

Abstract
Nuclear transport requires freely diffusing nuclear transport proteins to facilitate movement of cargo molecules through the nuclear pore. We analyzed dynamic properties of importin α, importin β, Ran and NTF2 in nucleus, cytoplasm and at the nuclear pore of neuroblastoma cells using fluorescence correlation spectroscopy. Mobile components were quantified by global fitting of autocorrelation data from multiple cells. Immobile components were quantified by analysis of photobleaching kinetics. Wild type Ran was compared to various mutant Ran proteins to identify components representing GTP or GDP forms of Ran. Untreated cells were compared to cells treated with nocodazole or latrunculin to identify components associated with cytoskeletal elements. The results indicate that freely diffusing importin α, importin β, Ran and NTF2 are in dynamic equilibrium with larger pools associated with immobile binding partners such as microtubules in the cytoplasm. These findings suggest that formation of freely diffusing nuclear transport intermediates is in competition with binding to immobile partners. Variation in concentrations of freely diffusing nuclear transport intermediates among cells indicates that the nuclear transport system is sufficiently robust to function over a wide range of conditions.
Keywords: importin, Ran, NTF2, fluorescence correlation spectroscopy, nuclear pore complex
INTRODUCTION
Intracellular processes are mediated by systems of diffusing molecules that interact with each other and with immobile partners. Dynamic properties of macromolecules in cytoplasm can be strikingly different from unconstrained diffusion in vitro. Therefore, in order to understand how a particular intracellular system functions, it is necessary to analyze dynamic properties for each component of the system in live cells. Here we analyze dynamic properties of components of the nuclear transport system (importin α, importin β, Ran and NTF2) 1–3 in live neuroblastoma cells using fluorescence correlation spectroscopy (FCS) 4,5.
Molecules enter and leave the nucleus through nuclear pore complexes (NPC). Molecules < 40 kDa can move through the NPC by passive diffusion. Larger molecules require soluble nuclear transport proteins for transport across the NPC. Importin α, importin β, Ran and NTF2 were originally identified as soluble factors necessary to reconstitute nuclear transport activity in permeabilized cell preparations 6. In order to function in nuclear transport each of these proteins must diffuse freely in nucleus and cytoplasm and participate in a series of diffusion-limited interactions, as outlined in Fig. 8. Cargo molecules containing specific nuclear localization sequences (NLS) bind to karyopherins, such as importin β and importin α, to form complexes (importin β::importin α::cargo) 7,8 that are transported into the nucleus through interactions with nucleoporins in the NPC 9. In the nucleus, importin β dissociates from importin α and forms a complex with RanGTP (importin β::RanGTP) 10, while importin α dissociates from cargo and forms a complex with the exportin, CAS and RanGTP (importin α::CAS::RanGTP) 11,12. These newly formed complexes are then transported back to the cytoplasm, where RanGTP is converted to RanGDP causing the complexes to dissociate. In the nucleus Ran is predominantly bound to GTP; in the cytoplasm Ran is predominantly bound to GDP. The nuclear/cytoplasmic RanGTP/RanGDP gradient is established through differential subcellular distributions of specific regulators of Ran. Ran guanine nucleotide exchange factor (RanGEF), RCC1, is localized to the nucleus 13–16, whereas Ran GTPase activating protein (RanGAP) and RanBP1 and 2, which stimulate GTP hydrolysis by Ran, are localized predominantly to the cytoplasm 17–22. RanGDP produced in the cytoplasm is imported into the nucleus by NTF2 23–26, which also interacts with nucleoporins in the NPC 27–29. In the nucleus, RanGDP dissociates from NTF2 and binds to RCC1, which facilitates guanine nucleotide exchange to form RanGTP. Mutations of Ran that affect binding of GTP and/or GDP restrict molecular interactions of the mutant proteins. RanE70A cannot exchange bound GDP for GTP and therefore cannot associate with karyopherins (importins or exportins) 16. RanQ69L cannot hydrolyze GTP and so cannot associate with NTF2 30. RanT24N cannot bind nucleotides and hence cannot associate with either karyopherins or NTF2 31,32. Here dynamic properties of wild type Ran are compared to various mutant Ran proteins to identify components representing GTP or GDP forms of Ran.
Figure 8. Molecular interactions of nuclear transport proteins.
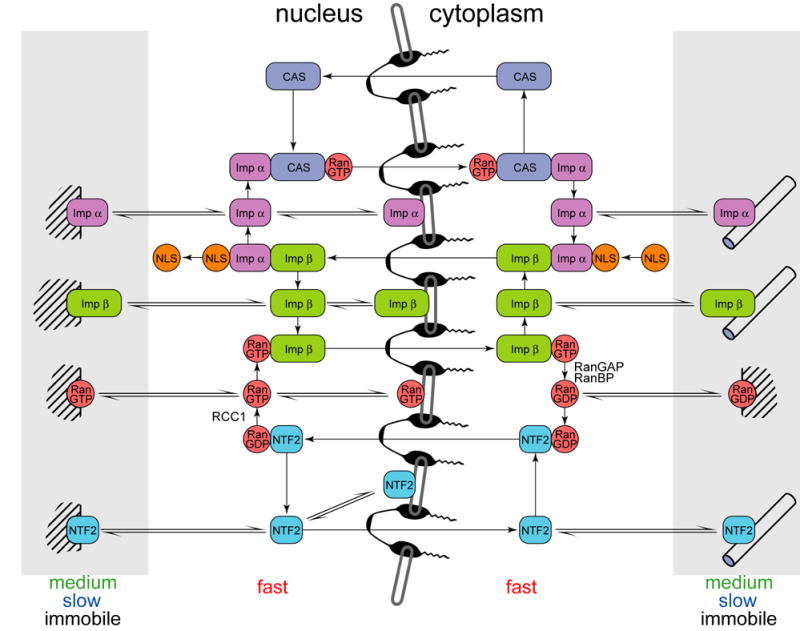
Molecular interactions during nuclear transport are shown for NLS-containing cargo molecule (orange), importin α (light green), importin β (dark green), Ran (red), NTF2 (cyan), CAS (blue), immobile binging partners (cross hatched), microtubules (cylinders) and NPC (black). Freely diffusing molecules are shown in unshaded regions, molecules bound to medium, slow or immobile partners are shown in shaded regions in nucleus and cytoplasm.
Nuclear transport proteins are also involved in other cell biological processes. Importins function as chaperones for highly charged nuclear proteins 33, adapters for motor driven movement along microtubules 34–36, and signal transducers from neurite to nucleus in neurons 37,38. During mitosis, spatial gradients of RanGTP facilitate spindle assembly, centrosome dynamics, nuclear envelope reformation and nuclear pore assembly 39,40. Molecules involved in non-nuclear transport functions may have different dynamic properties than molecules actively engaged in nuclear transport.
The nuclear transport system relies on movement of nuclear transport proteins in cytoplasm, nucleus, and across the nuclear envelope. Diffusion is required for many bimolecular reactions and may become rate-limiting especially if some components of the system have differential localizations. Therefore, intracellular mobilities of the components of the nuclear transport system must be determined for comprehensive understanding of regulation, sensitivity, and rate-limiting steps. Current models of nuclear transport 41–43 assume that the entire pool of nuclear transport proteins is freely diffusing in nucleus and cytoplasm. This assumption may be incorrect because cytoplasm provides target-rich environment for intermolecular interactions. Molecular movement in cytoplasm can be affected by: viscosity, macromolecular crowding, sieving effects of cytoskeleton, barriers presented by intracellular membranes, active transport by molecular motors, and association with partners of various mobilities, affinities, and specificities 44,45. These factors tend to reduce apparent diffusion coefficients 46–48 but can also result in behavior characterized as anomalous diffusion 49. Some factors affect all molecules nonspecifically, whereas others only affect specific molecules. For example, viscosity affects all molecules; sieving depends on molecular size; some immobile binding sites interact with molecules nonspecifically based on charge, while others recognize their partners more specifically. It is important to characterize both specific and nonspecific interactions affecting dynamic properties of each nuclear transport protein.
In this work dynamic properties of nuclear transport proteins were measured in live cells using fluorescence correlation spectroscopy (FCS), which analyzes fluctuations of light emitted by fluorescent particles moving through a small (0.25 fl) observation volume (defined by confocal optics or two photon excitation) positioned either in solution (in vitro) or inside a live cell (in vivo) 50. Fluorescently labeled importin β, importin α, Ran, and NTF2 were microinjected into B104 neuroblastoma cells and analyzed by FCS with the observation volume positioned in the nucleus, cytoplasm, or at the nuclear envelope as illustrated in Fig. 1. The autocorrelation function calculated from the recorded fluorescence fluctuation provides a measure of the number of fluorescent particles (concentration) and the length of time each particle remains inside the observation volume (dynamic properties). Continuous photobleaching during FCS measurements provides a measure of immobile protein 51. Autocorrelation data collected in live cells shows that nuclear transport proteins are involved in multiple interactions that dramatically alter their dynamic properteis. Specific interactions were identified by performing FCS measurements with mutant forms of Ran with reduced molecular interaction potentials and in cells treated with cytoskeletal disrupting agents.
Figure 1. Fluorescence correlation spectroscopy in live cells.
