

Abstract
Tethered particle experiments use light microscopy to measure the position of a micrometer-sized bead tethered to a microscope slide via an approximately micrometer-length polymer, to infer the behavior of the invisible polymer. Currently, this method is used to measure rate constants of DNA loop formation and breakdown mediated by repressor protein that binds to the DNA. We report a new technique for measuring these rates using a modified hidden Markov analysis that directly incorporates the diffusive motion of the bead, which is an inherent complication of tethered particle motion because it occurs on a timescale between the sampling frequency and the looping time. We compare looping lifetimes found with our method, which are consistent over a range of sampling frequencies, to those obtained via the traditional threshold-crossing analysis, which vary depending on how the raw data are filtered in the time domain. Our method does not involve such filtering, and so can detect short-lived looping events and sudden changes in looping behavior.
One mechanism for regulating DNA transcription is for a protein to bind to specific operator sites in the DNA sequence, thereby enhancing or diminishing the expression of adjacent genes. In an elaboration of this idea, multiple operators recruit copies of the repressor protein, bind to each other, and bend the DNA into a loop, for example in the λ-system (1). One goal of in vitro DNA looping experiments is to determine the rate constants for DNA loop breakdown/formation and gain insight into how the physical process of looping influences the biochemistry of transcription. The purpose of this letter is to present a new diffusive hidden Markov method (DHMM) for determining the looping kinetics from data measured in tethered particle experiments. The main advantage of our method is that by directly incorporating the dynamics of particle diffusion we do not need to filter the raw data. Consequently, DHMM has better time resolution and more consistent results than the traditional threshold-crossing analyses.
The tethered particle method (TPM, Fig. 1) consists of measuring the Brownian motion of a small bead attached to a microscope slide via a short polymer tether, to learn about the tether's behavior (2–6). Our setup uses DIC imaging of a 480-nm diameter polystyrene bead tethered to the slide via a 3477-bp DNA construct containing two sets of three wild-type λ-operator sites separated by 2317 bp, as described previously (7). The (x,y) coordinates of up to six well-spaced beads are recorded simultaneously with 20-ms time resolution using custom particle tracking software and a CCD camera with a 1-ms shutter to reduce blurring. Bead positions are first recorded for ∼10 min to ensure uniform behavior. Upon addition of 200 nM cI repressor protein, dynamic exchanges between unlooped and looped tether lengths—consistent with the known construct length and operator spacing—are observed. After recording for 30–60 min, the data are corrected for microscope drift and screened for anomalous sticking events using methods described previously (8).
FIGURE 1.

Schematic showing DNA loop formation and breakdown in a tethered bead. The character ρ denotes the plane-projected distance from the attachment point to the bead center.
After drift correction, transitions are clearly visible when the data are plotted as the radial distance from the anchor point 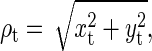 where t is an index indicating which video frame (Fig. 2). The equilibrium distributions of ρ are well understood (8,9), but the large overlap between unlooped and looped distributions at small values of ρ prevents us from unambiguously determining the state of the DNA at particular times—loop formation is not directly observable. Typically, this ambiguity is reduced by filtering ρ (we find the variance over windows of time width W); however, the time resolution is then degraded by at least the same amount (6,7). Filtering helps remove false events at very short times introduced by natural Brownian motion of the bead, but actual events are missed due to the reduced time resolution. Unfortunately, we show below that in our system, looping lifetimes determined by this technique depend strongly on the chosen value of W.
where t is an index indicating which video frame (Fig. 2). The equilibrium distributions of ρ are well understood (8,9), but the large overlap between unlooped and looped distributions at small values of ρ prevents us from unambiguously determining the state of the DNA at particular times—loop formation is not directly observable. Typically, this ambiguity is reduced by filtering ρ (we find the variance over windows of time width W); however, the time resolution is then degraded by at least the same amount (6,7). Filtering helps remove false events at very short times introduced by natural Brownian motion of the bead, but actual events are missed due to the reduced time resolution. Unfortunately, we show below that in our system, looping lifetimes determined by this technique depend strongly on the chosen value of W.
FIGURE 2.

Dynamic looping seen in tethered particle time-series data. The region from time 200–1200 s shows dynamic looping behavior and was used in the subsequent analysis. The ▾ indicates a brief 4.5 s sticking event that was omitted from the analysis by concatenating the drift-corrected data. The inset shows the corresponding filtered time series with window size W = 4 s. Red lines are inferred transition sequences for DHMM and threshold-crossing analyses, respectively. Dashed line = 270 nm threshold.
