

Abstract
Histone proteins play essential structural and functional roles in the transition between active and inactive chromatin states. Although histones have a high degree of conservation due to constraints to maintain the overall structure of the nucleosomal octameric core, variants have evolved to assume diverse roles in gene regulation and epigenetic silencing. Histone variants, post-translational modifications and interactions with chromatin remodeling complexes influence DMA replication, transcription, repair and recombination. The authors review recent findings on the structure of chromatin that confirm previous interparticle interactions observed in crystal structures.
Keywords: chromatin structure, histone variants, nucleosomes
The eukaryotic cell stores its genetic information in DNA molecules that can be over 1 m in length. The DNA is hierarchically packed in the nucleus (up to ~2 × 10−5-times smaller in length) with the aid of proteins to form a complex called chromatin. The nucleosome core particle represents the first level of chromatin organization and is composed of two copies of each of histones H2A, H2B, H3 and H4, assembled in an octameric core with 146-147 bp of DNA tightly wrapped around it [1,2]. Nucleosome cores are separated by linker DNA of variable length and are associated with the linker histone H1. The next level of chromatin organization is the 30-nm fiber, which is composed of packed nucleosome arrays recently found to be arranged as a two-start helical model [3], and mediated by core histone internucleosomal interactions. Earlier models proposed by several groups have not been extensively tested (reviewed in [4]), probably due to technological limitations. Chromatin compaction can also be achieved by the polycomb repression complex (PRC); a multi-protein complex recently described by Francis and colleagues that can alter the chromatin structure of nucleosomal arrays [5].
The overall structural state of chromatin and its domains controls both DNA replication and the expression of genes. Core histones are characterized by the presence of a histone fold domain [6] and N-terminal tails of variable length that are the subject of extensive post-translational modifications (PTMs), which have been implicated in transcriptional activation, silencing, chromatin assembly and DNA replication (reviewed in [7]). PTMs are a component of the epigenome that includes changes to DNA and its connected proteins. These epigenetic modifications are switches for the regulation of gene expression and are chemical modifications of the DNA and histones that do not result in changes to the DNA sequence.
Several chromatin states are likely to be regulated and maintained in a tissue-specific manner, making the DNA accessible to the transcription machinery during specific periods of time and at precise locations. Recent efforts to characterize the epigenome include the characterization of methylated cytosines in CpG dinucleotides [8], and global patterns of histone modifications [9–12].
This review focuses on the histones including structural information, sequence variants, PTMs, interactions and contributions to chromatin organization. Three main topics are addressed:
Functional information obtained from recent structural studies
Functional roles of core histone variants in chromatin
Changes in chromatin structure due to PTMs of core histones
Nucleosome core particle organization
Core histones play structural roles in chromatin assembly and compaction by forming the nucleosome. Nucleosomes were first proposed by Kornberg and Thomas [13,14]. They described a particle that is a heterotypic tetramer (H3–H4)2 with two associated dimers (H2A–H2B) [15] in the form ([H2A–H2B][{H3-H4}2] [H2A–H2B]). Associated with this octamer are about 147 bp of DNA wrapped in 1.7 superhelical turns. Nucleosomes are connected by a DNA linker of variable length that forms a 10-nm beads-on-a-string array [16,17]. Structural details of the nucleosome core particle were obtained from a 7 Å resolution crystallographic analysis [18], with an (H3)2(H4)2 tetramer and two H2A-H2B dimers at the core, and the DNA wrapped around it.
Each of the core histones contain the histone fold domain, composed of three α-helices connected by two loops [6,19,20], which allow heterodimeric interactions between core histones known as the handshake motif. Additionally, each core histone also contains an N-terminal tail that is subjected to a wide variety of PTMs (reviewed in [7,21]). The core histone tails play important roles in nucleosome stability [22], and may contribute to define the condensed state of the chromatin fiber and higher order structures [23] by facilitating nucleosome assembly or disassembly.
The 2.8 Å resolution structure of the nucleosome core particle revealed molecular details of the nucleosome core, particularly the interactions between each of the histone chains [1]. In FIGURE 1A, each of the histones is depicted as a ribbon with different colors for each histone amino acid chain. The α-helices are helically depicted and the unstructured N-terminal tails can be seen on the outside of the DNA surface as unstructured lines. Histones H3 and H4 form a tetramer through an H3–H3′ four-helix bundle. The two H2A-H2B dimers inter-act with the H3–H4 tetramer, via two H2B–H4 associations, to complete the octamer that is assembled in the presence of DNA or high salt concentration. In addition to the histone fold domain, the four core histones have tails that appear as disordered structures protruding from the center of the nucleosome core. Moreover, the N-terminal H4 histone tail has internucleosomal interactions with histone H2A (α-helix 2) that could stabilize higher order structures. Internucleosomal interactions mediated by opposite charges were first observed in crystal structures [1, 2]. Their biologic relevance was confirmed when Dorigo and collaborators showed that nucleosome arrays could be stabilized by using disulfide crosslinks between the N-terminal H4 histone tail and α-helix 2 of histone H2A [3,24].
Figure 1. Post-translational modifications in the nucleosome core particle near the protein-DNA interface.
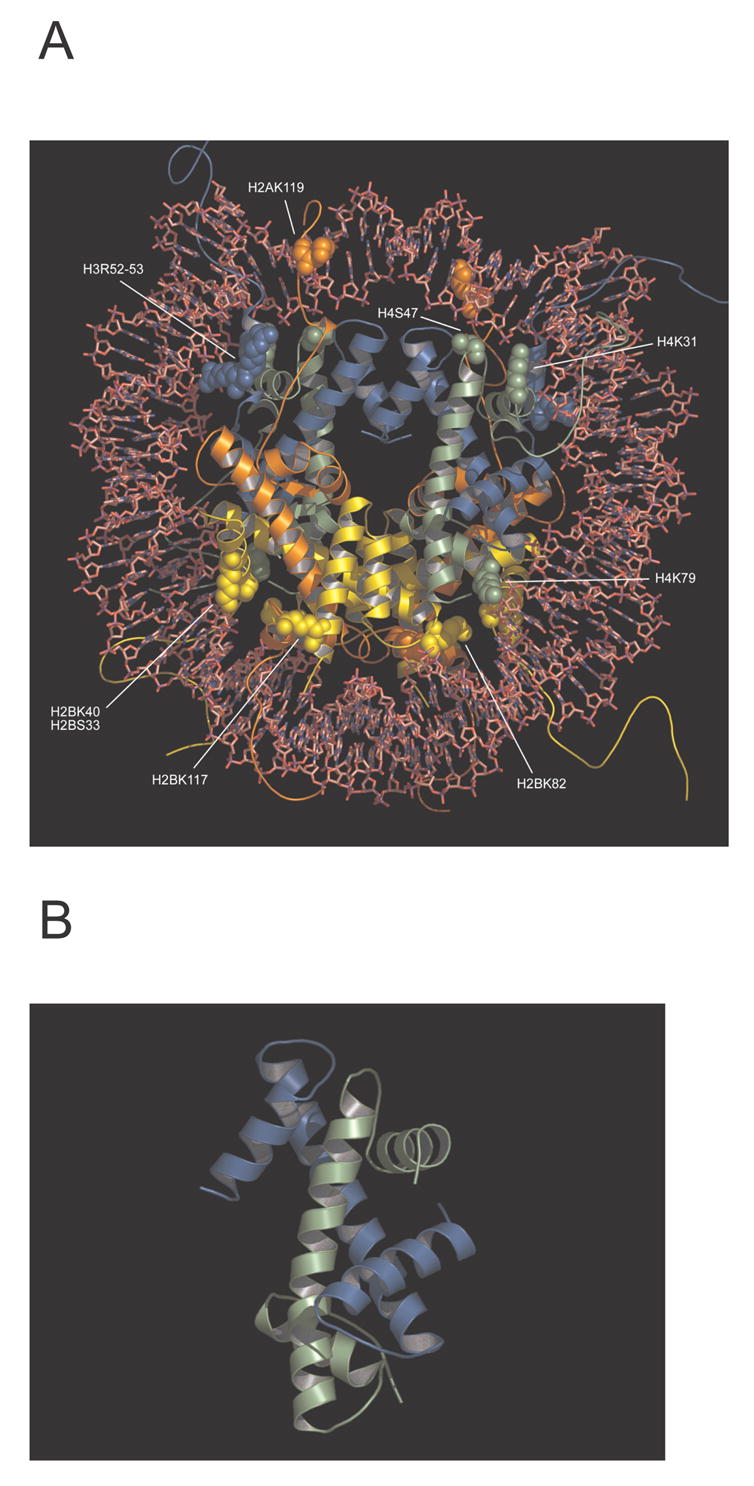
(A) The nucleosome core particle structure (Protein Data Bank accession code 1KX5) with the histone modifications that could be involved in increasing nucleosome mobility. The colours represent different histones H3 (blue), H4 (light green), H2A (orange) and H2B (yellow); the histone modifications are shown as spheres. (B) The histone fold domain of H3and H4 interacting in the characteristic handshake motif. The figure was prepared with PyMCL [102].