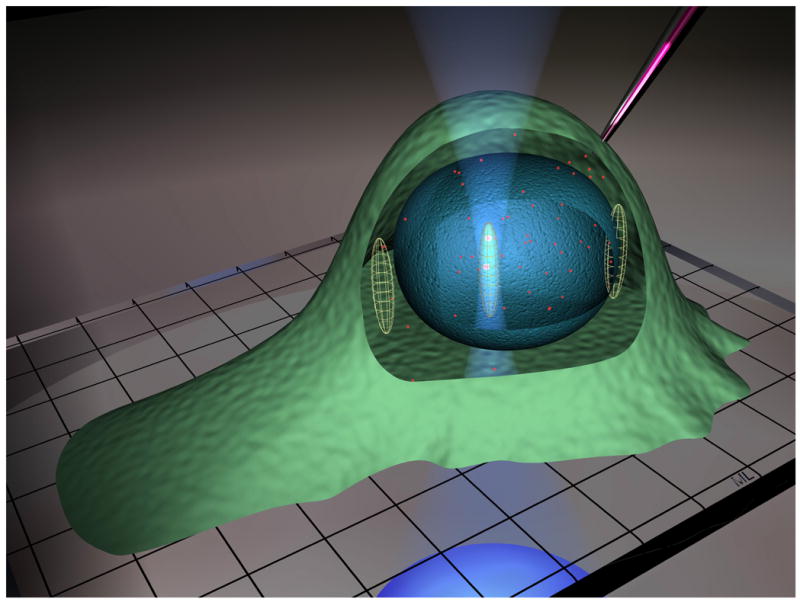
The diagram illustrates fluorescent protein microinjected into a live cell positioned on a microscope cover slip. The inverted objective focuses the laser beam to form the observation volume. . Ellipsoids drawn approximately to scale, indicate the FCS observation volume positioned in cytoplasm, nucleus, or at the nuclear envelope.
RESULTS
FCS analysis of nuclear transport proteins in vitro
In order to understand how intracellular environment affects protein mobility, it was first necessary to analyze dynamics of fluorescently labeled nuclear transport proteins (importin β, importin α, Ran and NTF2) in vitro for comparison (Table I). In each case autocorrelation data could be fitted with a single autocorrelation decay time (τD) corresponding to the diffusion coefficient (D) expected from the size of the monomeric protein, indicating that fluorescently labeled recombinant nuclear transport proteins behave as single molecules and do not form large aggregates in vitro. NTF2 is known to dimerize in vitro (KD ~ 1 μM) 52. However, since FCS measurements were performed at nanomolar concentrations, the proportion of NTF2 dimer at these concentrations is too small to be detected by FCS. Furthermore, a two-fold difference in molecular mass between monomeric and dimeric NTF2 would result in ~ 1.25 fold difference in diffusion coefficient, which is difficult to resolve by FCS.
Table I.
FCS analysis of nuclear transport proteins in vitro
| MW (kD) | τ (μs) | D (μm2/s) | |
|---|---|---|---|
| Importin β | 97 | 460 | 51 |
| Importin α | 58 | 349 | 68 |
| Ran | 23 | 227 | 104 |
| NTF2 | 15 | 218 | 108 |
Since FCS analyzes dynamic properties of fluorescently labeled proteins it was important to compare the properties of labeled and unlabeled proteins to determine if fluorescent labeling affects protein function. To analyze effects of fluorescent labeling on binding specificity importin β, importin α, Ran and NTF2 were differentially labeled with either Alexa Fluor 488 or Alexa Fluor 647 and tested for binding interactions in pairwise combinations, using dual channel cross correlation FCS in vitro 53. The following specific interactions were demonstrated: importin β::importin α, importin β::RanQ69L, NTF2::RanE70A, RanQ69L::RCC1, RanE70A::RCC1, and RanT24N::RCC1 (data not shown). These results indicate that binding specificities of fluorescently tagged proteins are comparable to unlabeled proteins. To determine the effect of fluorescent labeling on nuclear transport function, fluorescent and non-fluorescent importin β, importin α, Ran and NTF2 were tested for their ability to mediate nuclear uptake of fluorescent cargo in a permeabilized cell assay (Figure S1). In the absence of added protein cargo was not taken up into the nucleus. In the presence of either fluorescent or non-fluorescent importin β, importin α, Ran and NTF2, cargo was taken up into the nucleus. This indicates that fluorescently labeling does not interfere with the functions of importin β, importin α, Ran and NTF2 in nuclear transport.
Although fluorescent labeling does not completely inactivate importin β, importin α, Ran or NTF2 it is possible that a fraction of the protein(s) is inactivated. Fluorescent labeling was performed chemically, which means that fluorophores may be conjugated to different residues in different molecules. If fluorophore conjugation to some residues does not affect protein function while conjugation to other residues interferes with protein function a fraction of the labeled protein may be inactive. To test this possibility binding properties of labeled and unlabeled proteins were compared quantitatively using surface plasmon resonance analysis. In this technique one binding partner (ligand) is immobilized on a gold chip and the other partner (analyte) in solution is flowed across the chip in a microfluidic flow cell. If analyte binds to ligand the mass on the chip increases which is detected as a change in refractive index. Association of analyte with ligand over time and dissociation of analyte from ligand when buffer is passed through the flow cell are recorded as a sensorgram, which provides a quantitative measure of on rates and off rates. Comparing sensorgrams for labeled and unlabeled proteins provides a sensitive and quantitative way to determine if a fraction of the protein is inactivated by labeling.
Sensorgrams for different combinations of labeled and unlabeled proteins are shown in Figure S2. Panel A shows sensorgrams for unlabeled (blue) and labeled (red) importin β (analyte) binding to unlabeled importin α (ligand). The amplitudes of the sensorgrams are almost identical in both association and dissociation phases indicating that fluorescent labeling does not inactivate a significant fraction of importin β. The reciprocal experiment (with importin β as ligand) could not be performed because importin β is inactivated when immobilized on the chip. However we were able to repeat the experiment with immobilized labeled importin α as ligand (panel B). The amplitudes of the sensorgrams in panels A (unlabeled importin α as ligand) and B (labeled importin α as ligand) are comparable indicating that fluorescent labeling does not inactivate a significant faction of importin α. Panel C shows sensorgrams for unlabeled (blue) and labeled (red) Ran (analyte) binding to unlabeled NTF2 (ligand). The amplitude of the sensorgram for labeled Ran is slightly reduced compared to unlabeled Ran indicating that a small fraction (<25%) of Ran protein is inactivated by fluorescent labeling. As before, the reciprocal experiment (with Ran as ligand) could not be performed because Ran is inactivated when immobilized on the chip. However, the experiment was repeated with labeled NTF2 as ligand (PanelD). The amplitudes of the sensorgrams in panels C (unlabeled NTF2 as ligand) and D (labeled NTF2 as ligand) are comparable indicating that fluorescent labeling does not incativate a significant fraction of NTF2. These results indicate that conjugation with fluorophore does not significantly affect binding properties of nuclear transport proteins except for Ran, where conjugation with fluorophore interferes with binding to NTF2 in < 25% of labeled molecules. This means that fluorescent importin β, importin α, NTF2 and Ran should exhibit the same dynamic properties in live cells as their unlabeled counterparts.
FCS analysis of nuclear transport proteins in vivo
To measure protein dynamics in vivo, fluorescently tagged nuclear transport proteins were microinjected into the cytoplasm of B104 cells. After injected proteins reached steady state distribution within the cell, FCS measurements were performed with the observation volume positioned in nucleus, cytoplasm, or on the nuclear envelope (Fig. 1). Concentrations and autocorrelation decay times were determined by global fitting of FCS data from many injected cells. Immobile fractions were determined from photobleaching kinetics.
To distinguish effects of specific and nonspecific interactions on protein mobility, importin β, a protein with multiple specific interactions was compared with allophycocyanin (APC), a protein of similar molecular weight (100 kDa), but with no known specific interactions in mammalian cells. Both proteins exhibited normal single component diffusion in vitro with diffusion coefficients of 67 for APC and 70 μm2/s for importin b (Fig 2). To study their dynamics in vivo, the proteins were microinjected into either cell cytoplasm or the nucleus. APC remained in the injection compartment because the protein lacks nuclear localization signals and is too large to pass through the NPC by passive diffusion, whereas importin β equilibrates between nucleus and cytoplasm. FCS autocorrelation data for APC in vivo, in either nucleus or cytoplasm, could be fitted with a single autocorrelation decay time (τD = 1000 μs) corresponding to a slower diffusion coefficient (D = 24 μm2/s) than in vitro. This is consistent with previous observations that diffusion of macromolecules in vivo is non-specifically slowed by viscosity of cytoplasm relative to diffusion in vitro 46,54. We conclude that non-specific factors (viscosity, macromolecular crowding, sieving effects of cytoskeleton, etc.) reduce the diffusion coefficient in vivo but do not necessarily produce apparent multicomponent or anomalous diffusion. The autocorrelation function for importin β had a more complex shape and could be fit with either a multicomponent diffusion model or an anomalous diffusion model. We attribute the complex shape of the measured autocorrelations to specific interactions of nuclear transport proteins with cellular component of different mobilities.
Figure 2. FCS analysis of allophycocyanin and importin β in vitro and in vivo.
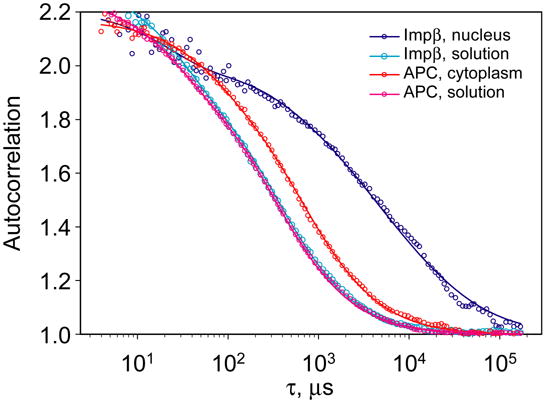
Autocorrelation functions for APC and importin β labeled with Alexa Fluor 647 were measured in vitro and inside live cells and normalized. Autocorrelation data for both APC and importin β in vitro were fitted with a single decay time τ = 300 μs. Autocorrelation data for APC in vivo was fitted with a decay time τ = 1000 μs. Autocorrelation data for importin β in vivo required multiple decay times for an adequate fit.
The in vivo autocorrelation data for nuclear transport proteins carries information about specific interactions of these proteins. Unfortunately, no method for rigorous analysis of such data is presently available. Nuclear transport proteins are known to form complexes that are immobile, have reduced mobilities, or are actively transported. Furthermore, the frequencies of binding/unbinding events, depending on the kinetic rates and concentrations, are broadly distributed. Despite this complexity, it is still possible to extract useful information from the data.