Hidden Markov methods (10,11), however, do not require such smoothing. These methods allow for analysis of the unfiltered data, once we overcome one obstacle: In traditional HMM applications, the uninteresting part of the observed signal (or “noise”) has no correlations apart from those introduced by the underlying hidden process. Unfortunately, the tethered Brownian motion of our bead has an intrinsic timescale that is slow compared with our 20-ms sampling frequency, but faster than the looping lifetime. The basic physics is easily reproduced by a particle diffusing in a harmonic potential well (a problem similar to TPM motion), with diffusion constant D = 480,000 nm2/s and spring constant κ = 0.65 × 10−3 pN/nm obtained from fits to the autocorrelation of the measured position, the characteristic decay time τD = kBT/Dκ is ∼140 ms (12). This diffusive motion not only prevents efficient filtering, but also the direct application of traditional hidden Markov methods to TPM.
More precisely, standard HMM supposes that an observed signal reflects two processes (10): A hidden process that generates a time series {qt} according to an autonomous Markov process with some time-step distribution D(qt+1|qt), and an observed signal {rt} that, at each instant t, is drawn from a probability distribution P(rt|qt), which depends only on the current value of qt. This framework is appropriate for the case where qt is the internal state of an ion channel and rt is the instantaneous current through the channel. We might be tempted to apply it to our case as well, letting qt denote the looping state of our DNA tether and rt = (xt, yt). But the ability to form a loop depends on the location of the bead: For example, if the bead is too far from the attachment point, then loop formation is impossible until the bead has wandered closer, invalidating the assumption made in standard HMM. Moreover, the next bead location rt+1 depends not only on the present looping state, but also on the present bead location (if the chosen time step is not much longer than the bead diffusion time). For both of these reasons, we must modify the usual formulation of HMM.
To find the required modification, we first note when no cI protein is present, the bead executes tethered Brownian motion, and this motion is itself a Markov process: The bead's displacement rt+1 depends only on rt, not on earlier positions. We extracted the “unlooped” probability distribution for the next position, Dun(rt+1|rt), from observed time series in a control experiment. Then we found the analogous distribution Dloop(rt+1|rt) for permanently looped tethers. Our two distributions Dun and Dloop were thus determined phenomenologically, with no attempt to model the dynamical details of tethered Brownian motion near a wall. As functions of rt+1, the distributions Dun and Dloop are both roughly two-dimensional Gaussians centered about a point that depends on rt, with widths that reflect the random excursions of Brownian motion in one time step. We checked that simulating Markov processes with these distributions gave good agreement with the control data, both for the probability distributions of the radial distance ρ, and for the autocorrelation functions of x and y, two nontrivial consistency checks on our data and theory.
To incorporate the hidden state dependence, we constructed a heuristic joint distribution function DDHMM(qt+1, rt+1|qt, rt), the probability of observing qt+1, rt+1 given qt, rt, as follows. If the DNA is initially unlooped (qt = 1) and ρt is too large to permit loop formation, then the DNA must remain unlooped in the final state: DDHMM = Dun(rt+1|rt) for qt+1 = 1 and DDHMM = 0 for qt+1 = 2.
However, if qt =1 and ρt is less than the maximum excursion observed for beads with a permanently looped tether (observed in a separate experiment and verified via Monte Carlo simulation (9)), then both final states are allowed, and we take DDHMM = (1−Δt/τLF)Dun(rt+1|rt) for qt+1 = 1 and DDHMM = (Δt/τLF)Dloop(rt+1|rt) for qt+1 = 2. The rate constant 1/τLF is a parameter of the model, the probability per time to form a loop when permitted. A similar construction gives the case when the DNA is initially looped (qt = 2), in terms of a second unknown rate constant 1/τLB for loop breakdown.
We repeated the above calculation for all pairs of data points and summed over all possible sets of the hidden variables qt (10), resulting in a likelihood function
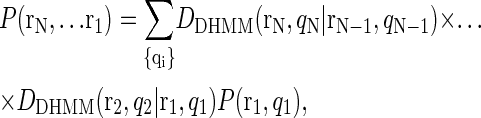 |
with two unknown fit parameters, τLF and τLB, which are the quantities of interest to us. We evaluated the likelihood for various values of the parameters, expressing it as a two-dimensional surface on a logarithmically spaced grid. The resulting surface is smooth, so the peak likelihood can be determined by fitting a two-dimensional quadratic in the neighborhood of the optimum lifetimes, including error bars corresponding to a range of lifetimes enclosing the maximum with 97% confidence. A computer code implementing our algorithm is available as Supplementary Material.