Structural characterization of nucleosome core particles from a variety of organisms reveals striking similarities despite histone tail and core variation (reviewed in [25, 26]). The sequence variability in the histone fold domain was recently reanalyzed in a novel fashion using the structural information [27]. Histone H2B and H2A were found to be the most variable pair, whereas H4 and H3 were the most conserved. Interestingly a large fraction of the positions where sequence variation in the histone fold domain was observed had similar physicochemical properties (i.e., changes in sequence were mostly to physicochemically conserved amino adds; e.g., leucine to isoleucine or valine, or lysine to arginine). Additionally they found that the degree of conservation for a particular position was correlated with the number of contacts observed in the crystal structures of the nucleosome core particles [27]. Therefore, sequence variation in the histone fold domain appears to be functionally constrained.
Nucleosome core particle high-resolution structures reveal functional detail
Early structural determinations of the nucleosome core particle at 22 Å [28] and at 7 Å[18] exposed features about peptide chain orientations and positions in the core that were well supported by earlier biochemical analyses. The high-resolution structure of the nucleosome core particle determined by Luger and collaborators revealed significantly more detail of the interactions within the nucleosome and between nucleosomes in the crystal [1]. Some of the key features of this structure allow for the identification of multiple contacts between protein-protein and protein-DNA interfaces. The core histones make contact with the DNA primarily through the phosphodiester backbone. The lack of specific contacts between the core histones and the DNA bases explains how nucleosomes can pack DNA in a sequence-independent fashion. A higher resolution structure of the nucleosome core par-tide revealed that DNA binding to the octameric core is accomplished by mainly three to six hydrogen bonds between the protein main chain amides and the DNA phosphate backbone [2]. The core histone tails are exposed on the outside of the DNA and appear in the crystal structure as random coils, and may be involved in internucleosomal interactions. Electrostatic potential calculations of the nucleosome core particle surface (FIGURE 2A) reveals positively charged regions (blue shading) located in the tails and along the surface where the DNA makes nonspecific contacts with the octamer. In contrast, the electrostatic surface of the octamer near the center of the exposed face is negatively charged (red shading in FIGURE 2B). The difference in electrostatic surface potential is likely to play an important role in internucleosomal interactions via the core histone tails and may also contribute to the structural plasticity of the nucleosome core particle. Indeed, Luger and colleagues observed internucleosomal interactions in crystals between oppositely charged surfaces of the core particle (see arrow in FIGURE 2) [1].
Figure 2. Electrostatic surface potential of the nucleosome core particle (Protein Data Bank accession code 1KX5).
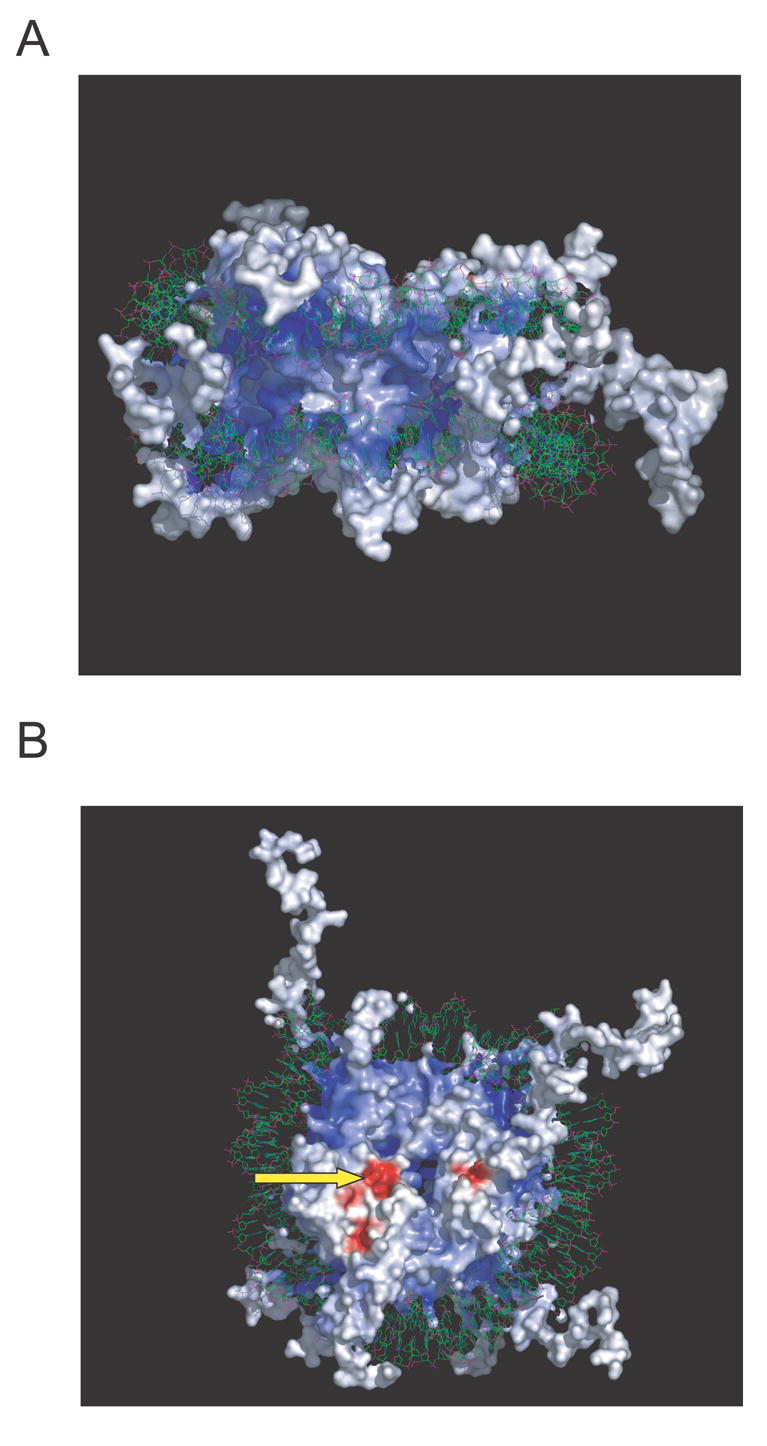
The electrostatic potential ranges from +20 (blue) to −20 (red) kTe−1. (A) Side view of the nucleosome core particle. Note the distribution of positive charges (blue) around the DNA interface and the core histone tails. (B) Top view of the nucleosome core particle. This surface has a number of negative charges (red) indicated by a green arrow in the exposed protein surface of the histone octamer. The electrostatic potential mapped to the molecular surface was calculated using Graphical Representation and Analysis of Structural Properties (GRASP) [69], and the figures were prepared with the Swiss-PDB Viewer [70].