Complex autocorrelation functions can be analyzed by curve fitting with either anomalous or multicomponent diffusion models. One criticism of the latter is that many particles undergo binding/release reactions while passing through the FCS observation volume. Therefore apparent diffusion times cannot be interpreted as diffusion coefficients. On the other hand, multicomponent diffusion models can estimate the percentage of freely diffusing molecules while giving overall information about the dynamic properties of the remaining species.
Compared to the multi-component diffusion model, analysis with anomalous diffusion model is somewhat less informative. Since anomalous diffusion is an apparent phenomenon, a result of heterogeneous interactions of the protein of interest with the intracellular environment, the diffusion coefficient and the anomalous exponent produced by the analysis are only useful as overall descriptors of protein mobility and do not provide insight into specific interactions. Therefore FCS data was analyzed using the multicomponent normal diffusion model with up to three components.
Interpretation of in vivo FCS data is based on the assumption that dynamic properties of injected fluorescent proteins reflect the behavior of corresponding endogenous proteins. Concentrations of endogenous nuclear transport proteins in B104 cells measured by quantitative western blotting were: [importin β] = 9.7, [importin α] = 4.4, [Ran] = 8.2 and [NTF2] = 1.1 μM. By comparison, concentrations of exogenous fluorescent proteins in injected cells, determined from the amplitudes of the FCS autocorrelation functions, varied from cell to cell but were always in the low nanomolar range. Since concentrations of exogenous proteins are much lower than concentrations of corresponding endogenous proteins, exogenous protein is unlikely to affect the overall distribution of the corresponding endogenous protein.
Subcellular distributions of fluorescent proteins microinjected into B104 cells, determined by confocal microscopy (Figure 3), were comparable to distributions of corresponding endogenous proteins, determined by immunofluorescence (data not shown, and reported previously in other cell types 16,30,42,55,56), indicating that exogenous fluorescent proteins and corresponding endogenous proteins have comparable steady-state subcellular distributions. In addition, time-lapse analysis of uptake kinetics in injected cells (data not shown) revealed rapid nuclear uptake for each of the microinjected proteins, indicating that the labeled proteins are recognized by the nuclear transport system. Fuorescent importin β, NTF2, Ran, and Ran Q69L were concentrated in the vicinity of the nuclear envelope, as were the corresponding endogenous proteins, indicating that these proteins have affinity for nucleoporins or other components of the nuclear envelope. Fluorescent importin α, Ran E70A, and Ran T24N were not concentrated in the vicinity of the nuclear envelope indicating that this is not a general phenomenon due to fluorophore conjugation. Ran E70A accumulates in the nucleus because it can bind NTF2 for nuclear import but cannot bind karyopherins for nuclear export. RanQ69L does not accumulate in the nucleus because it cannot bind NTF2 for nuclear import but can bind karyopherins for nuclear export. The reason for nuclear accumulation of RanT24N is not clear. The protein is small enough to equilibrate between nucleus and cytoplasm by passive diffusion through the NPC but since it does not bind to either karyopherins or NTF2 it must bind to some other binding partner(s) concentrated in the nucleus. We conclude that subcellular distributions of microinjected fluorescent proteins and corresponding endogenous proteins are comparable, and that FCS measurements of microinjected fluorescent proteins accurately reflect the dynamic behavior of the corresponding endogenous proteins. Accordingly, relative proportions of components with different autocorrelation decay times (determined for fluorescent proteins by FCS) were used to calculate absolute concentrations of the endogenous counterparts, based on the total concentrations of each protein in the cell (determined by quantitative western blotting). In the case of mutant Ran proteins, relative proportions of different components are expressed as fractions since mutant proteins do not have endogenous counterparts.
Figure 3. Confocal imaging of cells injected with fluorescently labeled nuclear transport proteins.
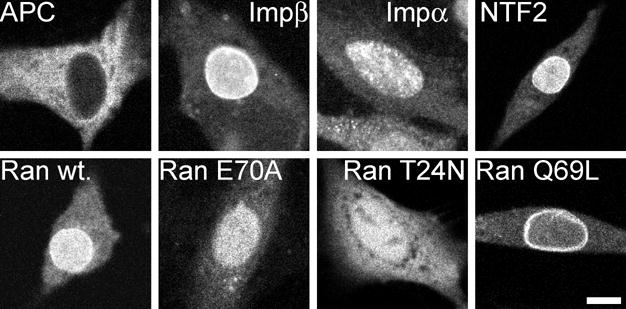
Intrinsically fluorescent APC and Alexa Fluor 647 labeled importin β, importin α, NTF2, Ran, RanE70A, RanT24N and RanQ69L were microinjected into the cytoplasm of B104 cells. After 30–60 minutes the injected cells were imaged by confocal microscopy. Scale bar indicates 10 μm.
Dynamics of nuclear transport proteins in nucleus and cytoplasm
FCS analysis of nuclear transport proteins in the nucleus and cytoplasm is summarized in Figures 4 and 5, respectively. Autocorrelation data for each of the proteins were globally fitted with three autocorrelation decay times: fast (τD ~ hundreds of microseconds), medium (τD ~ milliseconds) and slow (τD ~ tens of milliseconds). Photobleaching data were fitted to single exponential kinetics to obtain the immobile fraction for each protein. The fact that FCS autocorrelation data for a particular protein can be fitted with discrete τD values does not imply that every molecule in the cell has dynamic properties characterized by one of the specific τD values. Each τD value should be considered to represent a range of dynamic behaviors.
Figure 4. FCS analysis of nuclear transport proteins in nucleus.
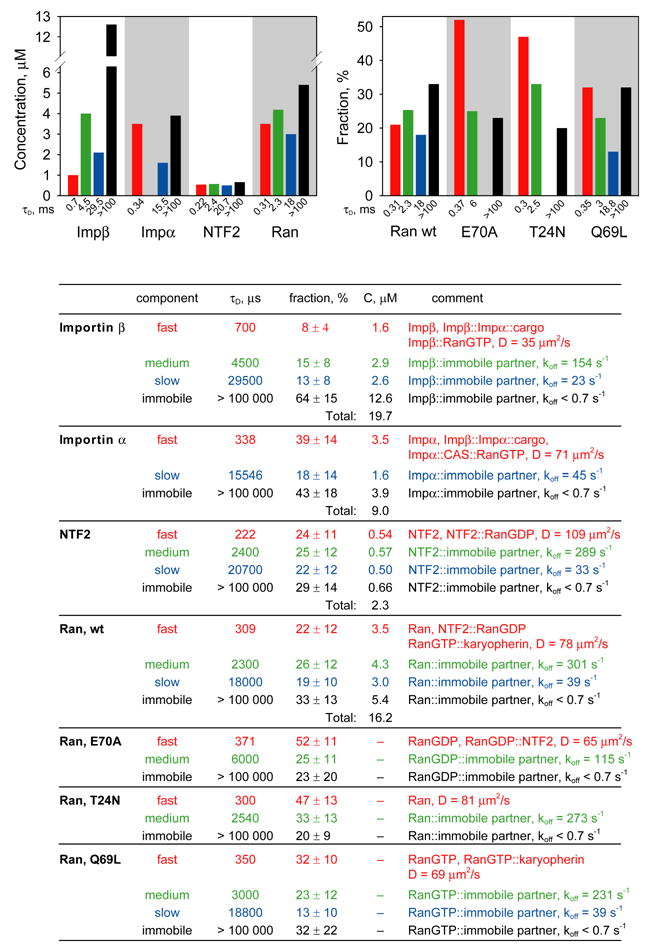
Fluorescently labeled importin β, importin α, NTF2, Ran, RanE70A, RanT24N and RanQ69L labeled with Alexa Fluor 647 were microinjected into cytoplasm of B104 cells. After injected protein achieved steady state nucleocytoplasmic distribution, FCS measurements were performed with confocal volume positioned inside nucleus. For each protein at least 30 cells were analyzed. Autocorrelation data from all cells were globally fitted to resolve protein populations with different mobilities. Immobile fraction for each protein was determined from photobleaching amplitudes. Concentrations for each population were calculated based on total concentration and nucleocytoplasmic distribution of endogenous protein. Concentrations of each component are shown on bar graphs and in the table. Fast, medium, slow and immobile populations are labeled with red, green, blue, and black colors respectively. For mutant Ran proteins, the fraction of each population is shown compared with wt Ran. The table shows autocorrelation decay times (τD), fractions, and concentrations for each population. Molecular species comprising each population and calculated diffusion coefficients or off rates are listed.
Figure 5. FCS analysis of nuclear transport proteins in cytoplasm.
