

Abstract
Myeloperoxidase (MPO) is an oxidant-generating enzyme expressed in macrophages and implicated in atherosclerosis and cholesterol homeostasis. LXRα and PPARα regulate genes involved in cholesterol metabolism and the inflammatory response in macrophages. Here we examine the effect of LXR and PPARα ligands on MPO expression. LXR and PPARα, as heterodimers with RXR, are shown to bind overlapping sites in an Alu receptor response element (AluRRE) in the MPO promoter. The LXR ligand T0901317 suppresses MPO mRNA expression in primary human macrophages, and in bone marrow cells and macrophages from huMPO transgenic mice. The PPARα ligand GW9578 downregulates MPO expression in GMCSF- macrophages, while upregulating in MCSF-macrophages. In contrast, the mouse MPO gene, which lacks the primate-specific AluRRE, is not regulated by LXR or PPARα ligands. These findings identify human MPO as a novel LXR and PPARα target gene, consistent with the role of these receptors in regulation of proinflammatory genes in macrophages.
Keywords: myeloperoxidase, MPO, atherosclerosis, nuclear receptors, LXR, PPARα, PPARγ, transgenic, macrophage, cholesterol
Introduction
Atherosclerosis is a chronic inflammatory disease in which macrophages accumulate in the arterial wall [1–3]. An initiating event is the entrapment of LDL-cholesterol particles in the vascular intima at sites of shear stress. Oxidation of the LDL particles signals the overlying endothelial cells to express cytokines and cell surface receptors, inducing circulating monocytes to bind and traverse the endothelial layer to the intima. The monocytes thereupon differentiate into macrophages, engulfing large amounts of oxidized LDL (oxLDL) to become lipid-laden foam cells. Eventual apoptosis of foam cells contributes oxidized lipids and cell debris to the necrotic core of the plaque whose rupture releases tissue factor, causing coagulation and thrombosis.
Myeloperoxidase (MPO) has been implicated in the initiation and progression of atherosclerotic plaques. MPO catalyzes a reaction between hydrogen peroxide and chloride to generate potent oxidants including hypochlorous acid (HOCl), while reacting with nitric oxide and nitrite to produce reactive nitrating species (RNS)[4, 5]. MPO and its oxidant byproducts are detected in lesions, colocalizing with foam cell macrophages [6, 7], and are especially abundant at sites of thrombosis [8], suggesting MPO-oxidants destabilize vulnerable plaques. MPO oxidizes LDL to create a more atherogenic form taken up more effectively by macrophage scavenger receptors, CD36 and SR-A1, generating foam cells. MPO further enhances foam cell formation by oxidizing ApoA1, the major protein component of HDL, impairing ABCA-1 mediated cholesterol efflux from foam cells [9, 10]. In addition, MPO scavenges nitric oxide, inhibiting the induction of iNOS gene expression [11] and impairing vasodilation [4].
Higher levels of MPO gene expression are linked to increased risk of atherosclerosis. A functional MPO promoter polymorphism, −463 G/A, alters expression levels [12, 13], and is associated with increased incidence of CAD [14–16] and severity of atherosclerosis [17, 18]. Elevated serum MPO is an indicator for heightened risk of coronary artery disease (CAD) events [19, 20], while individuals with inherited MPO deficiencies have reduced cardiovascular disease [21]. Members of the nuclear receptor superfamily, notably PPARγ and estrogen receptor, have been found to play key roles in regulating human MPO gene expression [12, 13].
Mouse models greatly facilitate investigations into the regulation of genes involved in atherosclerosis. However, unlike human MPO, the mouse MPO gene is not expressed in foam cell macrophages at lesions in murine models for atherosclerosis, such as LDL receptor deficient mice (LDLR−/−)[22]. One possible explanation for this species difference is the presence of a PPARγ binding site in an Alu element preceding the human MPO gene. PPARγ is a member of the nuclear receptor superfamily of ligand-dependent transcription factors, expressed in foam cell macrophages at lesions, and known to regulate genes involved in the macrophage inflammatory response including MPO[13], inducible nitric oxide synthase (iNOS), TNFα, and interleukin IL-1B, as well as genes involved in cholesterol efflux and uptake, including ApoE, LXR, and CD36 [23–27]. PPARγ ligands strongly induce (~20 fold) human MPO expression in MCSF- Mφ, while suppressing MPO in GMCSF-Mφ (~20 fold)[13]. This opposite regulation may reflect differences in the constellation of coactivators and corepressors in MCSF- versus GMCSF-derived macrophages. Estrogen receptor binds an adjacent site in the AluRRE, and its ligand 17β-estradiol (E2) blocks the effects of PPARγ, especially on the MPOA allele in which the −463A mutation creates a stronger ER binding site[13, 28]. Because Alus are primate specific, the mouse MPO gene lacks the Alu encoded PPARγ site, and is not regulated by PPARγ ligands.