The large number of nucleosome core particle structures determined provides a wealth of information and insight into chromatin function (reviewed in [25]). Interestingly, the nucleosome core particle can accommodate a variety of sequence changes at protein-DNA interfaces and protein-protein interfaces without major structural changes. In addition, the crystal structures of several Swi-INdependent (Sin) mutants, which were originally isolated because they alleviate the transcriptional defects caused by SWI/SNF inactivation, reveal only small changes in the overall structure of the nucleosome. Therefore, Sin mutations facilitate nucleosome sliding by localized changes in protein-DNA interactions while leaving the nucleosome core particle structure mostly unchanged [29].
The structural data available for nucleosome core particles indicate that even more substantial sequence changes introduced by histone variants like H2AZ can be accommodated without major changes in the nucleosome structure [30]. An interesting question would be which structural changes are responsible for active and inactive chromatin states? It has been proposed that the core histone tails and their PTMs could help define chromatin states via changes in internucleosomal interactions effecting changes in higher order structures that are not evident at the nucleosome core particle level [23]. Additionally, PTMs found at the protein-DNA interfaces (FIGURE 2) could increase the nucleosome mobility as suggested by Cosgrove and colleagues [31].
Conservation of histone interactions in the nucleosomal core particle
While histone structures are similar, it is useful to know what structural variation, if any exists at the interfaces between histone pairs and to group these interfaces by structural similarity The authors performed a systematic comparison of interface surfaces amongst all pairs of histones in 26 structures present in the Protein Data Bank (PDB) that contained more than one histone fold domain. Each pair of histones with at least five interfacial residue contacts (Cα–Cα distance within 8 Å) was compared against all other pairs in all other structures using structural alignments between corresponding domains. Once two pairs were aligned, residues at the interaction surface were compared using the alignment. If at least half of these interacting residues occupied the same positions in the structural alignment, the pairs were considered to interact in the same mode and were grouped together with single linkage clustering [SHOEMAKER & COWORKERS, UNPUBLISHED DATA].
The histone interfaces fall into eight distinct modes that are listed for a representative nucleosome core particle structure, (PDB:1KX5), in TABLE 1 and FIGURE 3 [2]. As expected, the two modes with the largest interaction surfaces group the H3–H4 and H2A–H2B heterotypic dimer interactions. Each of the other interaction combinations forms a unique structural interface that is different from the first two modes, with specific contacts between particular peptide chains Across the complete set of 26 structures, each of these binding modes was conserved in each of the structures with only one exception. This exception occurs with the H2A–H2A (Mode 8 in TABLE 1) interface in the nucleosome core particle structure containing H2AZ (PDB:1F66) [30]. This structure has a modification in the loop at this interface that excludes it from clustering with the same interface in all of the other structures. This is not surprising since the H2AZ variant has a different function in transcriptional activation.
Table 1.
Binding modes between core histories in a representative nucleosome core particle (Protein Data Bank identifier 1KX5).
| Mode | Histones | No. of interface contacts |
|---|---|---|
| 1a | H3|H4 | 135 |
| 1b | H3|H4 | 138 |
| 2a | H2A|H2B | 133 |
| 2b | H2A|H2B | 132 |
| 3a | H3|H2A | 30 |
| 3b | H2A|H3 | 30 |
| 4a | H4|H2A | 22 |
| 4b | H2A|H4 | 22 |
| 5 | H3|H3 | 19 |
| 6a | H4|H2B | 15 |
| 6b | H2B|H4 | 12 |
| 7a | H4|H2B | 8 |
| 7b | H4|H2B | 6 |
| 8 | H2A|H2A | 8 |
Note: Core histone interfaces were grouped into binding modes using structural alignment. All interfaces and modes for the structure are listed including chain locations, domains participating and the number of residue contacts at the interface. The Protein Data Bank structures used in this study were: 1AOI, 1EQZ, 1F66, 1HIQ 1HQ3, 1ID3, 1KX3, 1KX4, 1KX5, 1M18, 1M19, 1M1A, 1P34, 1P3A, 1P3B, 1R3F, 1P3G, 1P31, 1P3K 1P3L, 1P3M, 1P3Q, 1P3P, 1S32, 1TZY and 2HIQ
Figure 3. Schematic representation of the regions of the core histone sequences in pairs of interacting chains.
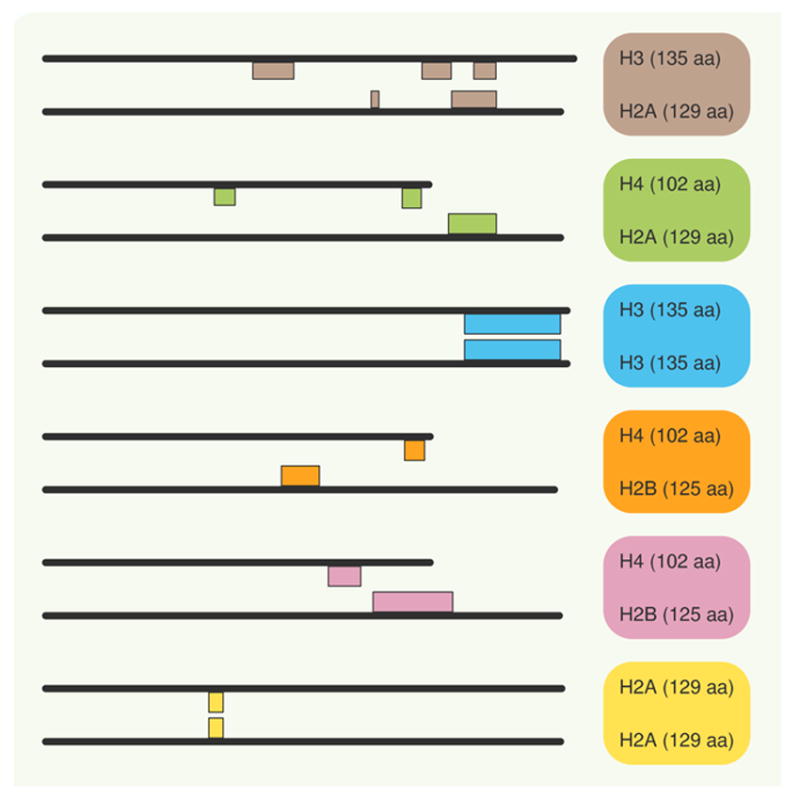
Each core histone is represented by a black line drawn to sequence length scale, the location of interacting residues is indicated by colored boxes depending on the interacting mode (see TABLE 2 for details; each mode in TABLE 2 is identified). The box encompasses all residues in the region of contact. Specific residues are identified in TABLE 2.
aa: Amino acids.
