements decreased from 0.51 ± 0.11 μm/s and −0.55 ± 0.09 μm/s to 0.22 ± 0.07 μm/s (n = 21) and −0.21 ± 0.08 μm/s (n = 28), respectively. After ADP injections, we did not observed significant differences in the transport of mitochondria in the perisynaptic and other regions that can be caused by smoothing of [ADP]i gradients within the cells. Applications of hypoxia after ADP injection did not inhibit the mitochondrial motility (Fig. 3 C, n = 4).
Application of SPT to analyze mitochondrial trajectories in the presence of ADP did not reveal significant changes in the frequency of transport events or in the run times. For 12 mitochondria in three injected cells, the respective values were 1/(7 ± 2 s) vs. 1/(8 ± 3 s) and 8.7 ± 1.0 vs. 8.6 ± 1.2 s. The run lengths decreased from 4.9 ± 1.1 to 2.6 ± 0.4 μm. The data thus indicate that the occurrence and duration of the episodes of the directed transport in the presence of ADP were unchanged, whereas the velocity of mitochondria decreased ∼2-fold.
Injections of ADP together with 10 μM diadenosine pentaphosphate (Ap5a, inhibitor of AK) produced similar effects (n = 3). This speaks against substantial activity of AK in neurons during hypoxia (see Methods) and is also is in line with the recent data (26) showing that disproportionation of ADP by AK during hypoxia can take tens of minutes.
To explain differential effects of ATP and ADP on mitochondrial transport, we modeled the dependence of mitochondrial velocity on [ATP] and [ADP] levels. The mean [ATP]i profile around the synapse was obtained from luminescence measurements (Fig. 4 A) and complemented the corresponding [ADP]i gradient given by Eq. 3. At constant resting [ATP]i = 1 mM and [ADP]i = 0.1 mM, the average motor speed was 0.5 μm/s. In nonuniform profiles of [ATP]i and [ADP]i, the transport velocity in the vicinity of a “synapse” reversibly decreased (Fig. 4 B). To determine which nucleotide, ATP or ADP, has more influence on the transport of mitochondria, the concentration of one nucleotide was fixed. In the field of [ATP]i gradient and at a constant [ADP]i = 0.1 mM, the transport velocity was the same as in the control. At constant [ATP]i = 1 mM and nonuniform [ADP]i, the velocity was two times smaller, and its changes resembled the results obtained by considering both [ATP]i and [ADP]i gradients. The simulations were then extended by considering the ensemble of 16 mitochondria. The particles were allowed to switch between the modes of stochastic motion and antero- and retrograde transport (4) (see the pauses and continuous movements in Fig. 3) and to “feel” the gradients of [ATP]i and [ADP]i. During the simulations that corresponded to ∼2 h of a cell's life (Fig. 4 C), we often observed a dynamic formation of long-living linear clusters in the proximity of a synapse.
FIGURE 4.

Mitochondrial transport in nonuniform fields of [ATP]i and [ADP]i. (A) Mean [ATP] profile in dendrites. The data were obtained from the luminescence scans (21 in total), which were aligned according to the maxima of FM 1-43 fluorescence in dendrites identified as synaptic locations (see Methods). The experimental data points were approximated by Eq. 2 with the parameters C0 = 1 mM (the resting [ATP]i), C1 = 0.2 mM ([ATP]i at synapse), and the space constant 1/λ = 6 μm. (B) Monte Carlo simulations of the directed transport of mitochondria. The four kymographs show the results of simulations at resting [ATP]i = 1 mM and [ADP]i = 0.1 mM (denoted as “rest”), in varying profiles of [ATP]i and [ADP]i (denoted as “var”), and by using the profiles of either [ADP]i or [ATP]i with the concentration of another nucleotide fixed (the conditions of simulations are indicated near each curve). (C) Monte Carlo simulations of 16 mitochondria. The kymograph shows a 2-h-long simulation run performed using prescriptions described in Methods. Mitochondria were assumed to move bidirectionally in nonuniform profiles of [ATP]i and [ADP]i (shown in the lowermost panel), which models the concentration gradients in the vicinity of persistently active synapses.
