

Abstract
The electrochemical behavior of guanine, guanosine, and guanosine monophosphate (GMP) at redox polymer film modified indium tin oxide electrodes is examined by voltammetry and redox titration. Utilizing the redox polymer-coated electrodes as indicator electrodes, a new method for measuring the oxidation potentials, based on monitoring their catalytic oxidation by different redox polymer coated electrodes at different pH, was proposed in this work. The oxidation potentials of 0.81 V and 1.02 V versus normal hydrogen electrode were determined for guanine and guanosine/GMP under physiological conditions, the lowest oxidation potentials ever reported, to our knowledge.
The first to oxidize the base of DNA is guanine, oxidized either directly or through hole transfer along the DNA π-stack to the radical (1). Its oxidation has been extensively studied in the context of DNA damage, associated with mutation and aging (2,3). The oxidation potentials of guanine and guanosine were measured by pulse radiolysis and by cyclic voltammetry (4,5). Pulse radiolysis, the measurement technique of choice when the redox reaction involves unstable radicals in the presence of an internal reference (6), registered values of the one-electron oxidation potentials of guanine and guanosine, which varied between 0.63 and 0.83 V versus normal hydrogen electrode (NHE) at pH 13 (4,7). The electrochemically measured direct oxidation potentials were ∼0.9 V versus NHE at physiological pH (8). High overpotentials make difficult the accurate direct determination of the oxidation potentials (8). Guanine bases in DNA were also catalytically oxidized by 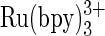 and polyvinylpyridine (PVP)-bound [Ru(bpy)2]2+ (9,10). The Rusling group observed voltammetric responses to the catalytic guanine oxidation in DNA on pyrolytic graphite electrode covered with PVP-
and polyvinylpyridine (PVP)-bound [Ru(bpy)2]2+ (9,10). The Rusling group observed voltammetric responses to the catalytic guanine oxidation in DNA on pyrolytic graphite electrode covered with PVP-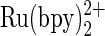 film at 0.99 V versus NHE (10). Thorp et al. measured the oxidation potential of guanine in double helical DNA indirectly, by using trans-[Re(O)2(4-Ome-py)4]+ and related dioxorhenium (V) complexes as mediators, reporting a potential between 1.1 and 1.2 V versus NHE at pH 7 (9).
film at 0.99 V versus NHE (10). Thorp et al. measured the oxidation potential of guanine in double helical DNA indirectly, by using trans-[Re(O)2(4-Ome-py)4]+ and related dioxorhenium (V) complexes as mediators, reporting a potential between 1.1 and 1.2 V versus NHE at pH 7 (9).
Our interest in sensitive and selective electrochemical nucleic acid sensors led us to search for electrocatalysts, lowering the potential at which DNA is electrooxidized: the lower the potential, the better is its detectivity. Previously we reported that guanine is catalytically oxidized already at 0.84 V versus NHE at pH 7.4 by the threading intercalator N,N′-bis[3-propylimidazole]-1,4,5,8-naphthalene diimide complexed with Ru(bpy)2Cl (11), well below the earlier measured potential. Here we report the systematic determination of the apparent oxidation potentials of guanine, guanosine, and guanosine monophosphate (GMP) in aqueous saline solutions, by monitoring their catalytic oxidation currents. At the physiological pH of 7.4, guanine electrooxidation is first observed on a 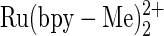 -grafted redox polymer catalyst-modified indium tin oxide (ITO) electrode at 0.83 ± 0.01 V (NHE). Catalyzed guanosine and GMP electrooxidations become observable at 1.03 ± 0.01 V (NHE). They establish that in a pH 7.4 saline aqueous solution, guanine and guanosine are catalytically oxidized at potentials much more reducing than previously reported.
-grafted redox polymer catalyst-modified indium tin oxide (ITO) electrode at 0.83 ± 0.01 V (NHE). Catalyzed guanosine and GMP electrooxidations become observable at 1.03 ± 0.01 V (NHE). They establish that in a pH 7.4 saline aqueous solution, guanine and guanosine are catalytically oxidized at potentials much more reducing than previously reported.