TABLE 2 lists the principal residues involved in the interfaces for modes 3–8 of the representative nucleosome core particle structure (PDB: 1KX5) in TABLE 1. The Interface 1 column lists the residues from the first histone and Interface 2 lists the residues from the second histone of an interface. Only structurally conserved interfacial residues are listed to highlight the common features of a mode. Modes 1 and 2 are not shown due to the extensive nature of their contacts. FIGURE 5A–F shows the regions of contact between each histone peptide in the structure (data from TABLE 2). These interactions are all symmetrical, as expected, and are highly conserved.
Table 2.
Interface core histone residues in a representative nucleosome core particle (Protein Data Bank identifier 1KX5).
| Mode | Histones | Interface 1 | Interface 2 |
|---|---|---|---|
| 3a/b | H3|H2A | L48,I51,R52,Q55-L60,E94,A95,A98,I112,V117 | R81,T101,A103-I111 |
| 4a/b | H4|H2A | R40,G42,K44,G94-G99 | R99-A103,V107,N110,I111 |
| 5 | H3|H3 | D106,L109,C110,H113-K115,L126,A127,I130,R131 | D106,L109,C110,H113-K115,L126,A127,I130,R131 |
| 6a/b | H4|H2B | Y98-F100 | K57,A58,S60,I61,S64,F65 |
| 7a/b | H4|H2B | D68,T71,Y72,H75,A76 | L80,N84,T96,L100 |
| 8 | H2A|H2A | N38-E41 | N38-E41 |
Note: The smaller histone interfaces with fewer numbers of contacts are listed in greater detail. Structurally conserved residues at each interface are listed under Interface 1 for the first histone listed and under Interface 2 for the second histone listed.
Figure 5. Conserved binding modes in a representative nucleosome core particle structure.

TABLE 2 describes the specific interface residues involved in the conserved interaction. Core histones are represented as molecular surf aces and are colored the same as for FIGURE 1: histone H3 (blue), H4 (light green), H2A (orange) and H2B (yellow). The crystallographic dyad is indicated by a dyad symbol. (A) Conserved binding mode 3, interaction between H3 and H2A (B) Conserved binding mode 4, interaction between H4 and H2A (C) Conserved binding mode 5, interaction between H3 and H3. (D) Conserved binding mode 6, interaction between H4 and H2B (E) Conserved binding mode 7, interaction between H4 and H2B (F) Conserved binding mode 8, interaction between H2A and H2A The figure was prepared with PyMOL[102].
It is interesting to note that a couple of the binding modes show common structural elements between them. Modes 1 and 2 are distinct, but a superposition of the longest helix in chains A and C of the representative nucleosome core particle structure (1KX5) shows some similarity between the H3–H4 and H2A–H2B interfaces. These are contacts in the known handshake interaction between two histone fold proteins (FIGURE 1B) and distinguish these two modes as one group distinct from the rest [6]. The other groups have a lower number of contacts and are not involved in similar handshake interactions. There is also some similarity between Modes 5 and 7, which are both positioned between the two types of histone fold dimers; however, an adequate amount of structural variation is present to classify them separately This comparison shows that, while all histone domains share a similar structural motif, they form interfaces with each other in a number of unique ways. This interface specificity is consistent across many variant histone sequences, demonstrating the flexible and adaptable architecture of histone complexes.
FIGURE 4 shows the conserved regions of peptide sequences of histones H4 and H2Athat form the internucleosomal contacts (these are contacts not shown in TABLES 1 & 2). The residues K16, R19, K20 and R23 in H4 interact with residues E56, E61, E64, D90, E91 and E92 in H2A K16 and K20 can be acetylated and would result in an alteration of the ionic interactions between these two peptides and thus could modify neighboring internucleosomal contacts, potentially providing a path for the opening of the chromatin.
Figure 4. Conserved regions of histories H4 andH2A, which are the contacts for internucleosomal interactions.
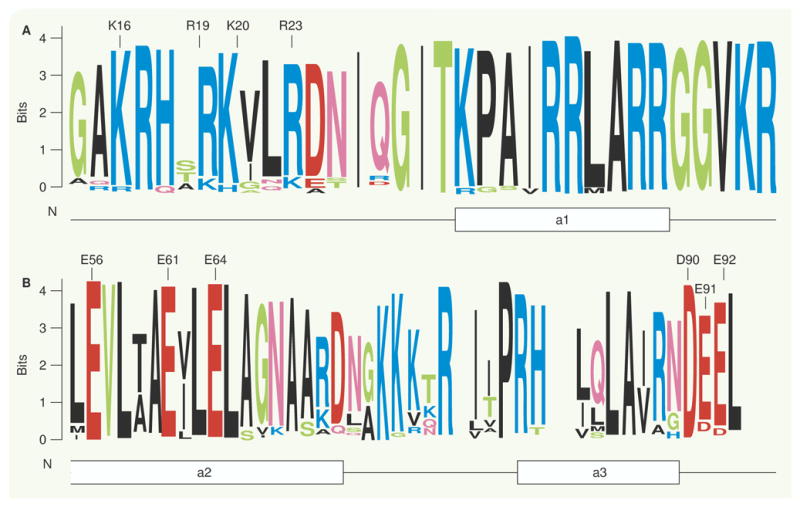
Sequence logo representation of multiple sequence alignments of histoneH4 and H2A. The multiple sequence alignments were obtained from the Histone Database [101]; secondary structure elements are shown as lines (loops) and boxes (helices). The amino adds that make contacts in the crystal structures are marked above the sequence logos. (A) Histone H4 alignment around the basic region that mediates internucleosomal interactions. Lysine 16 and 20 can be subject to post-translational modifications. (B) HistoneH2Aalignment around the addle region that mediates internucleosomal interactions. The figure was prepared with WebLogo [71].
Histone variants
Eukaryotic chromatin is a highly dynamic macromolecular assembly. Nucleosome particles can be modified in their composition, structure and location by chromatin remodeling complexes that introduce PTMs to the core histones (reviewed in [7,21]) and leads to nucleosome removal and reassembly (reviewed in [31,32]). Additionally, the chromatin fiber can be modified by the incorporation of histone variants (reviewed in [33–35]) FIGURE 4. This would change local chromatin structure by promoting nucleosome subunit exchange to facilitate cellular processes such as transcription [36] or development [37].
Most eukaryotic organisms have multiple copies of histone genes; see the histone database for a comprehensive collection of histone sequences [101]. The major histone protein genes are mostly present in gene clusters and are expressed primarily during the S phase of the cell cycle. These histones are used for nucleosome assembly and packing of newly synthesized DNA. In contrast, some histone variants are expressed throughout the cell cycle and their expression is not restricted to the S phase (reviewed in [34]).