Liver X receptor alpha (LXRα) is expressed in macrophages, and along with PPARγ, regulates genes involved in cholesterol homeostasis and inflammation[29, 30]. LXRs are cholesterol sensors whose natural ligands are cholesterol metabolites, oxysterols, such as 22(R)-hydroxycholesterol [31]. In the liver, LXRs (LXRα and LXRβ) upregulate genes involved in cholesterol metabolism [32, 33] inducing cholesterol 7-hydroxylase (Cyp-7a), which promotes conversion of cholesterol into bile acids for excretion. LXR also increases expression of hepatic transporters ABCAG5/ABCG8 which excrete cholesterol through bile [34]. LXR agonists have broad anti-inflammatory effects, repressing genes involved in the immune response of macrophages, including inducible nitric oxide synthase (iNOS), interleukins IL-1 and IL-6, monocyte chemotactic protein (MCP)-1, and cyclooxygenase-2 (COX-2)[29, 33]. In macrophages, LXRs also modulate expression of genes involved in lipid metabolism, increasing expression of ApoE and the transporter ABCA-1, thereby enhancing ApoA-1 mediated cholesterol efflux to HDL[32]. In LDLR−/ − mice fed a high fat diet, the synthetic LXR agonist T0901317 increased ABCA1 expression in existing atherosclerotic lesions, causing regression of plaques [35]. Macrophage-LXR expression was shown to be required for the atheroprotective effects of LXR agonists [30, 35].
PPARα regulates genes involved in lipid metabolism and transport, and has anti-atherogenic effects in macrophages [27, 36]. Endogenous ligands include polyunsaturated fatty acids and eicosanoids, as well as oxidized LDL. In foam cell macrophages, PPARα enhances cholesterol efflux by increasing expression of ApoA1 as well as LXR, which promotes expression of ABCA1, enhancing cholesterol efflux to HDL[37].
To facilitate investigation of the role of MPO in murine models of atherosclerosis, we generated transgenic mice expressing the human MPO gene under control of the native human MPO promoter. In a previous study, these huMPO transgenic mice were crossed onto the LDLR−/− background and fed a high fat Western diet, resulting in larger atherosclerosis lesions, as well as hypercholesterolemia, hypertriglycerolemia, and obesity in males[38]. Considering the central role of LXR and PPARα in regulation of genes involved in lipid metabolism [32, 33, 36], we investigated the ability of these receptors to regulate MPO gene expression. Findings show that LXR-RXR and PPARα-RXR bind overlapping sites in the AluRRE. The synthetic LXR agonist T0901317 strongly downregulated the human MPO gene in human monocyte-macrophages, as well as in huMPO transgenic mouse macrophages. PPARα ligand GW9578 upregulated MPO in MCSF-Mφ while downregulating in GMCSF-Mφ. In contrast, LXR and PPARα ligands had no effect on the mouse MPO gene which lacks the primate specific AluRRE with LXR binding site.
Materials and Methods
Isolation and Culture of Human Peripheral Blood Mononuclear Cells
Peripheral blood mononuclear cells (PBMC) were isolated from 500 mls of whole blood from healthy donors. A blood bank provided the cells without personal identifiers as a 50 ml concentrate obtained by low speed centrifugation. Collection of samples from human donors and anonymous samples from the blood bank were approved by our affiliated IRB (institutional review board). The concentrated leukocytes in 20 ml volume were layered over 20 ml of Ficoll Hypaque (Lymphoprep, Axis Shield) and centrifuged for 30 min at 900g. The interphase cell layer was again centrifuged over Lymphoprep, and the interphase layer was diluted with three volumes of RPMI medium 1640 (GIBCO) and collected by centrifugation for 30 min at 900g. The cell isolate includes monocytes, macrophages, and lymphocytes, and is largely depleted of neutrophil/granulocytes. The cells were resuspended in 40 ml of RPMI medium supplemented with 200 mM L-glutamine, 10,000U/ml PenicillinG, 10,000ug/ml streptomycin, non-essential amino acids (Irvine Scientific), 1X Fungizone (GIBCO), and 10% fetal calf serum (Hyclone). The human serum for each donor was heat inactivated (55°C, 30 min) and added to the culture medium at 10% volume. The cells were plated in 24-well tissue culture plates (106 cells/well) in 400 μl volume with human GMCSF (Sigma) (10 ng/ml) or human MCSF (R&D Systems) (10 ng/ml) in a humidified CO2 (5%) incubator at 37°C. The cells were incubated for 24h prior to addition of PPARγ ligands for 24 additional hours, followed by harvesting.