The catalysts we used were redox polymer films with polycationic backbones, varying in their redox potentials, comprising rapidly electron exchanging [Ru(bpy)2Cl]+/2+, where bpy is 2,2′-bipyridine or a subsitituted 2,2′-bipyridine. The backbones, to which the [Ru(bpy)2Cl]+/2+ was coordinatively bound, were PVP or poly(vinylimidazole) (PVI) copolyacrylamide (PAA) (12,13). The redox polymer films were immobilized on ITO-coated glass electrodes. Ruthenium complexes containing polymers were earlier studied for their electron transfer, photosensitization, diode-like behavior, and redox catalysis (14–16). Fig. 1 shows cyclic voltammograms of the redox polymer-coated ITO electrodes, their redox potentials ranging from 0.6 to 1.2 V versus NHE. When guanine was added to their phosphate-buffered saline (PBS) (physiological buffer, 0.14 M NaCl, 20 mM phosphate, pH 7.4) solutions, their reversible voltammograms changed to voltammograms characteristic of irreversible electrocatalytic oxidations. For example, in the case of PVIPAA-Ru(bpy)2Cl, a rise in anodic current and a decrease in cathodic current were observed (Fig. 2, traces a and b), indicative of catalytic guanine electrooxidation, not observed on the bare ITO electrode (17).
FIGURE 1.

Cyclic voltammograms of redox polymer thin film-coated ITO electrodes in PBS. From left to right: PVPPAA-Ru(bpy-OMe)2Cl, PVPPAA-Ru(bpy-Me)2Cl, PVPPAA-Ru(bpy)2Cl, PVIPAA-Ru(bpy-COOMe)2Cl, and PVPPAA-Ru(bpy-COOMe)2Cl.
FIGURE 2.

Cyclic voltammograms of a PVIPAA-Ru(bpy)2Cl thin film-coated ITO electrode in (a) PBS and (b) with guanine added to 20 μM concentration. (c) Cyclic voltammogram of a bare ITO electrode in 50 μM guanine in PBS. Scan rate, 100 mV/s.
The rates of catalytic oxidation of organic compounds, including guanine, are pH dependent. Because the oxidations are proton-releasing, −ΔG, the Gibbs free energy release driving the reactions, increases at higher pH. Mechanistically, the electron transfer in the guanine-Ru(III) complex is proton-coupled, the abstraction of the first guanine electron being concomitant with the deprotonation of guanine (18). We measured the catalytic oxidation currents of guanine and guanosine across the 2–12 pH range in a stirred four-electrode cell, containing a pH electrode, the redox polymer coated working electrode, a reference, and a counter electrode. With the working electrode poised at the formal potential of its redox polymer, we increased the pH stepwise while monitoring the electrooxidation current. The guanine within the redox polymer films was promptly consumed upon applying the formal potential. Fig. 3 shows acid-base titration curves for the electrooxidation of the guanine in the films. Each titration curve reveals a pH threshold and an upper limit, where the electrooxidation rate is no longer pH dependent. The classical S-curves, approximated by straight lines connecting these two points, establish that near the formal potentials of the polymers, guanine electrooxidation involves a proton-generating step. The slopes for polymers 2, 3, 4, and 5 of the closest to neutral pH domain (pH 5–10), are similar, the current increasing 10-fold for a 2-pH unit increase. With each pH unit translating to 59 mV, the behavior is Tafel-like, i.e., a 10-fold current increase is observed upon increasing the potential driving the reaction by 118 mV (19).
FIGURE 3.

Titration curves showing the increase in the electrocatalytic guanine oxidation current when the pH is raised at redox polymer film-coated ITO electrodes. From left to right, 1), PVIPAA-Ru(bpy-COOMe)2Cl; 2), PVPPAA-Ru(bpy)2Cl; 3), PVPPAA-Ru(bpy-Me)2Cl; 4), PVIPAA-Ru(bpy)2Cl; 5), PVPPAA-Ru(bpy-OMe)2Cl; and 6), PVIPAA-Ru(bpy-OMe)2Cl.