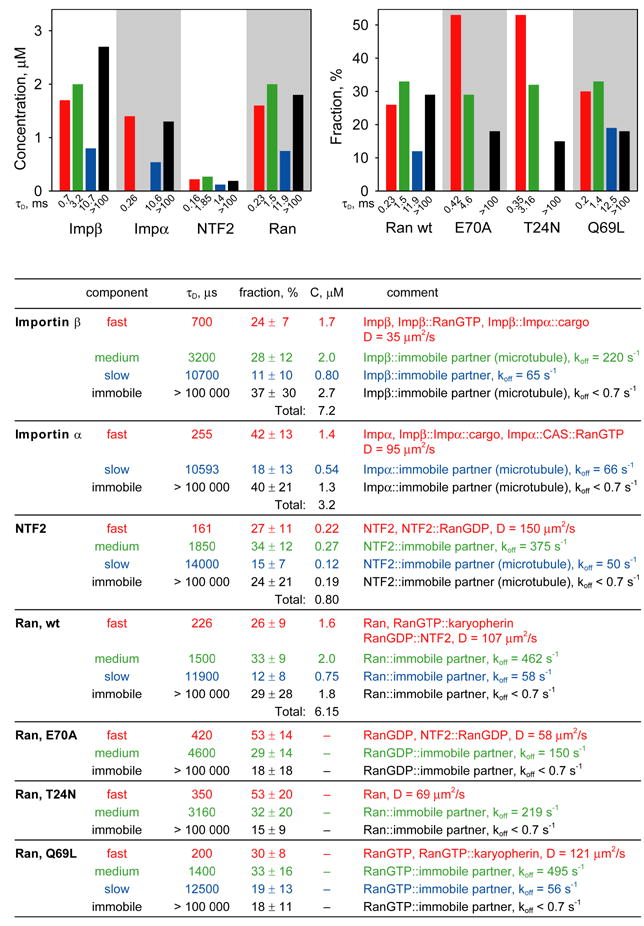
Mobilities of microinjected nuclear transport proteins in cytoplasm were analyzed by FCS. For each protein at least 20 cells were analyzed. Autocorrelation data from all cells were globally fitted to resolve protein populations with different mobilities. Immobile fraction for each protein was determined from photobleaching amplitudes. Concentrations for each population were calculated based on total concentration and nucleocytoplasmic distribution of endogenous protein. Concentrations of each component are shown on bar graphs and in the table. Fast, medium, slow and immobile populations are labeled with red, green, blue, and black colors respectively. For mutant Ran proteins, the fraction of each population is shown compared with wt Ran. The table shows autocorrelation decay times (τD), fractions, and concentrations for each population. Molecular species comprising each population and calculated diffusion coefficients or off rates are listed.
Fast autocorrelation decay times (τD ~ hundreds of microseconds) generally represent freely diffusing protein, including various nuclear transport intermediates. Accordingly, fast component for importin β likely represents monomeric importin β as well as importin β::importin α::cargo and importin β::RanGTP. Fast component for importin α represents monomeric importin α as well as importin β::importin α::cargo and importin α::CAS::RanGTP. Fast component for NTF2 represents monomeric and dimeric NTF2 as well as NTF2::RanGDP. Fast component for Ran represents monomeric Ran as well as RanGDP::NTF2 and RanGTP::karyopherins. Mutant forms of Ran (E70A, T24N and Q69L) exhibited increased proportions of fast component compared to wild type Ran, which is consistent with identification of fast component as freely diffusing protein since mutant Ran proteins have reduced potential for specific interactions with other proteins. These results indicate that effective concentrations of freely diffusing nuclear transport proteins and nuclear transport intermediates represent a relatively small proportion of total concentrations in nucleus and cytoplasm, which implies that the majority of each protein is involved in non-nuclear transport molecular interactions.
Medium and slow components represent non-freely diffusing nuclear transport proteins in nucleus and cytoplasm, which can either correspond to medium and slow rates of diffusion of large complexes in cytoplasm or binding of fluorescent molecules to immobile binding partners in nucleus or cytoplasm, followed by dissociation with medium and slow off rates. Medium and slow autocorrelation decay times (1–30 milliseconds) would correspond to diffusion of relatively large objects (10–100 nm) in diameter) in cytoplasm. If one assumes that each fluorescent molecule binds only once to an immobile partner as it traverses the FCS observation volume, apparent off rates can be calculated from autocorrelation decay times (as shown in Fig. 4 and 5).
In both nucleus and cytoplasm, each nuclear transport protein exhibited significant photobleaching during FCS measurement, indicating that these proteins are bound to immobile (koff < 0.7 sec−1) structures. By this criteria, more than 60% of importin β (19.5 μM) in the nucleus is immobile. Confocal imaging of cells after FCS measurements were taken, revealed a photobleached area corresponding to the FCS observation volume that did not recover up to 45 minutes post-bleach. Furthermore, immobile importin β did not appear until at least 30 minutes after injection (data not shown). These results indicate that the pool of immobile importin β in nucleus equilibrates relatively slowly with the pool of freely diffusing importin β. More than 40% of importin α (5.3 μM) in nucleus is also immobile. In this case the photobleached area in importin α injected cells recovered rapidly and immobile importin α appeared rapidly after microinjection, indicating that immobile importin α in nucleus and cytoplasm equilibrates relatively rapidly with mobile importin α. Immobile fractions of importins α and β in nucleus may reflect binding to immobile nuclear structures. Immobile importins α and β in cytoplasm may reflect binding to immobile cytoskeletal elements such as microtubules (see below). Significant proportions of NTF2 (0.90 μM), and Ran (4.0 μM) were also immobile in the nucleus. In both cases the photobleached area recovered rapidly and the immobile component appeared rapidly after microinjection, indicating that immobile Ran and NTF2 equilibrate relatively rapidly with mobile protein in the nucleus and cytoplasm. Since RanQ69L in nucleus has a higher proportion of immobile component than the other two mutants, it is likely that the immobile fraction of Ran in the nucleus is predominantly Ran GTP.
To test the possibility that medium, slow and immobile components detected by FCS reflect binding of nuclear transport proteins to cytoskeletal elements, cells were treated with either nocodazole (to disrupt microtubules) or latrunculin (to disrupt microfilaments) before injecting fluorescent protein and performing FCS. Disruption of microtubules or microfilaments was confirmed by immunofluorescence with antibody β tubulin or phalloidin, respectively (data not shown). For proteins associated with cytoskeletal elements autocorrelation decay times could represent either rates of motor-driven transport on microtubules or microfilaments or rates of dissociation from the cytoskeleton. Motor driven transport rates would result in very slow autocorrelation times, which cannot be accurately measured because they are obscured by photobleaching effects. Accordingly, autocorrelation decay times affected by nocodazole (or latrunculin) are interpreted as off rates for dissociation from immobile cytoskeletal elements, with the understanding that rates of motor-driven transport may also contribute to slow autocorrelation decay times.
In the nucleus autocorrelation decay times and photobleaching kinetics were not affected by either nocodazole or latrunculin (data not shown) indicating that proteins in the nucleus are not associated with cytoskeletal elements. In cytoplasm, latrunculin did not affect autocorrelation decay times or photobleaching kinetics for importin α, NTF2 or Ran (data not shown), indicating that mobility of these proteins is not affected by microfilaments. Latrunculin did cause a slight increase in the proportion of importin β slow component at expense of medium component (data not shown). Since disruption of microfilaments by latrunculin is expected to increase rather than decrease mobility, this may be an indirect effect of latrunculin. In cytoplasm of nocodazole-treated cells concentrations of slow and immobile components for importin α and NTF2 were reduced relative to untreated cells with concomitant increases in medium components (Figure 6). This indicates that in untreated cells importin α and NTF2 are associated with microtubules in the cytoplasm with slow off rates, and in the absence of microtubules associate with alternative immobile partners with medium off rates. In the case of importin β, nocodazole decreased medium and immobile components with concomitant increase in fast component, indicating that in untreated cells importin β associates with microtubules with medium off rate, and in the absence of microtubules is freely diffusing. In the case of Ran protein, nocodazole decreased fast and medium components with concomitant increase in slow and immobile components. Since nocodazole treatment is expected to increase mobility (as observed for importin α, importin β and NTF2), the effect on Ran protein probably does not indicate direct association with microtubules. Instead it suggests that Ran binds to slow or immobile binding partners that accumulate in nocodazole-treated cells.
Figure 6. FCS analysis of nuclear transport proteins in nocodazole treated cells.
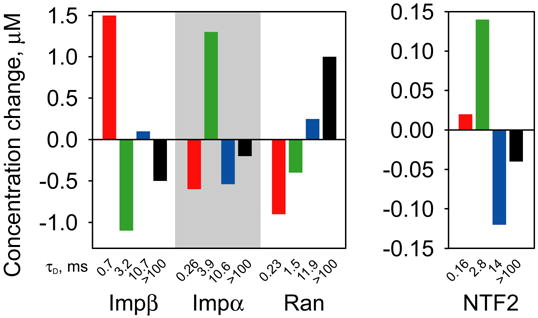
B104 cells were treated with nocodazole to disrupt microtubules followed by microinjection of nuclear transport proteins. Mobilities of each protein were analyzed by FCS with observation volume positioned in the cytoplasm. For each protein at least 20 cells were analyzed. Bar graph indicates relative changes in concentrations of fast (red), medium (green), slow (blue) and immobile (black) components in nocodazole treated cells compared to untreated cells.
Dynamics of nuclear transport proteins at the nuclear envelope
When the FCS observation volume was positioned over the nuclear envelope significant numbers of immobile molecules were detected for each nuclear transport protein (~104 importin β molecules/pore, ~48 importin α molecules/pore, ~43 Ran molecules/pore and ~6 NTF2 molecules/pore) (Figure 7). Immobile molecules could be associated with the inside of the pore, outside of the pore, nuclear envelope adjacent to the pore, or structures near the envelope. Nuclear uptake experiments (not shown) indicate that importin β gradually becomes concentrated in the vicinity of the nuclear envelope over a period of 30–45 minutes after injection, which is consistent with a pool of immobile molecules in the vicinity of the nuclear envelope, in slow equilibrium with nuclear and/or cytoplasmic pools. Other nuclear transport proteins (importin α, NTF2 and Ran) accumulate rapidly around the nuclear envelope, indicating more rapid equilibration with nuclear and/or cytoplasmic pools. Of the different Ran mutants, only Ran Q69L had a significant immobile fraction at the nuclear pore, suggesting that immobile Ran at the NPC is predominantly RanGTP. Immobile molecules of Ran E70A or Ran T24N mutant proteins were not detected at the NPC, consistent with their inability to be converted to RanGTP for export from the nucleus in association with importins or exportins.
Figure 7. FCS analysis of nuclear transport proteins in NPC.