Culture of Monocyte Derived Macrophages
To obtain monocyte-derived macrophages, PBMC were seeded at 106 cells/400 μl/well in 24 well plates in RPMI with 10%FCS and 10% autologous human serum, along with recombinant human GMCSF (10 ng/ml) or MCSF (10 ng/ml). Where indicated in the figures, 17β-estradiol (Sigma) (10−7M) was added. On days 2 and 3, the medium was supplemented with 100 μl of fresh medium containing GMCSF or MCSF (10 ng/ml). On days 4 through 6, medium was exchanged daily, without human serum, with GMCSF at 5 ng/ml or MCSF at 5 ng/ml, and where indicated, 17β-estradiol (10−7M). On day 7, fresh medium with ligands was added for 24 additional hours prior to harvesting. The resultant cell population was uniformly adherent, with macrophage morphology, and 98% were positive for CD68 by immunostaining.
RNA Isolation and Quantitation by TaqMan Real-time PCR
Total RNA was isolated from cells by Trizol reagent (Invitrogen). Medium was aspirated and 400 μl Trizol added directly to the adherent macrophages in 24 well plates. For PBMC, Trizol was added to combined nonadherent and adherent cells. The RNA was reverse transcribed with the Omniscript RT Kit (Qiagen) and random hexamer primers. Five μl of the 20 μl cDNA reaction volume was used in Real-time quantitative PCR using ABI PRISM 7900 (Perkin Elmer Applied Biosystems) using Taqman Master mix and primers. Normalization was to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) for human mRNA and beta 2-microglobulin for mouse mRNA.
Probes and primers were designed by ABI Primer Express software and obtained from PE Biosystems. To prevent amplification of genomic DNA, primer sequences were designed to cross exon-intron boundaries. PCR was performed in multiplex (both target and endogenous control coamplified in the same reaction). The sequences for primers and probes used in this study are as follows: Human MPO forward 5′ TTTGACAACCTGCACGATGAC-3′; reverse 5′-CGGTTGTGCTCCCGAAGTAA-3′, and probe 5′CCGTTCCAGTGAGATGCCCGAGC-3′. Mouse MPO forward 5′-AACATGCAGCGCAGCCGG-3′; reverse 5′-AGCCCACAAAAGCGTCTC-3′; and probe 5′-CCTCCCAGGATACAATGC-3′. Mouse beta-2-microglobulin. Forward 5′-CCGAGCCCAAGACCGTCTA-3′, reverse 5′-CTGGATTTGTAATTAAGCAGGTTCAA-3′, and probe 5′-TGATGCTTGATCACATGTCTCGATCCCA-3′. Primers and probe for human glyceraldehyde-3-phosphate dehydrogenase (GAPDH), used as endogenous control, were purchased as kits from Applied Biosystems (Assays on Demand).
Generation of human MPO -463 G and A Transgenic Mice
Transgenic mice carrying the human G or A alleles were created by microinjection of C57BL6/J eggs with a 32 kb BST11071 restriction fragment including the gene with 7 kb of upstream and 11 kb of downstream sequence. The G allele was isolated from a sequenced BAC clone (AC004687). The A allele was obtained by hybridization screening of a genomic BAC library from Research Genetics. Characterization of the transgenic mice has been previously reported [13, 38].
Isolation of Mouse Bone Marrow Cells and Cell Culture
Bone marrow cells were plated at a density of 106 cells/well in 24 well plates in RPMI with mouse GMCSF (R&D Systems) (10 ng/ml) or mouse MCSF (R&D Systems) (10 ng/ml). Cells were incubated for 24h prior to addition of ligands for 24 additional hours in the continued presence of GMCSF or MCSF.
To obtain bone marrow derived macrophages, bone marrow cells were incubated for 24 hours in 25cm2 flasks containing 5ml of RPMI with GMCSF at 5ng/ml or MCSF at 5ng/ml. Non-adherent cells were seeded at 106 cells/400 μl/well in 24 well plates and incubated for 7 days. The medium was supplemented on days 2 and 3 with 100 μl fresh medium with GMCSF or MCSF (10 ng/ml). The medium was exchanged on day 4, 5, and 6 to remove nonadherent cells. Where indicated, β-estradiol was present throughout at 10−7M. Adherent cells at day 7 were homogeneous in morphology, consisting of mononuclear cells with abundant cytoplasm.