DISCUSSION
The functioning of neurons is in many aspects determined by their ability to form and to maintain synapses, the reliable operation of which is dependent on an adequate energy supply. This can be provided by mitochondria, which are often positioned close to the active synapses (1,2,4). This arrangement may reflect an intense ATP consumption in these regions, which can potentially regulate mitochondrial motility according to the availability of fuel (ATP) for the molecular motors (1). Inhibition of mitochondrial movements by mitochondrial poisons in various cell types (5–8) supports this notion. Whether and how synaptic activity modifies the local levels of [ATP]i and how these changes are translated into changes in mitochondrial mobility are yet unknown. In solving this issue, the main obstacle was to measure [ATP]i levels in living cells. We imaged the luminescence of luciferase by expressing this enzyme in the respiratory neurons (Fig. 1) and directly showed the existence of a correlation between the motility of mitochondria and the local [ATP]i levels. Using the ATP analog TNP-ATP, we also monitored the binding of ATP to molecular motors during the directed transport of mitochondria (Fig. 2). Both hypoxia and glutamate inhibited transportation events. Injections of ATP to restore the measured [ATP]i depletion during hypoxia (0.4 mM) did not rescue the mobility of mitochondria (Fig. 3). Looking for another metabolic factor that could regulate the directed transport of mitochondria, we tested ADP, which is a natural product of ATP hydrolysis. Injections of ADP decreased the mobility of mitochondria, and subsequent hypoxia was without effect. We explain these data using the model described in Methods. According to the derived equations, the molecular motors that transport mitochondria should experience no shortage of fuel unless [ATP]i < 100 μM, which is unlikely to occur in a living cell. However, the motor velocity decreases when [ADP]i is even moderately elevated. This is caused by more frequent rebound of ADP to the motor, which slows down its turnover and correspondingly decreases the velocity.
A cytoplasm is often considered as a “well-mixed bag”, but this point has been recently challenged (32,33). We believe that [ATP]i levels in the vicinity of active synapses can be locally compromised (1), which would result in gradients of [ATP]i in dendrites. The perisynaptic regions can thus be considered as point “sinks” for ATP and point “sources” of ADP. In dendrites of persistently active neurons, steady-state gradients of [ATP]i and [ADP]i can exist (Fig. 4). The movements of mitochondria are modified according to the bursting activity of the respiratory neurons (see Fig. 7 of Mironov (4)). We previously observed periodic changes in the activity of KATP–sensitive channels in the respiratory neurons (13), which reflect cyclic changes in [ATP]i caused by bursts of activity. The rhythmic bursts of action potentials last for ∼1 s (Fig. 2 A), and when a single Na+ channel at a synapse is open for only 10 ms during the burst, this would increase intracellular [Na+] at a rate of 0.1 mM/s. Similar changes in [Ca2+]i can be expected (18,19). To restore resulting disturbances in ion homeostasis, submillimolar quantities of ATP are needed, whereby the equivalent amounts of ADP are also produced (20,34).
Mitochondria themselves generate most of the cellular ATP, and therefore, they can consume ADP. As a result, the [ATP] and [ADP] gradients that can be built in the vicinity of mitochondria can potentially influence the operation of motors. The effect of ADP on mitochondrial respiration is intimately regulated by ADP availability to an ADP-ATP carrier that transports ADP into mitochondria. The carrier has a very high affinity for this substrate, ∼10 μM (35). This means that at resting [ADP]i (100 μM), the transport of ADP into mitochondria is already activated by 90%, and it would change little on further increases in [ADP]i. Thus, mitochondria are probably unable to manipulate the local levels of [ADP]i in their immediate vicinity. This may make sense: otherwise, they would listen only to what is going inside their “body” (mitochondrial matrix) and would not respond to the needs of a cell.