We tested our DHMM by analyzing multiple data subsets obtained by thinning the data, by either a factor of two (Δt = 40 ms) or four (Δt = 80 ms). All computed lifetimes and cross validation of the likelihoods between independent data subsets agreed within uncertainty. To test the algorithm further, we generated a Monte Carlo simulation of the 40-ms looping data, assuming the values of τLB and τLF determined from the experimental data. Then we applied our DHMM method to the simulated data, and checked that it again found the known values and that the event detection corresponded to the time series of the hidden looping transitions (which were known in the simulated data). In contrast to these consistent results, we found that the threshold-crossing method resulted in a lifetime that depends on the filter window size W; see Fig. 3 where, for simplicity, only τLF is shown. (Additional tests and further mathematical details of the method will be discussed elsewhere.)
FIGURE 3.

Lifetime results for the threshold method for DNA loop formation (points) of the measured data shown in Fig. 2, as a function of filter window W. Dwell times between threshold crossings in the unlooped/looped state longer than twice the filter dead time, i.e., 2W, were fit to a histogram to determine τLF and τLB (13). Our DHMM method uses no window; its result is shown as the dashed line. The shaded region corresponds to the error estimate discussed in the text.
We have developed a new method for assessing DNA looping rates from data obtained by the tethered particle method. We tested it on actual and simulated data and determined lifetimes that were independent of sampling frequency. DHMM should improve TPM as a quantitative tool, providing results that are more consistent with improved time resolution compared to the threshold-crossing method.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We thank Rob Phillips for recommending HMM methods for this problem and Seth Blumberg, Lin Han, Randall Kamien, and Liam Paninski for useful discussions. We thank Yale Goldman and the anonymous referee for critical suggestions on the manuscript.
This work was supported in part by the Human Frontier Science Program and National Science Foundation grants No. DGE-0221664, DMR04-25780, and DMR-0404674. L.F. and P.N. acknowledge the hospitality of the Kavli Institute for Theoretical Physics, supported in part by the National Science Foundation under grant No. PHY99-07949.
References
- 1.Ptashne, M. 2004. A Genetic Switch. CSHL Press, New York.
- 2.Schafer, D. A., J. Gelles, M. P. Sheetz, and R. Landick. 1991. Transcription by single molecules of RNA polymerase observed by light microscopy. Nature. 352:444–448. [DOI] [PubMed] [Google Scholar]
- 3.Finzi, L., and J. Gelles. 1995. Measurement of lactose repressor-mediated loop formation and breakdown in single DNA molecules. Science. 267:378–380. [DOI] [PubMed] [Google Scholar]
- 4.Vanzi, F., S. Vladimirov, C. R. Knudsen, Y. E. Goldman, and B. S. Cooperman. 2003. Protein synthesis by single ribosomes. RNA. 9:1174–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van den Broek, B., F. Vanzi, D. Normanno, F. S. Pavone, and G. J. L. Wuite. 2006. Real-time observation of DNA looping dynamics of type IIE restriction enzymes NaeI and NarI. Nucleic Acids Res. 34:167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanzi, F., C. Broggio, L. Sacconi, and F. S. Pavone. 2006. Lac repressor hinge flexibility and DNA looping. Nucleic Acids Res. 34:3409–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zurla, C., A. Franzini, G. Galli, D. D. Dunlap, D. E. A. Lewis, S. Adhya, and L. Finzi. 2006. Novel tethered particle motion analysis of CI protein-mediated DNA looping in the regulation of bacteriophage-λ. J. Phys. Condens. Matter. 18:S225–S234. [Google Scholar]
- 8.Nelson, P. C., C. Zurla, D. Brogioli, J. F. Beausang, L. Finzi, and D. Dunlap. 2006. Tethered particle motion as a diagnostic of DNA tether length. J. Phys. Chem. B. 110:17260–17267. [DOI] [PubMed] [Google Scholar]
- 9.Segall, D. E., P. C. Nelson, and R. Phillips. 2006. Volume-exclusion effects in tethered-particle experiments. Phys. Rev. Lett. 96:088306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rabiner, L. R. 1989. A tutorial on hidden Markov models and selected applications in speech recognition. Proc. IEEE. 77:257–286. [Google Scholar]
- 11.McKinney, S. A., C. Joo, and T. Ha. 2006. Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys. J. 91:1941–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howard, J. 2001. Mechanics of Motor Proteins and the Cytoskeleton. Sinauer, Sunderland, MA.
- 13.Colquhoun, D., and F. J. Sigworth. 1983. Fitting and statistical analysis of single-channel records. In Single-Channel Recording. B. Sakmann, and E. Neher, editors. Plenum, New York. 191–263.