Electrophoretic Mobility Shift Assay (EMSA)
Human LXRα and RXRα proteins were synthesized in vitro from the corresponding expression plasmids in rabbit reticulocyte lysate by using the TNT transcription/translation system (Promega). To obtain an unprogrammed lysate as a negative control for EMSA, a reaction was performed with the empty vector, pCDNA3 (Invitrogen). Double stranded oligonucleotides were radiolabeled at overhanging ends using Klenow polymerase, and gel-purified. Proteins were incubated with poly (dI-dC) in binding buffer containing 20mM Hepes, pH 7.9, 50mM KCl, 1mM MgCl2, 1mM DTT, 10% glycerol, 0.2 μg/□l BSA for 10 min at 4°C. 32P labeled probe (100K cpm) was then added and incubation continued for 30min at 4°C. Samples were electrophoresed at 4°C in a 5% polyacrylamide gel in 0.5X TBE buffer (45mM Tris, 45mM boric acid, 1mM EDTA, pH 8.0). Oligonucleotide sequences for probes used in this assay have been described elsewhere [13]
Transient transfections and luciferase assay
The double stranded oligonucleotide containing the MPO-G AluRRE element with BamH1 overhang (gatccAGGCTGAGGCGGGTGGATCACTTGAGGTCAGGAGTTCAAACCAg; the hexamer sequences are underlined) was cloned in sense orientation into the Bgl II site of a pGL2-based luciferase reporter containing the minimal thymidine kinase promoter. Constructs were confirmed by sequencing. 5x104 CV-1 or HeLa cells were seeded in 24 well plates the day before transfection. Cells were co-transfected with 25 ng each of LXR/PPARα and RXR expressing pcDNA-derived vectors, 100 ng of MPO-G-luciferase reporter, 150 ng of β-galactosidase expression vector, and 700 ng pBluescript using a calcium phosphate precipitation procedure for 16 hours. After transfection, cells were incubated for an additional 24 hours in medium containing 10% charcoal-treated serum supplemented or not with the ligands indicated in the figures. Cell extracts were prepared and assayed for firefly luciferase, which was normalized with galactosidase activity.
Statistical Analysis
The real time PCR scores were normalized to endogenous control genes. Activated conditions were compared to non-activated conditions normalized to 1 X and compared by Student’s t test using Statview software. A value of P < 0.05 was considered significant.
RESULTS
LXRα-RXR and PPARα-RXR bind hexamer pairs in an AluRRE in the MPO Promoter
The Alu receptor response element (AluRRE) upstream of the MPO gene is typical of the major Alu subclass (Sx) with four hexamer halfsites related to the consensus AGGTCA, and recognized by members of the nuclear receptor superfamily of ligand-dependent transcription factors [12, 13, 39]. The hexamers are arranged as direct repeats with spacing of 2, 4, and 2 bp (36) (Fig. 1A). We previously showed that PPARγ-RXR heterodimers bind the third and fourth hexamers, while estrogen receptor (ER) homodimers bind the first two hexamers [13]. The −463A mutation in hexamer 1 enhances binding by ER [13, 28]. ER ligand, 17β-estradiol, blocks some of the effects of PPARγ ligands on MPO expression, especially on the MPO-463A allele with stronger ER binding site.