Obviously, a dynamic distribution of mitochondria within the cells is not controlled by ADP alone, and many other factors can be involved. One recently identified example is nitric oxide (36), which can be produced in a Ca2+-dependent way both in the cytoplasm and in mitochondria. Also important are such proteins as Milton (37) and Miro (38), which participate in the attachment of mitochondria to molecular motors and can mediate their interaction with cytoskeleton or other organelles (39,40). Such interactions are of the “all-or-nothing” type in the sense that they define whether mitochondria should go or stay. A short-range signaling via [ADP]i gradients in dendrites can provide an adjustable steering mechanism for slowing down mitochondria in the proximity of synapses that can increase the probability of their anchoring. The concerted interplay between metabolic and signaling factors would then help mitochondria to reside in perisynaptic regions.
We here examined only motile mitochondria (which in living neurons comprise ∼⅓ of all present (3–6)) and the mechanisms that can control their movements via local changes in [ATP]i and [ADP]i. The fission and the fusion of stationary mitochondria (41) are also important because they can lead to the formation of giant and/or reticulated mitochondria (7,8). A physical continuity within mitochondrial “filaments” (8,37–42) in the vicinity of synapses may be beneficial in terms of enhanced Ca2+ buffering and ATP production (43). Stationary mitochondria may eventually become nonfunctional, and such particles are delivered to the soma for “recycling” (7). The transportation of “fresh” mitochondria to dendrites and their proper targeting have to be controlled, for which the regulation of their transport by local [ADP]i levels may be well suited.
Acknowledgments
The author thanks N. Hartelt for excellent technical assistance, M. Müller and J. Schmidt for their help with a two-photon microscope, M. Ivannikov and T. Szellas for assistance with luciferase expression, and A. Woehler for critical reading of the manuscript and helpful suggestions.
References
- 1.Bereiter-Hahn, J., and M. Voth. 1994. Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microsc. Res. Tech. 27:198–219. [DOI] [PubMed] [Google Scholar]
- 2.Wong-Riley, M., and E. W. Carroll. 1984. Effect of impulse blockage on cytochrome oxidase activity in monkey visual system. Nature. 307:262–264. [DOI] [PubMed] [Google Scholar]
- 3.Morris, R. L., and P. J. Hollenbeck. 1995. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J. Cell Biol. 131:1315–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mironov, S. L. 2006. Spontaneous and evoked neuronal activities regulate movements of single neuronal mitochondria. Synapse. 59:403–411. [DOI] [PubMed] [Google Scholar]
- 5.Overly, C. C., H. J. Rieff, and P. J. Hollenbeck. 1996. Organelle motility and metabolism in axons vs. dendrites of cultured hippocampal neurons. J. Cell Sci. 109:971–980. [DOI] [PubMed] [Google Scholar]
- 6.Ligon, L. A., and O. Steward. 2000. Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J. Comp. Neurol. 427:351–361. [DOI] [PubMed] [Google Scholar]
- 7.Miller, K. E., and M. P. Sheetz. 2004. Axonal mitochondrial transport and potential are correlated. J. Cell Sci. 117:2791–2799. [DOI] [PubMed] [Google Scholar]
- 8.Muller, M., S. L. Mironov, M. V. Ivannikov, J. Schmidt, and D. W. Richter. 2005. Mitochondrial organization and motility probed by two-photon microscopy in cultured mouse brainstem neurons. Exp. Cell Res. 303:114–127. [DOI] [PubMed] [Google Scholar]