The rates of the five steps (reactions 1–5) of the one-electron electrocatalytic oxidation of guanine or guanosine denoted as GH are, by definition, equal at steady state:
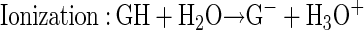 |
(1) |
 |
(2) |
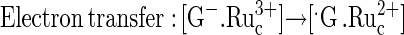 |
(3) |
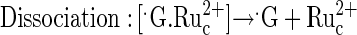 |
(4) |
Bulk electrooxidation of the Ru complex:
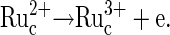 |
(5) |
Reaction 1 explains the pH dependence of the guanine electrooxidation current in Fig. 3, for the redox polymer electrocatalysts of Figs. 1 and 2. The rate, i.e., the current, reaches a plateau at the pH where the rate of formation of the ion pair [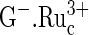 ] no longer depends on the G− concentration, because all the
] no longer depends on the G− concentration, because all the 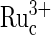 is exhausted. The concentration of
is exhausted. The concentration of 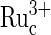 in the film is a function of the rate of electrooxidation of
in the film is a function of the rate of electrooxidation of 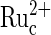 (reaction 5) in the bulk of the film, determined by the redox potential of the
(reaction 5) in the bulk of the film, determined by the redox potential of the 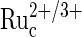 redox couple and by the electron diffusion coefficient in the film, which in turn depends on the rate of collisional electron exchange between the redox centers.
redox couple and by the electron diffusion coefficient in the film, which in turn depends on the rate of collisional electron exchange between the redox centers.
In Fig. 4, the potential at which catalytic oxidation by a particular redox polymer is plotted again the onset pH for the electrooxidation of guanine, guanosine, or GMP. The slopes are found of 60 mV per pH unit. Thus when normalized for pH, all potentials at which the electrooxidations are observed are the same. For example, the 0.81 ± 0.01 V (NHE) value at pH 7.4 for guanine is also obtained when the measured threshold potential is adjusted by 0.059 × [threshold pH − 7.4] V. These threshold potentials are neither reversible potentials nor thermodynamic values, but are practical values (apparent oxidation potential).
FIGURE 4.

pH dependence of the threshold-potentials of (a) guanine (•), (b) guanosine (▪), and (c) GMP (○) electrooxidation, catalyzed by different polymers.
Significantly for biological considerations, the electrooxidation potentials of guanosine are 0.21 ± 0.01 V higher than those of guanine (Fig. 4). Whereas the threshold for the catalytic electrooxidation of guanine at pH 7.4 is 0.81 ± 0.01 V (NHE), that of guanosine is 1.02 ± 0.01 V (NHE). The difference reflects, at least in part, the difference in the energetics of forming the G− anion in reaction 1 by deprotonation the imidazole of guanine versus by deprotonation of guanosine, which does not have an imidazole proton. In GMP and at physiological pH, the anionic proximal phosphate could make, at low ionic strength where the phosphate anion's charge is not screened by Na+ cations, the forming of G− energetically unfavorable and raise the threshold potential of oxidation of G of GMP to above that of guanosine. We found, however, the pH-dependent catalytic electrooxidation currents of GMP are indistinguishable from those of the guanosine. The value of 1.02 V (NHE) oxidation potential measured for guanosine and GMP in physiological buffer solution is considerably lower than the earlier reported 1.29 V (NHE) (20), as is this study's 0.81 V (NHE) oxidation potential for guanine at pH 7.4, which may be compared with the reported value of 1.17 V (NHE) (21).
Acknowledgments
A.H. thanks the Welch Foundation and the U.S. Office of Naval Research and H.X. thanks the Institute of Bioengineering and Nanotechnology for financial support.