Figure 1. LXRα-RXR and PPARα-RXR heterodimers bind the AluRRE in the MPO promoter.
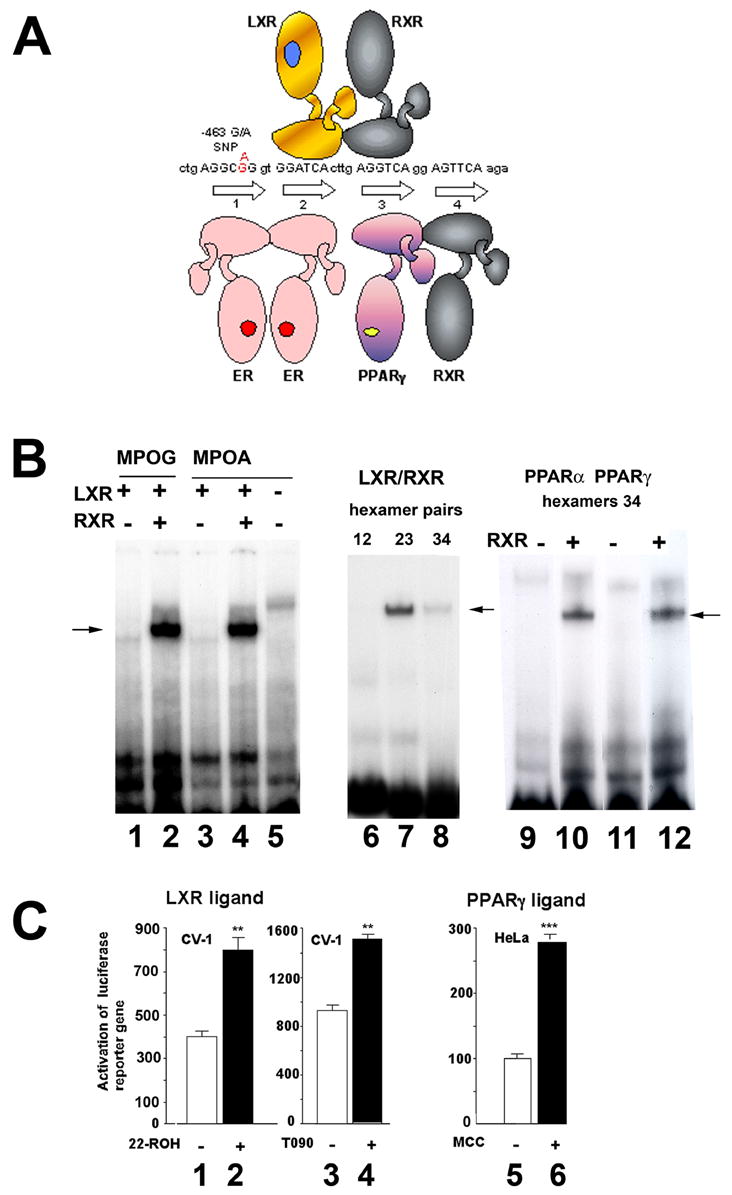
A. Diagrammatic representation of nuclear receptors bound to the MPO AluRRE. DNA sequence shows the four hexamer halfsites with binding sites for estrogen receptor homodimers (ER), LXR-RXR and PPARγRXR heterodimers. The −463G/A polymorphism (red) is in hexamer 1.
B. Double stranded DNA oligonucleotides encoding the four hexamers of the AluRRE with −463G or A in hexamer 1 were incubated with rabbit reticulocyte lysate programmed with in vitro synthesized mRNA encoding LXRα and/or RXR□ (lanes 1–4), or nonprogrammed lysate (lane 5). The arrow indicates the position of a retarded complex for the MPO-G and MPO-A promoter sequences. In lanes 6–8, oligonucleotides (22 bp) encoding the individual hexamer pairs 1/2, 2/3, or 3/4 were incubated with lysates programmed with LXR and RXR expression vectors. In lanes 9–12, hexamers 3/4 were incubated with lysates programmed with PPARα (lanes 9,10) or PPARγ (lanes 11,12), and RXR expression vectors as indicated. Arrows indicate the position of the retarded complexes.
C. CV-1 cells were cotransfected with 100 ng of luciferase reporter constructs containing the AluRRE sequence along with expression vectors for LXRα (25 ng) and RXRα (25 ng) (lanes 1–4). HeLa cells were cotransfected with 100 ng of luciferase reporter containing the AluRRE along with expression vectors for PPARγ (25 ng) and RXRα (25 ng) (lanes 5,6). After 24 h, cells were untreated (lanes 1, 3, 5) or treated with LXR ligands, 25 μM 22-ROH (lane 2) or 1 μM T0901317 (lane 4), or PPARγ ligand, 10 μM MCC-555 (lane 6) for 24 h, and then assayed for luciferase activity. Average values and standard error measurements were calculated from triplicate measurements. Statistically significant differences between the control and ligand-activated conditions were determined by Student’s t test (**p<0.01; ***p<0.005).
The middle hexamer pair with DR-4 spacing presented a potential site for LXR-RXR binding. To test for binding by LXR to the MPO AluRRE, electrophoretic mobility shift assays (EMSA) were carried out with LXRα produced in rabbit reticulocyte lysates programmed with in vitro synthesized mRNA. Synthesized receptors were incubated with 32P- labeled oligonucleotides including the AluRRE with the four hexamers (Fig.1A). LXR was found to bind the AluRRE with either −463G or A, requiring co-presence of RXR (Fig. 1B, lanes 1–5). To further define the recognition site, we tested binding to the individual hexamer pairs, and found that LXR-RXRbinds the DR-4 hexamer pair 2/3, with little binding to hexamers 3/4, and no binding to hexamers 1/2 (lanes 6–8).
We previously reported PPARγ-RXR binding to hexamer 3/4 of the MPO AluRRE[13]. Here we compared binding by PPARα and PPARγ and found that both bound to hexamer pair 3/4, requiring RXR (lanes 9–12).