- 9.Lopez-Barneo, J., R. del Toro, K. L. Levitsky, M. D. Chiara, and P. Ortega-Saenz. 2004. Regulation of oxygen sensing by ion channels. J. Appl. Physiol. 96:1187–1195. [DOI] [PubMed] [Google Scholar]
- 10.Brockhaus, J., K. Ballanyi, J. C. Smith, and D. W. Richter. 1993. Microenvironment of respiratory neurons in the in vitro brainstem-spinal cord of neonatal rats. J. Physiol. 462:421–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mironov, S. L., and K. Langohr. 2005. Mechanisms of Na+ and Ca2+ influx into respiratory neurons during hypoxia. Neuropharmacology. 48:1056–1065. [DOI] [PubMed] [Google Scholar]
- 12.Mironov, S. L., and D. W. Richter. 2001. Oscillations and hypoxic changes of mitochondrial variables in neurons of the brainstem respiratory centre. J. Physiol. 533:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haller, M., S. L. Mironov, A. Karschin, and D. W. Richter. 2001. Dynamic activation of KATP channels in rhythmically active neurons. J. Physiol. 537:69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mironov, S. L., K. Langohr, M. Haller, and D. W. Richter. 1998. Hypoxia activates ATP-dependent potassium channels in inspiratory neurons of neonatal mice. J. Physiol. 509:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Betz, W. J., and G. S. Bewick. 1992. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science. 255:200–203. [DOI] [PubMed] [Google Scholar]
- 16.Hiratsuka, T. 2003. Fluorescent and colored trinitrophenylated analogs of ATP and GTP. Eur. J. Biochem. 270:3479–3485. [DOI] [PubMed] [Google Scholar]
- 17.Mironov, S. L. 1990. Theoretical analysis of propagation of [Ca2+]i wave along the surface of intracellular stores. J. Theor. Biol. 146:87–97. [DOI] [PubMed] [Google Scholar]
- 18.Mironov, S. L., Y. M. Usachev, and H. D. Lux. 1993. Spatial and temporal control of intracellular free Ca2+ in chick sensory neurons. Pflugers Arch. 424:183–191. [DOI] [PubMed] [Google Scholar]
- 19.Mironov, S. L. 1995. Plasmalemmal and intracellular Ca2+ pumps as main determinants of slow Ca2+ buffering in rat hippocampal neurons. Neuropharmacology. 34:1123–1132. [DOI] [PubMed] [Google Scholar]
- 20.Vendelin, M., O. Kongas, and V. Saks. 2000. Regulation of mitochondrial respiration in heart cells analyzed by reaction-diffusion model of energy transfer. Am. J. Physiol. 278:C747–C764. [DOI] [PubMed] [Google Scholar]
- 21.Rosenfeld, S. S., and H. L. Sweeney. 2004. A model of myosin V processivity. J. Biol. Chem. 279:40100–40111. [DOI] [PubMed] [Google Scholar]
- 22.Yajima, J., M. C. Alonso, R. A. Cross, and Y. Y. Toyoshima. 2002. Direct long-term observation of kinesin processivity at low load. Curr. Biol. 12:301–306. [DOI] [PubMed] [Google Scholar]
- 23.Cross, R. A. 2004. The kinetic mechanism of kinesin. Trends Biochem. Sci. 29:301–319. [DOI] [PubMed] [Google Scholar]
- 24.Clay, J. R., and L. J. DeFelice. 1983. Relationship between membrane excitability and single channel open-close kinetics. Biophys. J. 42:151–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carrasco, A. J., P. P. Dzeja, A. E. Alekseev, D. Pucar, L. V. Zingman, M. R. Abraham, D. Hodgson, M. Bienengraeber, M. Puceat, E. Janssen, B. Wieringa, and A. Terzic. 2001. Adenylate kinase phosphotransfer communicates cellular energetic signals to ATP-sensitive potassium channels. Proc. Natl. Acad. Sci. USA. 98:7623–7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andjus, R. K., Z. Dzakula, J. L. Markley, and S. Macura. 2005. Brain energetics and tolerance to anoxia in deep hypothermia. Ann. N. Y. Acad. Sci. 1048:10–35. [DOI] [PubMed] [Google Scholar]
- 27.Plaxton, W. C. 2004. Principles of metabolic control. In Functional Metabolism: Regulation and Adaptation. K. B. Storey, editor. John Wiley & Sons, New York. 1–24.