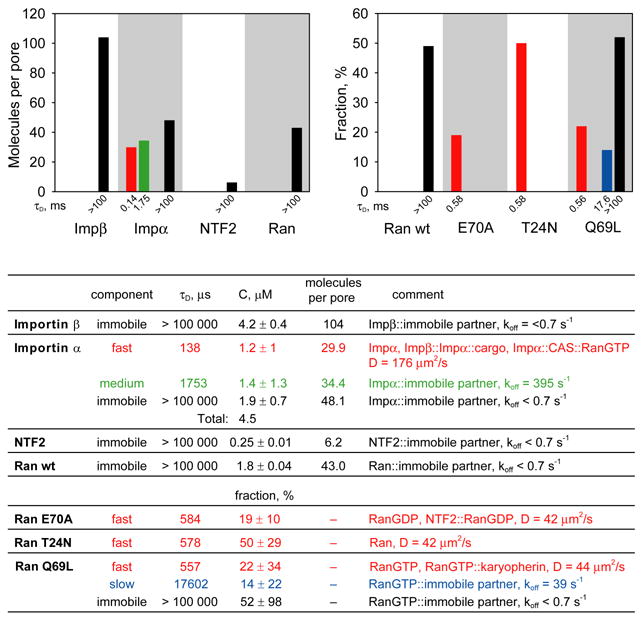
Importin β, importin α, NTF2, Ran wt, RanE70A, RanT24N and RanQ69L labeled with Alexa Fluor 647 were microinjected into the cytoplasm of B104 cells. Cells were incubated for 30–60 minutes after injection and analyzed by FCS with observation volume positioned over the nuclear envelope. For each protein at least 20 cells were analyzed. To resolve protein populations specific to NPC and nuclear envelope, contributions from adjacent nucleoplasm and cytoplasm were subtracted by a global fitting procedure. Number of molecules per NPC for each protein component was calculated based on number of pores encompassed by FCS observation volume. Distributions of each protein between populations with different mobilities are shown on bar graphs and in the table. Fast, medium, slow and immobile populations are labeled with red, green, blue, and black colors respectively. For mutant Ran proteins, the fraction of each component compared with wt Ran is shown. The table shows autocorrelation decay times (τD), concentrations, numbers of molecules per NPC, or fractions for each population. Molecular species comprising each population and calculated diffusion coefficients or off rates are listed.
In the case of importin α, a significant number of NPC-associated molecules had fast (~30 molecules/pore; τD = 138 μs) and medium (~34 molecules/pore; τD = 1.7 ms) autocorrelation decay times. These probably do not represent molecules in transit through the pore because importin β, Ran and NTF2, which also traverse the pore, did not exhibit fast and medium components at the nuclear envelope. Fast and medium components of importin α in the vicinity of the nuclear envelope may reflect molecules that dissociate from cargo and/or CAS protein at the nuclear and cytoplasmic faces of the NPC, respectively, and diffuse freely or bind to immobile partners with medium off rates. Each of the mutant forms of Ran shows accumulation of fast component at the nuclear envelope, which may reflect the inability of these proteins to interact with immobile partners in the vicinity of the NPC. RanQ69L also exhibits a small amount of slow component at the nuclear envelope, which may reflect binding of RanGTP to immobile partners with a slow off rate.
DISCUSSION
FCS analysis combined with quantitative western blotting was used to measure concentrations of fast, medium, slow and immobile components for nuclear transport proteins in nucleus, cytoplasm and NPC of live neuroblastoma cells. The results show that effective concentrations of importin α, importin β, Ran and NTF2 that are freely diffusing and therefore available for nuclear transport functions are much lower than overall concentrations because significant proportions of these proteins bind to microtubules and other immobile partners in cytoplasm and nucleus. As illustrated in Figure 8, importin α, importin β, Ran and NTF2 molecules can either bind to cargo or other nuclear transport protein molecules to form freely diffusing nuclear transport intermediates, or bind to immobile partners, such as microtubules, in which case the proteins are sequestered and unavailable for nuclear transport functions. This is consistent with previous reports that importins can function as adapters for motor-driven transport along microtubules 34–36. According to the model in Figure 8, nuclear transport intermediates and immobile partners (such as microtubules) compete for binding to freely diffusing nuclear transport proteins. This predicts that perturbation in microtubule concentration in the cell may affect nuclear transport. Consistent with the model, there are several reports that nuclear import of various cargoes is stimulated by disrupting microtubules and inhibited by stabilizing microtubules 57–60. The model also predicts competition between nuclear transport intermediates and immobile binding partners in the nucleus, which is more difficult to test since the nature of immobile binding partners in the nucleus is not known. Immobile nuclear structures consist of macromolecules that are imported into the nucleus as cargoes by the nuclear transport system. If such immobilized molecules retain affinity for nuclear transport proteins they may act as immobile binding partners in the nucleus. The differential between total concentration and effective concentration is most pronounced for importin β, where only ~ 5% of the protein in the nucleus is freely diffusing. One recent systems analysis of nuclear transport 43 indicates that excess importin β can inhibit transport by causing depletion of RanGTP through futile cycling. Thus, our finding that effective concentrations of importin β in the nucleus are reduced by binding to immobile partners, may actually result in increased nuclear transport by reducing futile cycling of RanGTP.
The global fitting approach used to determine concentrations of specific dynamic components assumes that autocorrelation decay times are similar in all cells (global parameters), but concentrations of individual components can vary among different cells (local parameters). Local parameters can be affected by variations in constitutive levels of expression of specific nuclear transport proteins among different cells. In addition, nuclear transport activity fluctuates during the cell cycle 61 and nuclear transport proteins are believed to participate in a variety of specific interactions prior to, and during mitosis 39. Therefore, concentrations of individual components may vary during the cell cycle. In fact, preliminary results suggest that variation in local parameters is reduced when FCS measurements are made in synchronized cells (data not shown). In any case, cell-to-cell variation in different components of individual nuclear transport proteins suggests that essential nuclear transport functions are maintained under a range of physiological conditions.
Transit times (~10 ms) for cargo molecules moving through the NPC have been measured by single molecule imaging techniques 62. The fact that FCS measurements at the NPC did not reveal increased amounts of components with autocorrelation decay times comparable to the previously measured NPC transit time suggests that concentrations of nuclear transport protein molecules in transit through the NPC are not sufficiently different from their concentrations in nucleus and cytoplasm surrounding the NPC to detect by FCS. Of the mutant Ran proteins, only RanQ69L exhibited a component with a slow autocorrelation time (τ = 17.6 ms) comparable to the previously measured NPC transit time. Since RanQ69L is unable to hydrolyze GTP it may be unable to dissociate from karyopherins, and thus may traverse the NPC repetitively in association with karyopherins. It is important to remember that τ values determined at the NPC were obtained by fitting autocorrelation data for the NPC with the same global parameters obtained for surrounding nucleus and cytoplasm and then subtracting nuclear and cytoplasmic contributions (see Materials and Methods). In other words, specific τ values determined for components at the NPC were not measured directly but, for the purposes of fitting the data, were assumed to be comparable to global τ values measured in nucleus and cytoplasm. Therefore, unless concentrations of molecules with specific τ values are higher in the NPC than in surrounding nucleus and cytoplasm FCS will not distinguish a pool of molecules in transit through the NPC from pools of molecules with comparable τ values in surrounding nucleoplasm and cytoplasm.
Current models for nuclear transport 41–43 are based on the assumption that the total pool of nuclear transport proteins in nucleus and cytoplasm is freely diffusing and available to participate in molecular interactions of nuclear transport. Our results indicate that effective concentrations of importin α, importin β, Ran and NTF2 available to participate in nuclear transport are much lower than their total concentrations in nucleus and cytoplasm. It is difficult to predict how this will affect sensitivity of nuclear transport to changes in intracellular concentrations of individual proteins. Riddick and Macara 43 have compared effects of changing intracellular concentrations of importin α, importin β, Ran or NTF2 on nuclear transport in vivo (where our results indicate that much of the protein is bound to immobile partners), and in silico (where all protein is assumed to be freely diffusing). Their results indicate that nuclear transport is slightly more sensitive to changes in concentrations of nuclear transport proteins in vivo compared to in silico. It will be important to incorporate effective concentrations of freely diffusing nuclear transport proteins and concentrations bound to immobile partners into a more complete reaction diffusion in silico nuclear transport model to analyze the behavior of the system under actual intracellular conditions.
FCS in live cells produces invaluable information about nuclear transport protein dynamics and interactions by gathering statistics about the fluorescent particles rapidly passing through the observation volume. Recent modifications of FCS provide access to additional information about fluorescent particles that is inaccessible by standard FCS. For example scanning FCS63–65 can capture a directional bias in particle movement, reduce photobleaching, and quantify the dynamics of slowly moving particles. Although in our experience, FCS measurements are reproducible at different positions in the same cellular compartment, scanning FCS could potentially provide a more comprehensive view of nuclear transport protein dynamics throughout the entire cell volume over an extended period of time.
The results presented here demonstrate that FCS can be used to analyze dynamic properties of a system of interacting proteins in live cells. In the case of importin α, importin β, Ran and NTF2, FCS analysis indicates that the majority of molecules in the cell are bound to immobile components and therefore not available for nuclear transport functions, which has important implications for understanding the nuclear transport system. It is likely that other systems of interacting proteins that mediate diffusion limited cellular processes are also affected by intracellular environment in ways that influence their dynamic properties and functions. In such cases FCS can provide quantitative information that is critical to understanding the system.
MATERIALS AND METHODS
Nuclear transport proteins
Recombinant human importin β, importin α, and NTF2 proteins were purchased from Sigma-Aldrich (St. Louis, MO). Recombinant human Ran, Ran E70A, Ran T24N, and Ran Q69L proteins were purchased from Jena Bioscience (Jena, Germany). Allophycocyanin (APC) was purchased from Molecular Probes (Eugene, OR). All proteins except APC, were fluorescently labeled using Alexa Fluor 647® carboxylic acid succinimidyl ester labeling kit (Molecular Probes) according to the company’s protocol. Briefly, the protein solution (1mg/mL) in 0.1M sodium bicarbonate was incubated with dye for 1 hour. Unincorporated dye was removed by gel filtration in PBS using Micro Bio-Spin P6 (for NTF2) or P30 (for all other proteins) spin columns (Bio-Rad Laboratories, Hercules, CA). Fluoresecently labeled proteins were microinjected into the cytoplasm of B104 neuroblastoma cells 66 grown in DMEM/F12 containing 5% fetal calf serum. Microinjections were performed with Eppendorf Micromanipulator 5171 system. Each protein was microinjected into approximately 250 cells.