To determine whether the LXR binding site in the AluRRE functions as a LXR response element (LXRE), the AluRRE was synthesized and inserted upstream of a luciferase reporter gene, and transiently transfected into CV-1 monkey kidney cells, in the presence of cotransfected expression vectors encoding LXRα and RXRα, and the presence or absence of LXR ligands, including the oxysterol, 22R-hydroxy-cholesterol (22-ROH) or the synthetic ligand T0901317 (T090) (Fig. 1C). 22-ROH increased expression of the reporter gene by 2 fold (lanes 1,2), while T090 increased expression by 1.7 fold (lanes 3,4). As a comparison, we assayed the effect in cotransfection assays of a PPARγ ligand of the thiazolidinedione class, MCC-555. PPARγ-RXR was previously demonstrated to bind hexamers 3/4 of the AluRRE, and PPARγ ligand rosiglitazone was shown to strongly upregulate the native human MPO gene in MCSF-Mφ, and downregulate in GMCSF-Mφ [13]. Here we tested whether hexamers 3/4 act as a PPAR response element (PPARE) when linked to a reporter gene in cotransfection assays. HeLa cells were transfected with expression constructs encoding PPARγ and RXRα, in the presence or absence of PPARγ ligand MCC-555. MCC-555 increased transcription of the reporter gene by 2.7 fold. These findings indicate the MPO AluRRE is able to function as an LXRE and a PPARE for a reporter gene construct in cotransfection assays.
LXR ligand T0901317 inhibits MPO expression in primary human monocytes and monocyte derived-macrophages
We next investigated whether LXR ligand T090 regulates expression of the human MPO gene in vivo. Our previous study revealed that PPARγ ligands downregulate MPO in GMCSF-Mφ, while upregulating in MCSF-Mφ[13]. Here we tested whether LXR ligand T090 had different effects in MCSF- versus GMCSF-treated monocytes or monocyte-derived macrophages (MD- Mφ). Primary human monocytes were cultured for 24 h in the presence of MCSF or GMCSF, prior to 24 h treatment with T090 (1 μM), prior to isolation of mRNA and quantitation of MPO mRNA levels by quantitative real time PCR. MD- Mφ were obtained by seven days culture in medium containing GMCSF or MCSF, prior to treatment for 24 h with T090 (1 μM). LXR ligand T090 consistently downregulated MPO mRNA levels by 7 to 15 fold (Fig 2, lanes 1–8), regardless of CSF. We previously reported that 17β-estradiol (E2) blocked the upregulation of MPO in MCSF-Mφ by PPARγ ligands [13]. In contrast, the addition of E2 did not block MPO suppression in MCSF-Mφ by LXR ligands (lanes 9,10). As a further control, we tested the effect of LXR ligand T090 on expression of ABCA1 in these primary MD-Mφ, and T090 was found to upregulate ABCA-1 by 8 fold (lanes 11,12), consistent with prior reports.
Figure 2. LXR ligand T0901317 downregulates human MPO mRNA expression in primary monocyte-macrophages.
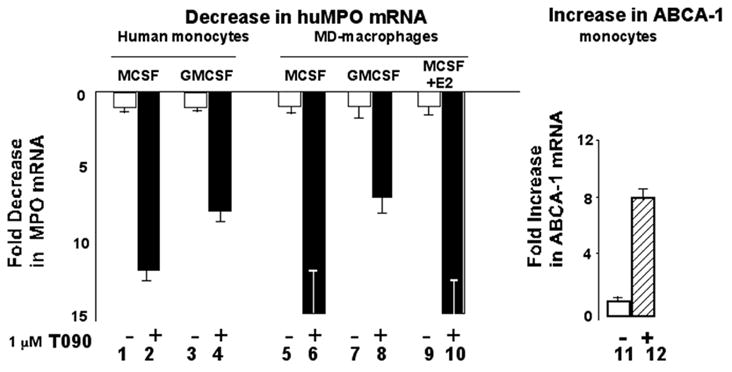
Human monocytes were cultured for 24 h in the presence of MCSF or GMCSF, prior to 24 h culture in the absence or presence of 1 μM T090. Monocyte derived macrophages were obtained by seven days culture in MCSF or GMCSF, prior to 24 h culture in the absence or presence of 1 μM T090. In lanes 9 and 10, 17β-estradiol (10−7M) was added to the MCSF-treated macrophages throughout treatment. MPO mRNA levels (lanes 1–10) or ABCA1 mRNA levels (lanes 11,12) were determined by quantitative real time PCR. Values shown are the means of two independent determinations ± SEM.
LXR ligand T0901317 suppresses human MPO mRNA expression in macrophages from transgenic huMPOG and A mice
We next examined the effects of LXR ligands on the human MPO −436G and A alleles in transgenic mice, TgMPO-G and TgMPO-A. The human MPO transgenes are each present in one copy in the transgenic mice, and are driven by the native MPO promoter, having been generated by microinjection of C57BL6/J eggs with a 32 kb restriction fragment centered on the intact MPO-463G or A genes (MPOG and MPOA), thus each transgene includes 4 to 11 kB of flanking regions[13]. The regulation of the huMPOG and A transgenes in the transgenic mice is like that observed in humans, with highest expression in bone marrow precursors.