Core histone variants
Histone H2A
H2A is the core histone with the largest number of variants. The histone H2A variants include H2AZ and H2AX, which are found in most eukaryotes, and H2A. Bbd and MacroH2A are only found in vertebrates. The H2A histone variants are characterized by their divergent C-terminal sequences and their genome localization. H2AZ has been implicated in transcriptional activation in yeast by preventing the spread of silent heterochromatin [38]. H2ABbd was recently described as a histone variant that is excluded from inactive X chromosomes [39]. H2ABbd has a truncated C-terminal tail and its localization correlates with transcriptionally active chromatin. Recent studies measured changes in nucleosome conformation as a function of variations in ionic strength, indicating that nucleosomes containing H2ABbd are less stable [40,41]. On the contrary, the MacroH2A histone variant has been found primarily in the chromatin of inactive X chromosomes [42] characterized by the presence of an additional large nonhistone macrodomain that is likely to have enzymatic activity [43]. However, it is not clear if chromatin inactivation is due to an enzymatic activity and/or a steric block that impedes access by transcription factors or the chromatin remodeling machinery [44]. H2AX is characterized by a C-terminal extension with the consensus SQ[E/D]Φ, where Φ represents a hydrophobic amino acid. The serine residue in the consensus is phosphorylated in response to double-strand DNA breaks [45]. Additionally, phosphorylation of H2AX has been found to aid in the recruitment of proteins involved in DNA repair [46,47].
Histone H2B
Histone H2B variants are few in number and those that have been documented have specialized roles in chromatin compaction during gametogenesis. The H2B variant documented in sea urchin has an N-terminal tail with a characteristic pentapeptide repeat that is highly charged [48,49]. Additional H2B variants have also been found in male gametic cells from lily [50] and, more recently, in bovine [51] and human spermatozoa [52], however, their specific roles remain to be elucidated.
Histone H3
Histone H3 variants include H3.3, CenHS and H3.4. H3.3 is a histone variant that is not S-phase regulated and is found in transcriptionally active chromatin [53]. H3.4 is a testis-specific H3 variant found in primary spermatocytes [54,55]. The CenH3 variant is localized in centromeric chromatin; their N-terminal tails are extremely divergent and share no sequence similarity with canonical H3 [33].
Histone H4
Histone H4 is the most highly conserved histone. H4 makes extensive contacts with the other three core histones in the nucleosome core particle and is thus constrained in its sequence variability. H4 has no known sequence variants; indeed, there are even identical sequence variants that are expressed in a cell cycle-independent manner as opposed to the predominant synthesis period for histones in the S phase of the cell cycle [56].
Histone modifications, structural changes & the molten globule state of the nucleosome core particle
Core histone tails can be reversibly modified by acetylation, methylation, ubiquitination and phosphorylation among others [7]. While many studies have identified the N- and C-terminal tails as the main targets for such modifications, recent analysis has shown several modifications within the histone core [57]. This finding has prompted further analysis of the role of these modifications at the histone-DNA interface in the chromatin. The precise mechanisms of how these modifications regulate changes in chromatin structure remain unclear.
The notion of the cell's ability to decipher histone modifications resulted in the concept of a histone code [21,58], which has also been subject of a controversial debate [59]. A recent study by Dion and colleagues discovered a simple histone acetylation code in H4 that has challenged this model [11]. Dion and colleagues found that H4 modifications are interpreted by two distinct mechanisms, one nonspecific (for lysines 5, 8 and 12) and one specific (for lysine 16). In the nonspecific mechanism, changes in gene expression of nearly 1200 genes were due to cumulative acetylation effects. Dion and colleagues demonstrated that there is a strong correlation between transcription and the total number of sites that can be acetylated in H4 tails. On the contrary, in the specific mechanism, the transcriptional changes did not follow a cumulative model and caused a unique transcriptional phenotype independent of the mutational state of the other lysine residues affecting approximately 100 genes. Interestingly, the specific mechanism involves histone H4 lysine 16, which is part of a region that makes intercore particle contacts with an H2A-H2B dimer of the adjacent particle [1,2], thus indicating that charge changes at interparticle interfaces or other key residues subject to PTMs may play critical regulatory roles. In addition, K56 in histone H3 is specifically acetylated in a subset of active genes [60]. Furthermore, several modification sites have been identified on the protein surface of the core particle using mass spectrometry (e.g., H4K79, H4K77 and H2BK120). These could well play a similar role to H3K56, but for different subsets of genes. Therefore, histone acetylation events alter specific interactions, thereby influencing nucleosome dynamics in a complex way.
This review will focus on several experiments that indicate the importance of charge-dependent interaction alterations resulting from hyperacetylated nucleosomes. Electron microscopy (EM) studies by Oliva and colleagues found that hyper-acetylated nucleosomes lose their compact shape and become elongated when bound to electron microscope grids at low ionic strength [61]. The alterations in shape observed by these authors could well represent the range of possible structures that result from different intranucleosomal dynamic interactions. Later, Dunker and colleagues studied the effect of hyperacetylation on nucleosome stability using atomic force microscopy [62]. They subjected nucleosomes to increasing forces until the nucleosomes were crushed, and observed that hyperacetylation makes nucleosome core particles less rigid. They proposed that charge changes due to hyperacetylation promote conversion of nucleosomes to a less rigid form or a molten globule state [63]. The molten globule state is a non-compact state with partial order and can be induced in some proteins under certain conditions such as changes in temperature, pH, charge imbalance or presence of protein denaturants. The molten globule state has some native secondary and tertiary structure but lacks well-packed side chains [63]. Numerous transitions from or to unstructured states have been reported in the literature to explain biologic processes that range from molecular recognition [64], molecular assembly and disassembly in viruses and phages [65] to several cellular control mechanisms [66,67]. Furthermore, Brower-Toland and colleagues examined the effect of core histone tail acetyiation on nucleosomal stability by studying the mechanical disruption of individual nucleosomes in a single chromatin fiber using an optical trap [22]. Nucleosome acetylation levels were controlled by using free core histones and recombinant histone acetyltransferase p300 followed by chromatin reconstitution. These mechanical disruption experiments showed that acetylation decreases the resistance of the nucleosome to mechanical unfolding, in addition to decreasing the average amount of DNA stably bound to the octameric core.
The data reviewed here on the effect of hyperacetylation on nucleosome stability provide support for the hypothesis raised by Dunker and colleagues that the molten globule state in nucleosomes could be induced by charge changes resulting from histone hyperacetylation [62]. Mechanistically, it is possible that nucleosome changes into the molten globule state could generate local deformations that facilitate access of regulatory factors to DNA without nucleosome disassembly. It is also entirely possible that chromatin and nucleosomes are in an equilibrium moving in and out of the molten globule state with only a few modifications necessary to result in an altered state. Simple and cumulative acetylation effects such as the ones observed by Dion and colleagues [11], as well as other recent studies [57,60], are also consistent with this theory. Therefore, understanding nucleosome structure in terms of the molten globule theory may provide further insight into the structural and functional changes that could be induced by histone hyperacetylation. However, extending the theory to higher order chromatin structure would indeed require new data. Assuming that chromatin is dynamic and adopts a variety of active and inactive states, the structural changes necessary to achieve these multiple states might be more open to several easily interchangeable molten globule-like states at energy minima, as opposed to the unyielding structures that are perceived to be similar to the rigidity of a metaphase chromosome. Thus, any chromatin state would not be fixed in an immovable, discrete low-energy trap.
Nucleosome arrays
Recent technologic advances are shedding light on the mechanism of nucleosome array folding into compact, higher order structures. Dorigo and colleagues assembled a well-defined nucleosome array composed of recombinant unmodified core histones and DNA with a strong nucleosome positioning sequence [24]. These nucleosome arrays were highly homogeneous and contained a saturated amount of histone octamers. Dorigo and colleagues evaluated the specific contribution of individual histone tails to the formation of higher order structures in chromatin fibers and found that the H4 N-terminus is necessary to obtain fully compacted chromatin fibers. In addition, the specific region of the histone H4 tail, which mediates compaction, was identified between amino acids 14–19. Luger and collaborators observed interparticle interactions in the crystal structure of nucleosome core particles between the basic side chains of amino acids 16–25 of the H4 tail and a highly acidic region of the H2A-H2B dimer [1,2]. Thus, the results from Dorigo and collaborators confirm the biologic relevance of the interparticle contacts observed in the crystal structures [3,24]. However, a recent low-resolution structure of a tetranucleosome suggests that nucleosome packing is polymorphic [68], therefore, the interfaces between nucleosomes are subject to change depending on the local level of chromatin compaction.