Laser scanning microscopy
Imaging and FCS measurements were performed with LSM 510 – ConfoCor 2 attached to Axiovert 100M microscope with C-Apochromat 40x, NA 1.2 water immersion objective and He-Ne (633 nm) laser (Carl Zeiss Jena GmbH, Jena, Germany). Fluorescence imaging of injected cells was performed 30 to 60 min. after microinjection, by which time injected protein had attained steady state subcellular distribution. Nuclear uptake kinetics were analyzed by time lapse imaging at 0.7 s/frame. Imaging was initiated several seconds before microinjection and continued for 60 s. Images were analyzed using MetaMorph software (Universal Imaging Center, Downing, PA). Fluorescence intensities in nuclear and cytoplasmic compartments were integrated for each cell as a function of time to determine when the protein reached steady state distribution in the cell.
Fluorescence correlation spectroscopy
For in vitro measurements, FCS data was collected for 3 x 60 s for each sample. Live cell data were collected for 5 x 5 s in the nucleus, nuclear envelope and cytoplasm. Analysis of FCS data was performed using MATLAB with Optimization toolbox (MathWorks, Natick, MA) and gfit 67. A reference solution of Alexa Fluor 647 was used to calibrate the instrument. Horizontal and vertical radii of the ellipsoidal confocal volume were determined to be 0.31 and 1.47 μm, respectively.
Immobile fraction was determined from photobleaching kinetics. Photon count rates for the first few measurements at each location were median filtered and fitted to single exponentials F (t) F0 − Ae−kt, where t is time and k is photobleaching rate, fixed to 0.5 s−1. The fraction of immobile particles was calculated as F0/(F0+A).
Autocorrelation data was analyzed by non-linear least-square curve fitting. Since the accuracy of autocorrelation measurements varies with the delay time τ and also between samples, individual autocorrelation points were statistically weighted by dividing them by their standard deviations 68. The standard deviations were calculated based on multiple autocorrelation traces collected from the same location. Traces were scaled to maximize the overlap, abnormal traces were eliminated, and mean and standard deviation were calculated for each delay time.
Autocorrelation data was fitted to the equation for three dimensional diffusion (Eq. 1).
| Eq. 1 |
where G(τ) is autocorrelation, T is triplet state fraction, τT is triplet state time, N is average number of particles inside the confocal volume, yi is fraction of component i, τDi is diffusion time of component i, and τDi is the ratio between the vertical and horizontal radii for the confocal volume. In addition to diffusion, observed autocorrelation function can reflect non-diffusional binding and transport processes. However, without knowing a priori the specific types of behaviors exhibited by each protein, we cannot define a more suitable equation for fitting the data. Furthermore, since most non-diffusional processes in the cell have time constants that are slower than diffusional processes, they tend to contribute predominantly to longer autocorrelation decay times, which inherently contain a larger amount of error 69,70, and also may be partially obscured by photobleaching effects, which makes it difficult to fit longer autocorrelation times with sufficient accuracy to distinguish diffusional from non-diffusional processes. Therefore, we believe that the equation for three dimensional diffusion (Eq. 1) provides a reasonable fit for in vivo autocorrelation data, with the understanding that components with slower autocorrelation decay times may actually represent non-diffusional processes such as dissociation from immobile binding partners or transport in the cell.
Eq. 1 can describe multiple homologous models with different complexity depending on the number of components. To ensure that curve fitting results are meaningful, it is important to select the model that produces a good fit for the experimental data without being excessively complex. A model with a large number of parameters is considered too complex for a given set of experimental data if the fitting does not result in a unique set of the parameter values. Standard methods for discriminating between different models (e.g. ANOVA, Akaike) cannot be used in FCS since individual points in the measured autocorrelation function are interdependent. Therefore, the number of components in Eq. 1 was chosen to provide a good fit as well as narrow confidence intervals for model parameters 50.
To improve the accuracy of parameter estimation, the autocorrelation data was analyzed globally71. During fitting, a single value for each global parameter (T, τT, τD1, …) was found that produced the optimal fit for all traces. At the same time, an optimal set of local parameters (N , y1,…) was found for each individual trace. The search for the global minimum was performed using random-restart and evolutionary algorithms. Confidence intervals for global parameters were obtained asymptotically and by bootstrapping. Particle numbers for different components of individual fluorescent proteins determined by FCS were used to calculate overall concentrations in different subcellular compartments based on concentrations of the corresponding endogenous proteins measured by quantitative western blotting.
FCS analysis of nuclear transport proteins in the NPC
FCS measurements were performed with the confocal volume positioned at the nuclear envelope. Transection of the confocal volume by the nuclear envelope, which represents a mostly impermeable barrier, could influence apparent diffusion times by restricting movement within the volume. However, computer simulations (MKL, unpublished data) indicate that a vertical reflecting plane transecting the FCS volume does not affect apparent diffusion times, which also follows from symmetry considerations. Since the nuclear radius is greater than the confocal radii, the angular size of intersection is not expected to be greater than 45°. Since the shape of the nuclear envelope intersection within the confocal volume is almost planar, the effect of the barrier is negligible.
The confocal volume positioned at the nuclear envelope encompasses NPCs as well as portions of surrounding nucleo- and cytoplasms. If the nuclear envelope is represented by a thin, non-interacting semi-permeable barrier dividing the confocal volume into halves, the measured FCS signal should represent an average of signals from nucleus and cytoplasm. However, in some cases we observed elevated concentrations of nuclear transport proteins near the nuclear envelope due to additional interactions with nuclear envelope structures. To analyze these interactions, we determined dynamics of the additional protein populations. Data from nucleus and cytoplasm were fitted globally to a set of three τD values. Nuclear envelope data was fit into the theoretical autocorrelation function with the first three components fixed to the average nucleocytoplasmic values. Additional components were added to accommodate excess concentration at the nuclear envelope and to determine its properties. The number of protein molecules per pore was calculated based on NPC density of 16 μm−2
Quantitative western blotting
B104 cells were harvested, counted in a hemacytometer, homogenized and analyzed by SDS PAGE. Nuclear transport proteins were quantitated by western blotting using specific antibodies, HRP-conjugated secondary antibody, and SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL). The average amounts of each protein per cell were determined from band intensities using known amounts of purified nuclear transport proteins for calibration. Average cell volume was assumed to be 10 pL. Relative nuclear and cytoplasmic concentrations for each protein were determined from immunofluorescent images assuming 1:9 nuclearcytoplasmic volume ratio.
Surface plasmon resonance analysis
Surface plasmon resonance (SPR) measurements were performed using the Biacore T100. Ligands (importin-α, NTF2, and their Alexa Fluor 647 labeled derivatives) were immobilized onto a carboxymethyl dextran sensor chip using the NHS/EDC coupling method provided by the Biacore manufacturer. The amount of ligand immobilized was 3000 RUs (Importin-α) or 1100 RUs (NTF2). Analytes (importin-β Ran and their Alexa Fluor 647 labeled derivatives) prepared at various concentrations in HBS buffer (10 mM Hepes, pH 7.4, containing 3 mM EDTA, 0.15 M NaCl, and 0.005% Surfactant P 20) and were injected over the sensor surface at a flow rate of 30 μl/min for 180 s. Dissociation was monitored in HBS buffer for 150 s at the same flow rate. The surface was regenerated between injections using 10 mM NaOH.
When importin-β was covalently linked to a carboxymethyl dextran sensor chip via amine coupling the resulting surface gave no specific signal upon importin-α injection, as previously reported 72. Likewise, immobilized RAN gave no specific signal upon NTF2 injection. This suggested that importin-β and Ran are not immobilized in an active orientation when amine coupling is used.
Nuclear Import Assay
The nuclear import assay was performed as described by Adam et al. 6 with some modifications. Briefly, B104 cells were grown on 8mm glass coverslips for 24 to 48 h to a subconfluent density. Cells were washed twice in transport buffer [20 mM Hepes•KOH, pH 7.3, 110 mM KOAc, 2 mM Mg(OAc)2, 1 mM EGTA, 2 mM DTT] and then treated with digitonin (70 μg/ml) for 6 min on ice. After permeabilization, cells were washed twice with transport buffer and overlayed with 50 μl import reaction consisting of transport buffer containing 1 μM fluorescent import ligand (Sulforhodamine B labeled BSA-NLS, Sigma I9906), an ATP-regenerating system [1 mM ATP/5 mM creatine phosphate/creatine kinase (10 units/ml)], 2 mg/ml BSA, 1 mM GTP, and protein mixture consisting of either all unlabelled or all Alexa Fluor 647 labeled importin-α, importin-β Ran, and NTF2 at 1 μM final concentration of each. The control assay excluded the protein mixture. The import assay was carried out in the permeabilized cells at room temperature for 3 h. Cells were washed twice in transport buffer and imaged with a Zeiss LSM confocal microscope equipped with 561 nm laser and 40x 1.2 N.A. objective.
Supplementary Material
Acknowledgments
This work was supported by NIH grants NS15190, RR13186 and RR22232 to JHC. Initial FCS experiments on Ran protein in B104 cells were performed by students (Nancy Skoura, Cheryl Habrukowich, Matt Buckwalter) in the Biochemistry II course at UCHC (Farmington, CT).