Bone marrow cells from the transgenic mice were cultured in RPMI medium with 10% FCS and T090 (1 μM) for 24 hours, prior to isolation of mRNA. Treatment with T090 downregulated MPO-G and MPO-A mRNA levels by approximately five fold (Fig. 3A, lanes 1–4). We tested the effect of T090 on MPO expression in macrophages, derived by culture of bone marrow cells for seven days with GMCSF or MCSF. In GMCSF -Mφ, T090 reduced MPOG mRNA expression by 7 ±1.3 fold, and MPO-A expression by 10 ±1.7 fold (lanes 5–8). In MCSF-Mφ, T090 reduced MPO-G mRNA expression by 12 ±2 fold, and MPO-A by 20 ±3 fold (lanes 9–12). Thus, T090 repressed both the MPOG and MPOA alleles, in either GMCSF or MCSF conditions. As with PPARγ, LXR ligand had no effect on mouse MPO mRNA expression in bone marrow cells or macrophages (Fig. 2A, lanes 13–16).
Figure 3. LXR and PPARγ/α ligands differentially regulate the human MPO gene in transgenic mice, but do not regulate the mouse MPO gene.
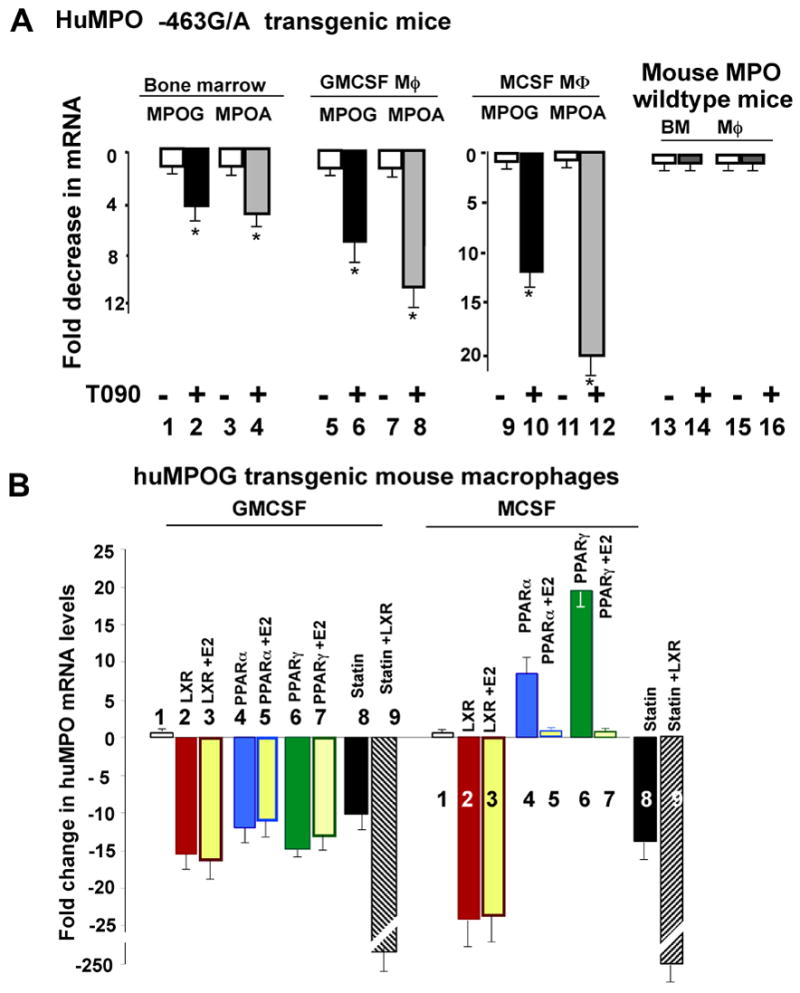
A. LXR ligand T090 (1 μM) suppresses human MPO gene expression in bone marrow cells (lanes 1–4), GMCSF- Mφ (lanes 5–8), and MCSF- Mφ (lanes 9–12) from TgMPO-G and TgMPO-A transgenic mice, but has no effect on expression of the mouse MPO gene (lanes 13–16). Statistically significant differences between the control and ligand-activated conditions were determined by Student’s t test (*p<0.05).
B. GMCSF –Mφ and MCSF- Mφ from human TgMPO-G transgenic mice were untreated (lanes 1) or treated with LXR ligand T090 (lanes 2,3), PPARα ligand GW9578 (lanes 4,5), PPARγ ligand rosiglitazone (5 μM) (lanes 6,7), simvastatin (25 μM) (lane 8), or simvastatin and T090 (lane 9). 17β-estradiol was added to the culture medium three hours prior to ligands in lanes 3,5,7.