The condensation state of the 30-nm fiber depends on internucleosomal interactions that appear to be modulated, at least in part, via charge-dependent interactions between the basic histone H4 N-terminal tail region (see residues K16, K19, K20 and K23 in FIGURE 4A.) that is highly conserved, and an acidic region in the exposed surface of the H2A-H2B dimer (red patch in FIGURE 1B) that is also highly conserved (see residues E56, E61 andE64 in FIGURE 4B).
Further structural characterization of nucleosome arrays stabilized by the introduction of specific disulfide crosslinks at the H4-H2A interparticle interface provided evidence for the structure of the 30-nm chromatin fiber in agreement with a two-start helical model [3]. Understanding the key surface contacts that create these higher order structures and how they are modulated is a challenge that has not been completely addressed with current technologies.
Expert commentary
Histones play critical roles in the establishment of the first and most basic level of chromosome organization, the nucleosome core particle. High-resolution structures of the nucleosome particle have revealed important details about protein-protein and protein-DNA interactions that explain why they are some of the most conserved protein families characterized to date (histone H4 in particular). Core histones share a structural motif known as the histone fold domain, formed by three α-helices connected by two loops. Nucleosome core particle assembly is a stepwise process that requires prior assembly of histone dimers H3-H4 and H2A-H2B. Specific constraints have been imposed on the core histones to maintain the overall structure of the nucleosome.
However, in some instances, histones have diversified throughout eukaryotic evolution to assume specialized roles to modulate chromatin function. These histone variants have specific expression patterns, chromosomal localization and can be exchanged with the major forms to effect changes in chromatin structure. Thus, histone variants have the potential to alter chromatin structure at the nucleosome core particle level and at higher order structure level.
Histones can also be subjected to a variety of PTMs that alter chromatin structure by charge-induced changes in the nucleosome core particle. Charge changes may have cumulative structural effects depending on the total number of PTMs at a particular chromatin location. On the contrary PTMs at key residues such as those involved in internucleosomal interactions, may lead to more targeted chromatin changes. PTMs can also promote nucleosome disassembly or assembly and may involve interactions with chromatin remodeling complexes and histone chaperones.
Five-year view
Nucleosome core particle structures available in public databases provide a wealth of information about the fundamental unit of chromatin organization. However, the detailed molecular arrangement of multiple nucleosome core particles in chromatin fibers remains unknown. Despite great advances from the Richmond Laboratory, who have contributed to our knowledge of the 30-nm chromatin fiber organization, structural details of nucleosome arrays remain to be elucidated [3,24]. The authors expect that nucleosome core particle structures for other model organisms will become available, as well as structures where additional histone variants have been incorporated. These studies will provide another insight into determining how transcription regulation is affected by the modification of nucleosome core particles. Additionally, the authors expect to see development of novel techniques or improvement of existing ones to probe the structure of the chromatin fiber or to obtain higher resolution images of nucleosome arrays.
Charge effects due to PTMs can have a great impact in gene expression [11]. The authors expect to see systematic studies to elucidate the effects of PTMs in the core histones on global gene expression. Such studies will identify residues that, when modified, will alter gene expression patterns in a collective fashion in addition to identifying key residues that may alter gene expression in a specific manner. Such key residues will be present at protein-protein interfaces and the charge-induced changes at these locations may be sufficient to significantly affect both intra- or internucleosome interactions.
In addition, atomic force microscopy experiments on reconstituted nucleosomes with variously acetylated histones would shed further light on the global effect of these histone modifications on nucleosomal stability and higher order structures.
Finally little is known about the mechanistic aspects of how chromatin can be assembled in vivo and how core subunit exchange can occur in addition to the proteins involved in these processes. Such studies will also shed light on the mechanisms of epigenetic inheritance.
Key issues
Core histone proteins are some of the most conserved protein families known to date due to their critical role in chromatin structure and function.
Histone variants, post-translational modifications and interactions with chromatin remodeling complexes facilitate changes in chromatin structure and function that are also regulated throughout the life of an organism and at specific chromosomal locations.
Histone variants have evolved to assume specialized roles in different chromatin functions, such as the control of gene expression, epigenetic silencing and centromere integrity. The number of histone variants appears to be greater in higher eukaryotes.
Charge-dependent interactions at key residues that are subject to post-translational modifications can have profound effects in chromatin function by affecting internucleosomal interactions or by inducing structural changes at protein-protein or protein-DNA interfaces.
Acknowledgments
The authors thank Michael Bustin and Marc Landsman for critically reviewing this manuscript.
Affiliations
Leonardo Mariño-Ramírez, PhD. National Institutes of Health, Computational Biology Branch, National Center for Biotechnology Information, National Library of Medicine, Building 38A, Room 6S614M, 8600 Rockville Pike, Bethesda, MD 20894, USA, Tel.: +1 301 402 3708, Fax+1 301 480 2288, marino@ncbi.nlm.nih.gov
Maricel G Kann, PhD. National Institutes of Health, Computational Biology Branch, National Center for Biotechnology Information, National Library of Medicine, Building 38A, Room 8N811R, 8600 Rockville Pike, Bethesda, MD 20894, USA, Tel: +1 301 402 3010, Fax: +1 301 480 9241, kann@ncbi.nlm.nih.gov
Benjamin A Shoemaker, PhD. National Institutes of Health, Computational Biology Branch, National Center for Biotechnology Information, National Library of Medicine, Building 38A, Room 8N811Q, 8600 Rockville Pike, Bethesda, MD 20894, USA, Tel: +1 301 594 8093, Fax: +1 301 480 9241, shoemake@ncbi.nlm.nih.gov
David Landsman, PhD. National Institutes of Health, Computational Biology Branch, National Center for Biotechnology Information, National Library of Medicine, Building 38A, Room 6N601, 8600 Rockville Pike, MSC 6075, Bethesda, MD 20894-6075, USA, Tel: +1 301 435 5981, Fax: +1 301 480 2288, landsman@ncbi.nlm.nih.gov
References
Papers of special note have been highlighted as
• of interest
•• of considerable interest
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389(6648):251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]; •• Contains high-resolution structures of the nucleosome that are key to understanding gene regulation and chromatin remodeling.
- 2.Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J Mol Biol. 2002 ;319(5):1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]; •• Contains high-resolution structures of the nucleosome that are key to understanding gene regulation and chromatin remodeling.
- 3.Dorigo B, Schalch T, Kulangara A, et al. Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science. 2004;306(5701):1571–1573. doi: 10.1126/science.1103124. [DOI] [PubMed] [Google Scholar]; • Provides evidence for a two-start helical organization of the 30-nm chromatin fiber and has helped resolve a controversy over how individual nucleosomes are packed in the chromatin fiber.