- 28.Pissarek, M., S. Garcia de Arriba, M. Schafer, D. Sieler, K. Nieber, and P. Illes. 1998. Changes by short-term hypoxia in the membrane properties of pyramidal cells and the levels of purine and pyrimidine nucleotides in slices of rat neocortex; effects of agonists and antagonists of ATP-dependent potassium channels. Naunyn Schmiedebergs Arch. Pharmacol. 358:430–439. [DOI] [PubMed] [Google Scholar]
- 29.Yi, M., D. Weaver, and G. Hajnoczky. 2004. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J. Cell Biol. 167:661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brough, D., M. J. Schell, and R. F. Irvine. 2005. Agonist-induced regulation of mitochondrial and endoplasmic reticulum motility. Biochem. J. 392:291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koop, A., and P. H. Cobbold. 1993. Continuous bioluminescent monitoring of cytoplasmic ATP in single isolated rat hepatocytes during metabolic poisoning. Biochem. J. 295:165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kennedy, H. J., A. E. Pouli, E. K. Ainscow, L. S. Jouaville, R. Rizzuto, and G. A. Rutter. 1999. Glucose generates sub-plasma membrane ATP microdomains in single islet beta-cells. J. Biol. Chem. 274:13281–13291. [DOI] [PubMed] [Google Scholar]
- 33.Weiss, J. N., and P. Korge. 2001. The cytoplasm: no longer a well-mixed bag. Circ. Res. 89:108–110. [PubMed] [Google Scholar]
- 34.Kargacin, M. E., and G. J. Kargacin. 1997. Predicted changes in concentrations of free and bound ATP and ADP during intracellular Ca2+ signaling. Am. J. Physiol. 273:C1416–C1426. [DOI] [PubMed] [Google Scholar]
- 35.Vignais, P. 1976. Molecular and physiological aspects of adenine nucleotide transport in mitochondria. Biochim. Biophys. Acta. 456:1–38. [DOI] [PubMed] [Google Scholar]
- 36.Zanelli, S. A., P. A. Trimmer, and N. J. Solenski. 2006. Nitric oxide impairs mitochondrial movement in cortical neurons during hypoxia. J. Neurochem. 97:724–736. [DOI] [PubMed] [Google Scholar]
- 37.Stowers, R. S., L. J. Megeath, J. Gorska-Andrzejak, I. A. Meinertzhagen, and T. L. Schwarz. 2002. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 36:1063–1077. [DOI] [PubMed] [Google Scholar]
- 38.Guo, X., G. T. Macleod, A. Wellington, F. Hu, S. Panchumarthi, M. Schoenfield, L. Marin, M. P. Charlton, H. L. Atwood, and K. E. Zinsmaier. 2005. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 47:379–393. [DOI] [PubMed] [Google Scholar]
- 39.Mironov, S. L., and N. Symonchuk. 2006. ER vesicles and mitochondria move and communicate at synapses. J. Cell Sci. 119:4926–4934. [DOI] [PubMed] [Google Scholar]
- 40.Mironov, S. L., M. V. Ivannikov, and M. Johansson. 2005. [Ca2+]i signaling between mitochondria and endoplasmic reticulum in neurons is regulated by microtubules. From mitochondrial permeability transition pore to Ca2+-induced Ca2+ release. J. Biol. Chem. 280:715–721. [DOI] [PubMed] [Google Scholar]
- 41.Meeusen, S., J. M. McCaffery, and J. Nunnari. 2004. Mitochondrial fusion intermediates revealed in vitro. Science. 305:1747–1752. [DOI] [PubMed] [Google Scholar]
- 42.Chang, D. T., A. S. Honick, and I. J. Reynolds. 2006. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J. Neurosci. 26:7035–7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skulachev, V. P. 2001. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem. Sci. 26:23–29. [DOI] [PubMed] [Google Scholar]