ABBREVIATIONS
- ANOVA
(analysis of variance)
- APC
(allophycocyanin)
- CAS
(cellular apoptosis susceptibility gene)
- FCS
(fluorescence correlation spectroscopy)
- NLS
(nuclear localization signal)
- NPC
(nuclear pore complex)
- NTF2
(nuclear transport factor 2)
- Ran
(Ras-related nuclear protein)
- RanGEF
(Ran guanine nucleotide exchange factor)
- RanGAP
(Ran GTPase activating protein)
- RanBP
(Ran binding protein)
- RCC1
(regulator of chromosome condensation)
- SPR
(surface plasmon resonance)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dasso M. The Ran GTPase: theme and variations. Curr Biol. 2002;12:R502–8. doi: 10.1016/s0960-9822(02)00970-3. [DOI] [PubMed] [Google Scholar]
- 2.Quimby BB, Dasso M. The small GTPase Ran: interpreting the signs. Curr Opin Cell Biol. 2003;15:338–44. doi: 10.1016/s0955-0674(03)00046-2. [DOI] [PubMed] [Google Scholar]
- 3.Stochaj U, Rother KL. Nucleocytoplasmic trafficking of proteins: with or without Ran? Bioessays. 1999;21:579–589. [Google Scholar]
- 4.Berland KM, So PT, Gratton E. Two-photon fluorescence correlation spectroscopy: method and application to the intracellular environment. Biophys J. 1995;68:694–701. doi: 10.1016/S0006-3495(95)80230-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Magde D, Elson E, Webb WW. Thermodynamic Fluctuations in a Reacting System---Measurement by Fluorescence Correlation Spectroscopy. Physical Review Letters. 1972;29:705–708. [Google Scholar]
- 6.Adam SA, Sterne-Marr R, Gerace L. Nuclear protein import using digitonin-permeabilized cells. Methods Enzymol. 1992;219:97–110. doi: 10.1016/0076-6879(92)19013-v. [DOI] [PubMed] [Google Scholar]
- 7.Cingolani G, Petosa C, Weis K, Muller CW. Structure of importin-beta bound to the IBB domain of importin-alpha. Nature. 1999;399:221–9. doi: 10.1038/20367. [DOI] [PubMed] [Google Scholar]
- 8.Gorlich D, Henklein P, Laskey RA, Hartmann E. A 41 amino acid motif in importin-alpha confers binding to importin-beta and hence transit into the nucleus. Embo J. 1996;15:1810–7. [PMC free article] [PubMed] [Google Scholar]
- 9.Bayliss R, Littlewood T, Stewart M. Structural basis for the interaction between FxFG nucleoporin repeats and importin-beta in nuclear trafficking. Cell. 2000;102:99–108. doi: 10.1016/s0092-8674(00)00014-3. [DOI] [PubMed] [Google Scholar]
- 10.Fradin C, Zbaida D, Elbaum M. Dissociation of nuclear import cargo complexes by the protein Ran: a fluorescence correlation spectroscopy study. C R Biol. 2005;328:1073–82. doi: 10.1016/j.crvi.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Floer M, Blobel G. The nuclear transport factor karyopherin beta binds stoichiometrically to Ran-GTP and inhibits the Ran GTPase activating protein. J Biol Chem. 1996;271:5313–6. doi: 10.1074/jbc.271.10.5313. [DOI] [PubMed] [Google Scholar]
- 12.Rexach M, Blobel G. Protein import into nuclei: association and dissociation reactions involving transport substrate, transport factors, and nucleoporins. Cell. 1995;83:683–92. doi: 10.1016/0092-8674(95)90181-7. [DOI] [PubMed] [Google Scholar]
- 13.Bischoff FR, Ponstingl H. Catalysis of guanine nucleotide exchange of Ran by RCC1 and stimulation of hydrolysis of Ran-bound GTP by Ran-GAP1. Methods Enzymol. 1995;257:135–44. doi: 10.1016/s0076-6879(95)57019-5. [DOI] [PubMed] [Google Scholar]
- 14.Nemergut ME, Mizzen CA, Stukenberg T, Allis CD, Macara IG. Chromatin docking and exchange activity enhancement of RCC1 by histones H2A and H2B. Science. 2001;292:1540–3. doi: 10.1126/science.292.5521.1540. [DOI] [PubMed] [Google Scholar]
- 15.Ohtsubo M, Okazaki H, Nishimoto T. The RCC1 protein, a regulator for the onset of chromosome condensation locates in the nucleus and binds to DNA. J Cell Biol. 1989;109:1389–97. doi: 10.1083/jcb.109.4.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renault L, Kuhlmann J, Henkel A, Wittinghofer A. Structural basis for guanine nucleotide exchange on Ran by the regulator of chromosome condensation (RCC1) Cell. 2001;105:245–55. doi: 10.1016/s0092-8674(01)00315-4. [DOI] [PubMed] [Google Scholar]
- 17.Bischoff FR, Krebber H, Kempf T, Hermes I, Ponstingl H. Human RanGTPase-activating protein RanGAP1 is a homologue of yeast Rna1p involved in mRNA processing and transport. Proc Natl Acad Sci U S A. 1995;92:1749–53. doi: 10.1073/pnas.92.5.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahajan R, Delphin C, Guan T, Gerace L, Melchior F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell. 1997;88:97–107. doi: 10.1016/s0092-8674(00)81862-0. [DOI] [PubMed] [Google Scholar]
- 19.Matunis MJ, Coutavas E, Blobel G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J Cell Biol. 1996;135:1457–70. doi: 10.1083/jcb.135.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bischoff FR, Gorlich D. RanBP1 is crucial for the release of RanGTP from importin beta-related nuclear transport factors. FEBS Lett. 1997;419:249–54. doi: 10.1016/s0014-5793(97)01467-1. [DOI] [PubMed] [Google Scholar]
- 21.Floer M, Blobel G, Rexach M. Disassembly of RanGTP-karyopherin beta complex, an intermediate in nuclear protein import. J Biol Chem. 1997;272:19538–46. doi: 10.1074/jbc.272.31.19538. [DOI] [PubMed] [Google Scholar]
- 22.Kehlenbach RH, Assheuer R, Kehlenbach A, Becker J, Gerace L. Stimulation of nuclear export and inhibition of nuclear import by a Ran mutant deficient in binding to Ran-binding protein 1. J Biol Chem. 2001;276:14524–31. doi: 10.1074/jbc.M011087200. [DOI] [PubMed] [Google Scholar]
- 23.Quimby BB, Lamitina T, L’Hernault SW, Corbett AH. The mechanism of ran import into the nucleus by nuclear transport factor 2. J Biol Chem. 2000;275:28575–82. doi: 10.1074/jbc.M005055200. [DOI] [PubMed] [Google Scholar]
- 24.Ribbeck K, Lipowsky G, Kent HM, Stewart M, Gorlich D. NTF2 mediates nuclear import of Ran. Embo Journal. 1998;17:6587–6598. doi: 10.1093/emboj/17.22.6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith A, Brownawell A, Macara IG. Nuclear import of Ran is mediated by the transport factor NTF2. Curr Biol. 1998;8:1403–6. doi: 10.1016/s0960-9822(98)00023-2. [DOI] [PubMed] [Google Scholar]
- 26.Stewart M, Kent HM, McCoy AJ. The structure of the Q69L mutant of GDP-Ran shows a major conformational change in the switch II loop that accounts for its failure to bind nuclear transport factor 2 (NTF2) J Mol Biol. 1998;284:1517–27. doi: 10.1006/jmbi.1998.2204. [DOI] [PubMed] [Google Scholar]
- 27.Clarkson WD, Kent HM, Stewart M. Separate binding sites on nuclear transport factor 2 (NTF2) for GDP-Ran and the phenylalanine-rich repeat regions of nucleoporins p62 and Nsp1p. J Mol Biol. 1996;263:517–24. doi: 10.1006/jmbi.1996.0594. [DOI] [PubMed] [Google Scholar]
- 28.Hu T, Guan T, Gerace L. Molecular and functional characterization of the p62 complex, an assembly of nuclear pore complex glycoproteins. J Cell Biol. 1996;134:589–601. doi: 10.1083/jcb.134.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nehrbass U, Blobel G. Role of the nuclear transport factor p10 in nuclear import. Science. 1996;272:120–2. doi: 10.1126/science.272.5258.120. [DOI] [PubMed] [Google Scholar]
- 30.Lounsbury KM, Richards SA, Carey KL, Macara IG. Mutations within the Ran/TC4 GTPase. Effects on regulatory factor interactions and subcellular localization. J Biol Chem. 1996;271:32834–41. doi: 10.1074/jbc.271.51.32834. [DOI] [PubMed] [Google Scholar]
- 31.Klebe C, Bischoff FR, Ponstingl H, Wittinghofer A. Interaction of the nuclear GTP-binding protein Ran with its regulatory proteins RCC1 and RanGAP1. Biochemistry. 1995;34:639–47. doi: 10.1021/bi00002a031. [DOI] [PubMed] [Google Scholar]
- 32.Stewart M, Kent HM, McCoy AJ. Structural basis for molecular recognition between nuclear transport factor 2 (NTF2) and the GDP-bound form of the Ras-family GTPase Ran. J Mol Biol. 1998;277:635–46. doi: 10.1006/jmbi.1997.1602. [DOI] [PubMed] [Google Scholar]
- 33.Jakel S, Mingot JM, Schwarzmaier P, Hartmann E, Gorlich D. Importins fulfil a dual function as nuclear import receptors and cytoplasmic chaperones for exposed basic domains. Embo J. 2002;21:377–86. doi: 10.1093/emboj/21.3.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lam MH, Thomas RJ, Loveland KL, Schilders S, Gu M, Martin TJ, Gillespie MT, Jans DA. Nuclear transport of parathyroid hormone (PTH)-related protein is dependent on microtubules. Mol Endocrinol. 2002;16:390–401. doi: 10.1210/mend.16.2.0775. [DOI] [PubMed] [Google Scholar]
- 35.Giannakakou P, Sackett DL, Ward Y, Webster KR, Blagosklonny MV, Fojo T. p53 is associated with cellular microtubules and is transported to the nucleus by dynein. Nat Cell Biol. 2000;2:709–17. doi: 10.1038/35036335. [DOI] [PubMed] [Google Scholar]