- 4.van Holde KE. Chromatin. Springer-Verlag; London, UK: 1988. Chapter 7: Highter order structure; pp. 317–354. [Google Scholar]
- 5.Francis NJ, Kingston RE, Woodcock CL. Chromatin compaction by a polycomb group protein complex. Science. 2004;306(5701):1574–1577. doi: 10.1126/science.1100576. [DOI] [PubMed] [Google Scholar]
- 6.Arents G, Moudrianakis EN. The histone fold: a ubiquitous architectural motif utilized in DNA compaction and protein dimerization. Proc Natl Acad Sci USA. 1995;92(24):11170–11174. doi: 10.1073/pnas.92.24.11170. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Describes the histone fold and its widespread presence in eukaryotic proteins present in the nucleus.
- 7.Peterson CL, Laniel MA. Histories and histone modifications. Curr Biol. 2004;14(14):R546–R551. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 8.Rakyan VK, Hildmann T, Novik KL, et al. DNA methylation profiling of the human major histocomparibility complex: a pilot study for the human epigenome project. PLoS Biol. 2004;2(12):e405. doi: 10.1371/journal.pbio.0020405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roh TY, Cuddapah S, Zhao K. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes Dev. 2005;19(5):542–552. doi: 10.1101/gad.1272505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roh TY, Ngau WC, Cui K, Landsman D, Zhao K. High-resolution genome-wide mapping of histone modifications. Nature Biotechnol. 2004;22(8):1013–1016. doi: 10.1038/nbt990. [DOI] [PubMed] [Google Scholar]
- 11.Dion MF, Altschuler SJ, Wu LF, Rando OJ. Genomic characterization reveals a simple histone H4 acetylation code. Proc Natl Acad Sci USA. 2005 doi: 10.1073/pnas.0500136102. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Provides a systematic evaluation of histone H4 acetylation patterns and their effects on gene expression measured by microarray analysis.
- 12.Kurdistani SK, Tavazoie S, Grunstein M. Mapping global histone acetylation patterns to gene expression. Cell. 2004;117(6):721–733. doi: 10.1016/j.cell.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 13.Kornberg RD, Thomas JO. Chromatin structure; oligomers of the histones. Science. 1974;184(139):865–868. doi: 10.1126/science.184.4139.865. [DOI] [PubMed] [Google Scholar]
- 14.Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184(139):868–871. doi: 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- 15.Kelley RI. Isolation of a histone IIb1–IIb2 complex. Biochem Biophys Res Commun. 1973;54(4):1588–1594. doi: 10.1016/0006-291x(73)91168-6. [DOI] [PubMed] [Google Scholar]
- 16.Olins AL, Olins DE. Spheroid chromatin units (v bodies) Science. 1974;183(122):330–332. doi: 10.1126/science.183.4122.330. [DOI] [PubMed] [Google Scholar]
- 17.Woodcock CL. Ultrastructure of inactive chromatin. J Cell Biol. 1973;59(2):368a. [Google Scholar]
- 18.Richmond TJ, Finch JT, Rushton B, Rhodes D, Klug A. Structure of the nucleosome core particle at Å resolution. Nature. 1984;311(5986):532–537. doi: 10.1038/311532a0. [DOI] [PubMed] [Google Scholar]
- 19.Baxevanis AD, Arents G, Moudrianakis EN, Landsman D. A variety of DNA-binding and multimeric proteins contain the histone fold motif. Nucleic Acids Res. 1995;23(14):2685–2691. doi: 10.1093/nar/23.14.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kokubo T, Gong DW, Wootton JC, et al. Molecular cloning of Drosophila TFIID subunits. Nature. 1994;367(6462):484–487. doi: 10.1038/367484a0. [DOI] [PubMed] [Google Scholar]
- 21.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 22.Brower-Toland B, Wacker DA, Fulbright RM, et al. Specific contributions of histone tails and their acetylation to the mechanical stability of nucleosomes. J Mol Biol. 2005;346(1):135–146. doi: 10.1016/j.jmb.2004.11.056. [DOI] [PubMed] [Google Scholar]
- 23.Zheng C, Hayes JJ. Structures and interactions of the core histone tail domains. Biopolymers. 2003;68(4):539–546. doi: 10.1002/bip.10303. [DOI] [PubMed] [Google Scholar]
- 24.Dorigo B, Schalch T, Bystricky K, Richmond TJ. Chromatin fiber folding requirement for the histone H4 N-terminal tail. J Mol Biol. 2003;327(1):85–96. doi: 10.1016/s0022-2836(03)00025-1. [DOI] [PubMed] [Google Scholar]
- 25.Chakravarthy S, Park YJ, Chodaparambil J, Edayathumangalam RS, Luger K. Structure and dynamic properties of nucleosome core particles. FEBS Lett. 2005;579(4):895–898. doi: 10.1016/j.febslet.2004.11.030. [DOI] [PubMed] [Google Scholar]
- 26.Luger K. Structure and dynamic behavior of nucleosomes. Curr Opin Genet Dev. 2003;13(2):127–135. doi: 10.1016/s0959-437x(03)00026-1. [DOI] [PubMed] [Google Scholar]
- 27.Sullivan SA, Landsman D. Characterization of sequence variability in nudeosome core histone folds. Proteins. 2003;52(3):454–465. doi: 10.1002/prot.10441. [DOI] [PubMed] [Google Scholar]
- 28.Klug A, Rhodes D, Smith J, Finch JT, Thomas JO. A low resolution structure for the histone core of the nucleosome. Nature. 1980;287(5782):509–516. doi: 10.1038/287509a0. [DOI] [PubMed] [Google Scholar]
- 29.Muthurajan UM, Bao Y, Forsberg LJ, et al. Crystal structures of histone Sin mutant nucleosomes reveal altered protein-DNA interactions. EMBO J. 2004;23(2):260–271. doi: 10.1038/sj.emboj.7600046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suto RK, Clarkson MJ, Tremethick DJ, Luger K. Crystal structure of a nucleosome core particle containing the variant histone H2A. Z Nature Struct Biol. 2000;7(12):1121–1124. doi: 10.1038/81971. [DOI] [PubMed] [Google Scholar]
- 31.Cosgrove MS, Boeke JD, Wolberger C. Regulated nucleosome mobility and the histone code. Nature Struct Mol Biol. 2004;11(11):1037–1043. doi: 10.1038/nsmb851. [DOI] [PubMed] [Google Scholar]
- 32.Korber P, Horz W. SWRred not shaken; mixing the histones. Cell. 2004;117(1):5–7. doi: 10.1016/s0092-8674(04)00296-x. [DOI] [PubMed] [Google Scholar]
- 33.Malik HS, Henikoff S. Phylogenomics of the nucleosome. Nature Struct Biol. 2003;10(11):882–891. doi: 10.1038/nsb996. [DOI] [PubMed] [Google Scholar]
- 34.Kamakaka RT, Biggins S. Histone variants deviants? Genes Dev. 2005;19(3):295–310. doi: 10.1101/gad.1272805. [DOI] [PubMed] [Google Scholar]
- 35.Henikoff S, Furuyama T, Ahmad K. Histone variants, nucleosome assembly and epigenetic inheritance. Trends Genet. 2004;20(7):320–326. doi: 10.1016/j.tig.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 36.Park YJ, Chodaparambil JV, Bao Y, McBryant SJ, Luger K. Nucleosome assembly protein 1 exchanges histone H2A–H2B dimers and assists nucleosome sliding. J Biol Chem. 2005;280(3):1817–1825. doi: 10.1074/jbc.M411347200. [DOI] [PubMed] [Google Scholar]
- 37.Saeki H, Ohsumi K, Aihara H, et al. Linker histone variants control chromatin dynamics during early embryogenesis. Proc Natl Acad Sci USA. 2005;102(16):5697–5702. doi: 10.1073/pnas.0409824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meneghini MD, Wu M, Madhani HD. Conserved histone variant H2AZ protects euchromatin from the ectopic spread of silent heterochromatin. Cell. 2003;112(5):725–736. doi: 10.1016/s0092-8674(03)00123-5. [DOI] [PubMed] [Google Scholar]