References
- 1.Lewis, F. D., R. L. Letsinger, and M. R. Wasielewski. 2001. Dynamics of photoinduced charge transfer and hole transport in synthetic DNA hairpins. Acc. Chem. Res. 34:159–170. [DOI] [PubMed] [Google Scholar]
- 2.Steenken, S. 1989. Purine bases, nucleosides, and nucleotides: aqueous solution redox chemistry and transformation reactions of their radical cations and e− and OH adducts. Chem. Rev. 89:503–520. [Google Scholar]
- 3.Burrows, C. J., and J. G. Muller. 1998. Oxidative nucleobase modifications leading to strand scission. Chem. Rev. 98:1109–1151. [DOI] [PubMed] [Google Scholar]
- 4.Jovanovic, S. V., and M. G. Simic. 1986. One-electron redox potentials of purines and pyrimidines. J. Phys. Chem. 90:974–978. [Google Scholar]
- 5.Subramanian, P., and G. Dryhurst. 1987. Electrochemical oxidation of guanosine formation of some novel guanine oligonucleotides. J. Electroanal. Chem. 224:137–162. [Google Scholar]
- 6.DeFelippis, M. R., C. P. Murthy, M. Faraggi, and M. H. Klapper. 1989. Pulse radiolytic measurement of redox potentials: the tyrosine and tryptophan radicals. Biochemistry. 28:4847–4853. [DOI] [PubMed] [Google Scholar]
- 7.Faraggi, M., F. Broitman, J. B. Trent, and M. H. Klapper. 1996. One-electron oxidation reactions of some purine and pyrimidine bases in aqueous solutions. Electrochemical and pulse radiolysis studies. J. Phys. Chem. 100:14751–14761. [Google Scholar]
- 8.Brabec, V., and G. Dryhurst. 1978. Electrochemical behavior of natural and biosynthetic polynucleotides at the pyrolytic graphite electrode: A new probe for studies of polynucleotide structure and reactions. J. Electroanal. Chem. 89:161–173. [Google Scholar]
- 9.Johnston, D. H., C.-C. Cheng, K. J. Campbell, and H. H. Thorp. 1994. Trans-dioxorhenium (V)-mediated electrocatalytic oxidation of DNA at indium tin-oxide electrodes: voltammetric detection of DNA cleavage in solution. Inorg. Chem. 33:6388–6390. [Google Scholar]
- 10.Mugweru, A., B. Wang, and J. F. Rusling. 2004. Voltammetric sensor for oxidized DNA using ultrathin films of osmium and ruthenium metallopolymers. Anal. Chem. 76:5557–5563. [DOI] [PubMed] [Google Scholar]
- 11.Tansil, N. C., F. Xie, H. Xie, and Z. Gao. 2005. An ultrasensitive nucleic acid biosensor based on the catalytic oxidation of guanine by a novel redox threading intercalator. Chem. Commun. 8:1064–1066. [DOI] [PubMed] [Google Scholar]
- 12.Mabrouk, P. A., and M. S. Wrighton. 1986. Resonance Raman spectroscopy of the lowest excited state of derivatives of tris(2,2′-bipyridine)ruthenium(II): substitute effects on electron localization in mixed-ligand complexes. Inorg. Chem. 25:526–531. [Google Scholar]
- 13.Forster, R. J., and J. G. Vos. 1990. Synthesis, characterization, and properties of a series of osmium- and ruthenium-containing metallopolymer. Macromolecules. 23:4372–4377. [Google Scholar]
- 14.Chidsey, E. D., and R. W. Murray. 1986. Electroactive polymers and macromolecular electronics. Science. 231:25–31. [DOI] [PubMed] [Google Scholar]
- 15.Potts, K. T., and D. A. Usifer. 1988. Polymers and polymer-metal complexes containing pendent 2,2′:6′,2′′-terpyridinyl ligands. Macromolecules. 21:1985–1991. [Google Scholar]
- 16.Oh, S. M., and L. R. Faulkner. 1989. Luminescent and redox probes of structure and dynamics in quaternized poly(4-vinylpyridine) films on electrodes. J. Am. Chem. Soc. 111:5613–5618. [Google Scholar]
- 17.Gore, M. R., V. A. Szalai, P. A. Ropp, I. V. Yang, J. S. Silverman, and H. H. Thorp. 2003. Detection of attomole quantities of DNA targets on gold micro electrodes by electrocatalytic nucleobase oxidation. Anal. Chem. 75:6586–6592. [DOI] [PubMed] [Google Scholar]
- 18.Weatherly, S. C., I. V. Yang, and H. H. Thorp. 2001. Proton-coupled electron transfer in duplex DNA: driving force dependence and isotope effects on electrocatalytic oxidation of guanine. J. Am. Chem. Soc. 123:1236–1237. [DOI] [PubMed] [Google Scholar]
- 19.Bard, A. J., and L. R. Faulkner. 2001. Electrochemical Methods. John Wiley & Sons, New York.
- 20.Steenken, S., and S. Jovanovic. 1997. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc. 119:617–618. [Google Scholar]
- 21.Steenken, S., J. P. Telo, H. M. Novais, and L. P. Candeias. 1992. One-electron-reduction potentials of pyrimidine bases, nucleosides, and nucleotides in aqueous solution. Consequences for DNA redox chemistry. J. Am. Chem. Soc. 114:4701–4709. [Google Scholar]