- 36.Smith HM, Raikhel NV. Nuclear localization signal receptor importin alpha associates with the cytoskeleton. Plant Cell. 1998;10:1791–9. doi: 10.1105/tpc.10.11.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanz S, Perlson E, Willis D, Zheng JQ, Massarwa R, Huerta JJ, Koltzenburg M, Kohler M, van-Minnen J, Twiss JL, Fainzilber M. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron. 2003;40:1095–104. doi: 10.1016/s0896-6273(03)00770-0. [DOI] [PubMed] [Google Scholar]
- 38.Thompson KR, Otis KO, Chen DY, Zhao Y, O’Dell TJ, Martin KC. Synapse to nucleus signaling during long-term synaptic plasticity; a role for the classical active nuclear import pathway. Neuron. 2004;44:997–1009. doi: 10.1016/j.neuron.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 39.Harel A, Forbes DJ. Importin beta: conducting a much larger cellular symphony. Mol Cell. 2004;16:319–30. doi: 10.1016/j.molcel.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 40.Caudron M, Bunt G, Bastiaens P, Karsenti E. Spatial coordination of spindle assembly by chromosome-mediated signaling gradients. Science. 2005;309:1373–6. doi: 10.1126/science.1115964. [DOI] [PubMed] [Google Scholar]
- 41.Gorlich D, Seewald MJ, Ribbeck K. Characterization of Ran-driven cargo transport and the RanGTPase system by kinetic measurements and computer simulation. Embo J. 2003;22:1088–100. doi: 10.1093/emboj/cdg113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith AE, Slepchenko BM, Schaff JC, Loew LM, Macara IG. Systems analysis of Ran transport. Science. 2002;295:488–91. doi: 10.1126/science.1064732. [DOI] [PubMed] [Google Scholar]
- 43.Riddick G, Macara IG. A systems analysis of importin-{alpha}-{beta} mediated nuclear protein import. J Cell Biol. 2005;168:1027–38. doi: 10.1083/jcb.200409024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schnell S, Turner TE. Reaction kinetics in intracellular environments with macromolecular crowding: simulations and rate laws. Prog Biophys Mol Biol. 2004;85:235–60. doi: 10.1016/j.pbiomolbio.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 45.Savageau MA. Development of fractal kinetic theory for enzyme-catalysed reactions and implications for the design of biochemical pathways. Biosystems. 1998;47:9–36. doi: 10.1016/s0303-2647(98)00020-3. [DOI] [PubMed] [Google Scholar]
- 46.Verkman AS. Solute and macromolecule diffusion in cellular aqueous compartments. Trends Biochem Sci. 2002;27:27–33. doi: 10.1016/s0968-0004(01)02003-5. [DOI] [PubMed] [Google Scholar]
- 47.Dauty E, Verkman AS. Molecular crowding reduces to a similar extent the diffusion of small solutes and macromolecules: measurement by fluorescence correlation spectroscopy. J Mol Recognit. 2004;17:441–7. doi: 10.1002/jmr.709. [DOI] [PubMed] [Google Scholar]
- 48.Dauty E, Verkman AS. Actin cytoskeleton as the principal determinant of size-dependent DNA mobility in cytoplasm: a new barrier for non-viral gene delivery. J Biol Chem. 2005;280:7823–8. doi: 10.1074/jbc.M412374200. [DOI] [PubMed] [Google Scholar]
- 49.Banks DS, Fradin C. Anomalous diffusion of proteins due to molecular crowding. Biophys J. 2005;89:2960–71. doi: 10.1529/biophysj.104.051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levin MK, Carson JH. Fluorescence correlation spectroscopy and quantitative cell biology. Differentiation. 2004;72:1–10. doi: 10.1111/j.1432-0436.2004.07201002.x. [DOI] [PubMed] [Google Scholar]
- 51.Wachsmuth M, Weidemann T, Muller G, Hoffmann-Rohrer UW, Knoch TA, Waldeck W, Langowski J. Analyzing intracellular binding and diffusion with continuous fluorescence photobleaching. Biophys J. 2003;84:3353–63. doi: 10.1016/S0006-3495(03)70059-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chaillan-Huntington C, Butler PJ, Huntington JA, Akin D, Feldherr C, Stewart M. NTF2 monomer-dimer equilibrium. J Mol Biol. 2001;314:465–77. doi: 10.1006/jmbi.2001.5136. [DOI] [PubMed] [Google Scholar]
- 53.Schwille P, Meyer-Almes FJ, Rigler R. Dual-color fluorescence cross-correlation spectroscopy for multicomponent diffusional analysis in solution. Biophys J. 1997;72:1878–86. doi: 10.1016/S0006-3495(97)78833-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luby-Phelps K. Cytoarchitecture and physical properties of cytoplasm: volume, viscosity, diffusion, intracellular surface area. Int Rev Cytol. 2000;192:189–221. doi: 10.1016/s0074-7696(08)60527-6. [DOI] [PubMed] [Google Scholar]
- 55.Kodiha M, Chu A, Matusiewicz N, Stochaj U. Multiple mechanisms promote the inhibition of classical nuclear import upon exposure to severe oxidative stress. Cell Death Differ. 2004;11:862–74. doi: 10.1038/sj.cdd.4401432. [DOI] [PubMed] [Google Scholar]
- 56.Steggerda SM, Black BE, Paschal BM. Monoclonal antibodies to NTF2 inhibit nuclear protein import by preventing nuclear translocation of the GTPase Ran. Mol Biol Cell. 2000;11:703–19. doi: 10.1091/mbc.11.2.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pockwinse SM, Rajgopal A, Young DW, Mujeeb KA, Nickerson J, Javed A, Redick S, Lian JB, van Wijnen AJ, Stein JL, Stein GS, Doxsey SJ. Microtubule-dependent nuclear-cytoplasmic shuttling of Runx2. J Cell Physiol. 2006;206:354–62. doi: 10.1002/jcp.20469. [DOI] [PubMed] [Google Scholar]
- 58.Gleason EL, Hogan JC, Stephens JM. Stabilization, not polymerization, of microtubules inhibits the nuclear translocation of STATs in adipocytes. Biochem Biophys Res Commun. 2004;325:716–8. doi: 10.1016/j.bbrc.2004.10.081. [DOI] [PubMed] [Google Scholar]
- 59.Trushina E, Heldebrant MP, Perez-Terzic CM, Bortolon R, Kovtun IV, Badger JD, 2nd, Terzic A, Estevez A, Windebank AJ, Dyer RB, Yao J, McMurray CT. Microtubule destabilization and nuclear entry are sequential steps leading to toxicity in Huntington’s disease. Proc Natl Acad Sci U S A. 2003;100:12171–6. doi: 10.1073/pnas.2034961100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brumwell C, Antolik C, Carson JH, Barbarese E. Intracellular trafficking of hnRNP A2 in oligodendrocytes. Exp Cell Res. 2002;279:310–20. doi: 10.1006/excr.2002.5604. [DOI] [PubMed] [Google Scholar]
- 61.Makhnevych T, Lusk CP, Anderson AM, Aitchison JD, Wozniak RW. Cell cycle regulated transport controlled by alterations in the nuclear pore complex. Cell. 2003;115:813–23. doi: 10.1016/s0092-8674(03)00986-3. [DOI] [PubMed] [Google Scholar]
- 62.Kubitscheck U, Grunwald D, Hoekstra A, Rohleder D, Kues T, Siebrasse JP, Peters R. Nuclear transport of single molecules: dwell times at the nuclear pore complex. J Cell Biol. 2005;168:233–43. doi: 10.1083/jcb.200411005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Digman MA, Brown CM, Sengupta P, Wiseman PW, Horwitz AR, Gratton E. Measuring fast dynamics in solutions and cells with a laser scanning microscope. Biophys J. 2005;89:1317–27. doi: 10.1529/biophysj.105.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Skinner JP, Chen Y, Muller JD. Position-sensitive scanning fluorescence correlation spectroscopy. Biophysical Journal. 2005;89:1288–1301. doi: 10.1529/biophysj.105.060749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jureller JE, Kim HY, Scherer NF. Stochastic scanning multiphoton multifocal microscopy. Optics Express. 2006;14:3406–3414. doi: 10.1364/oe.14.003406. [DOI] [PubMed] [Google Scholar]
- 66.Schubert D, Heinemann S, Carlisle W, Tarikas H, Kimes B, Patrick J, Steinbach JH, Culp W, Brandt BL. Clonal cell lines from the rat central nervous system. Nature. 1974;249:224–7. doi: 10.1038/249224a0. [DOI] [PubMed] [Google Scholar]
- 67.gfit, open-source software for global analysis of experimental dataavailable from the authors. and http://gfit.sf.net.
- 68.Wohland T, Rigler R, Vogel H. The standard deviation in fluorescence correlation spectroscopy. Biophys J. 2001;80:2987–99. doi: 10.1016/S0006-3495(01)76264-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Koppel DE. Statistical accuracy in fluorescence correlation spectroscopy. Physical Review A (General Physics) 1974;10:1938–1945. [Google Scholar]
- 70.Saffarian S, Elson EL. Statistical analysis of fluorescence correlation spectroscopy: the standard deviation and bias. Biophys J. 2003;84:2030–42. doi: 10.1016/S0006-3495(03)75011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Skakun VV, Hink MA, Digris AV, Engel R, Novikov EG, Apanasovich VV, Visser AJ. Global analysis of fluorescence fluctuation data. Eur Biophys J. 2005;34:323–34. doi: 10.1007/s00249-004-0453-9. [DOI] [PubMed] [Google Scholar]
- 72.Catimel B, Teh T, Fontes MR, Jennings IG, Jans DA, Howlett GJ, Nice EC, Kobe B. Biophysical characterization of interactions involving importin-alpha during nuclear import. J Biol Chem. 2001;276:34189–98. doi: 10.1074/jbc.M103531200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.