- 39.Chadwick BP, Willard HF. A novel chromatin protein, distantly related to histone H2A is largely excluded from the inactive X chromosome. J Cell Biol. 2001;152(2):375–384. doi: 10.1083/jcb.152.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bao Y, Konesky K, Park YJ, et al. Nucleosomes containing the histone variant H2A Bbd organize only 118 base pairs of DNA. EMBOJ. 2004;23(16):3314–3324. doi: 10.1038/sj.emboj.7600316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gautier T, Abbott DW, Molla A, et al. Histone variant H2ABbd confers lower stability to the nucleosome. EMBO J. 2004;5(7):715–720. doi: 10.1038/sj.embor.7400182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Costanzi C, Stein P, Worrad DM, Schultz RM, Pehrson JR. Histone macroH2A1 is concentrated in the inactive X chromosome of female preimplantation mouse embryos. Development. 2000;127(11):2283–2289. doi: 10.1242/dev.127.11.2283. [DOI] [PubMed] [Google Scholar]
- 43.Ladurner AG. Inactivating chromosomes a macro domain that minimizes transcription. Mol Cell. 2003;12(1):1–3. doi: 10.1016/s1097-2765(03)00284-3. [DOI] [PubMed] [Google Scholar]
- 44.Angelov D, Molla A, Perche PY, et al. The histone variant macroH2A interferes with transcription factor binding and SWI/SNF nucleosome remodeling. Mol Cell. 2003;11(4):1033–1041. doi: 10.1016/s1097-2765(03)00100-x. [DOI] [PubMed] [Google Scholar]
- 45.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273(10):5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 46.Celeste A, Femandez-Capetillo O, Kruhlak MJ, et al. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nature Cell Biol. 2003;5(7):675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- 47.Thiriet C, Hayes JJ. Chromatin in need of a fix: phosphorylation of H2AX connects chromatin to DNA repair. Mol Cell. 2005;18(6):617–622. doi: 10.1016/j.molcel.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 48.Strickland M, Strickland WN, Brandt WF, Von Holt C. The complete amino-acid sequence of histone H2B(1) from sperm of the sea urchin Parechinus angulosus. Eur J Biochem. 1977;77(2):263–275. doi: 10.1111/j.1432-1033.1977.tb11665.x. [DOI] [PubMed] [Google Scholar]
- 49.Lieber T, Weisser K, Childs G. Analysis of histone gene expression in adult tissues of the sea urchins Strongylocentrotus purpuratus and Lytechinus pictus. tissue-specific expression of sperm histone genes. Mol Cell Biol. 1986;6(7):2602–2612. doi: 10.1128/mcb.6.7.2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ueda K, Kinoshita Y, Xu ZJ, et al. Unusual core histones specifically expressed in male gametic cells of Lilium longiflorum. Chromosoma. 2000;108(8):491–500. doi: 10.1007/s004120050401. [DOI] [PubMed] [Google Scholar]
- 51.Aul RB, Oko RJ. The major subacrosomal occupant of bull spermatozoa is a novel histone H2B. Dev Biol. 2002;242(2):376–387. [PubMed] [Google Scholar]
- 52.Zalensky AO, Siino JS, Gineitis AA, et al. Human testis/sperm-specific histone H2B (hTSH2B). Molecular cloning and characterization. J Biol Chem. 2002;277(45):43474–43480. doi: 10.1074/jbc.M206065200. [DOI] [PubMed] [Google Scholar]
- 53.Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell. 2002;9(6):1191–1200. doi: 10.1016/s1097-2765(02)00542-7. [DOI] [PubMed] [Google Scholar]
- 54.Witt O, Albig W, Doenecke D. Testis-specific expression of a novel human H3 histone gene. Exp Cell Res. 1996;229(2):301–306. doi: 10.1006/excr.1996.0375. [DOI] [PubMed] [Google Scholar]
- 55.Albig W, Ebentheuer J, Klobeck G, Kunz J, Doenecke D. A solitary human H3 histone gene on chromosome 1. Hum Genet. 1996;97(4):486–491. doi: 10.1007/BF02267072. [DOI] [PubMed] [Google Scholar]
- 56.Akhmanova A, Miedema K, Hennig W. Identification and characterization of the Drosophila histone H4 replacement gene. FEBS Lett. 1996;388(2–3):219–222. doi: 10.1016/0014-5793(96)00551-0. [DOI] [PubMed] [Google Scholar]
- 57.Freitas MA, Sklenar AR, Parthun MR. Application of mass spectrometry to the identification and quantification of histone post-translational modifications. J Cell Biochem. 2004;92(4):691–700. doi: 10.1002/jcb.20106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turner BM. Decoding the nucleosome. Cell. 1993;75(1):5–8. [PubMed] [Google Scholar]
- 59.Henikoff S. Histone modifications combinatorial complexity or cumulative simplicity? Proc Natl Acad Sci USA. 2005;102(15):5308–5309. doi: 10.1073/pnas.0501853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu F, Zhang K, Grunstein M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell. 2005;121(3):375–385. doi: 10.1016/j.cell.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 61.Oliva R, Bazett-Jones DP, Locklear L, Dixon GH. Histone hyperacetylation can induce unfolding of the nucleosome core particle. Nucleic Aids Res. 1990;18(9):2739–2747. doi: 10.1093/nar/18.9.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dunker AK, Lawson JD, Brown CJ, et al. Intrinsically disordered protein. J Mol Graph Model. 2001;19(1):26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 63.Ptitsyn OB. Molten globule and protein folding. Adv Protein Chem. 1995;47:83–229. doi: 10.1016/s0065-3233(08)60546-x. [DOI] [PubMed] [Google Scholar]
- 64.Meador WE, Means AR, Quiocho FA. Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science. 1993;262(5140):1718–1721. doi: 10.1126/science.8259515. [DOI] [PubMed] [Google Scholar]
- 65.Tuma R, Coward LU, Kirk MC, Barnes S, Prevelige PE., Jr Hydrogen-deuterium exchange as a probe of folding and assembly in viral capsids. J Mol Biol. 2001;306(3):389–396. doi: 10.1006/jmbi.2000.4383. [DOI] [PubMed] [Google Scholar]
- 66.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293(2):321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 67.Iakoucheva LM, Radivojac P, Brown CJ, et al. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32(3):1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schalch T, Duda S, Sargent DF, Richmond TJ. X ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 2005 ;436(7047):138–141. doi: 10.1038/nature03686. [DOI] [PubMed] [Google Scholar]; •• First low-resolution structure of a compacted tetranucleosome provides additional evidence for the two-start model organization of the chromatin fiber first confirmed by [3].
- 69.Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11 (4):281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 70.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 71.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Websites
- 101.Marino-Ramirez L, Hsu B, Baxevanis AD, Landsman D NHGRI/NCBI. http://research.nhgri.nih.gov/histones (Viewed September 2005)
- 102.Delano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. http://www.pymol.org (Viewed September 2005) [Google Scholar]