In figure 3B, we contrast the regulation of MPO expression by LXR and PPAR α/γ ligands. LXR ligand T090 reduced MPO gene expression in either GMCSF or MCSF(lanes 2), and estrogen had no effect on this suppression (lanes 3). PPARα ligand GW9578 downregulated TgMPO-G in GMCSF-Mφ, while upregulating in MCSF-Mφ (lanes 4). The upregulation in MCSF was blocked by E2, but not the downregulation in GMCSF (lanes 5). The findings with PPARα ligand are thus consistent with prior findings with PPARγ ligand [13]. As shown here for comparison. PPARγ ligand rosiglitazone increased MPO expression in MCSF-Mφ and inhibited expression in GMCSF-Mφ (lanes 6), with E2 blocking the upregulation in MCSF-Mφ but not GMCSF (lanes 7).
LXR ligand T090 represses human MPO but does not affect mouse MPO gene expression, indicating this repression is mediated by promoter sites specific to human MPO. In contrast, statins downregulate both the human and mouse MPO genes[11], presumably acting through common promoter elements, therefore not the primate-specific AluRRE. Statins inhibit MPO gene expression by blocking HMG-CoA reductase, thereby blocking production of isoprenoid intermediates of the mevalonate pathway, including geranylgeranylpyrophosphate (GGPP) which is required for MPO gene expression[11]. Like LXR, statins inhibit MPO in either GMCSF or MCSF-treated cells, and this inhibition is not blocked by E2. This led us to examine whether there might be common pathways or transcription factors utilized by LXR and statins to suppress MPO. However, when macrophages were co-treated with T090 and simvastatin, the suppressive effect was more than additive, suggesting the mechanisms of suppression are independent and synergistic (Fig. 3B, lanes 8,9)
Discussion
These findings provide the first evidence that LXR and PPARα ligands inhibit expression of the human MPO gene. The synthetic LXRα ligand T0901317 and the PPARα ligand GW9578 repress MPO mRNA expression in primary human macrophages, and in huMPO transgenic bone marrow cells and BM-Mφ. These findings identify MPO as a novel member of the set of macrophage genes regulated by LXR and PPARα and involved in the macrophage inflammatory response and cholesterol metabolism.
These LXR and PPARα ligands downregulate human MPO, but not mouse MPO, indicating the relevant promoter elements are specific to the human MPO gene. One possibility is the LXR-RXR and PPARα-RXR binding sites in the AluRRE. Alu elements are primate specific, thus absent from the mouse MPO promoter. The MPO AluRRE contains four direct repeat (DR) hexamer halfsites related to the consensus AGGTCA, spaced by 2, 4, and 2 basepair (DR-2 or DR-4). LXR-RXR binds the central DR-4 pair (hexamers 2/3) while PPARγ/α-RXR binds the overlapping DR-2 pair (hexamers 3/4), and ER binds the first pair (hexamers 1/2) which includes the -463G/A polymorphism (Figure 1A). Cotransfection experiments showed the AluRRE functions as an LXRE when placed upstream of a minimal promoter and reporter gene, and cotransfected into CV1 cells along with LXR and RXR expression constructs. The AluRRE also functions as an PPARE in cotransfection experiments with PPARγ and RXR expression constructs. These cotransfection experiments showed modest ligand-dependent increases in reporter gene expression in CV-1 (monkey kidney) cells, in contrast to the strong repression by LXR ligands for the native MPO gene in primary macrophages. One possible explanation is that LXR ligand-dependent repression is mediated through sites other than the AluRRE. For example, LXR inhibits expression of iNOS and Cox-2, although these genes appear to lack LXR-RXR binding motifs, suggesting this repression is indirect. The suggested mechanism is transrepression, and refers to the ability of nuclear receptors to associate with other transcription factors, notably NF-κB or AP1, and recruit corepressors, such as N-CoR, which antagonize the activities of these factors[40, 41].
Our previous study showed that PPARγ ligands induce MPO in MCSF-Mφ, and downregulate in GMCSF-Mφ [13]. Estrogen blocks the induction in MCSF for both the MPO-G and MPO-A alleles, while blocking downregulation of MPO-A, but not MPO-G, in GMCSF -Mφ. The preferential block of PPARγ effects on MPOA correlates with the higher affinity ER binding site at −463A [13, 28]. These findings suggested that ER binding to hexamers 1/2 inhibits PPARγ-RXR binding or activity on the adjacent hexamer 3/4. In contrast, estrogen does not block suppression of MPO by LXR ligand, possibly due to the overlapping binding sites for LXR-RXR and ER in the AluRRE. ER binds hexamers 1/2 while LXR-RXR binds hexamers 2/3, thus sharing hexamer 2. Thus, LXR-RXR binding may preclude ER binding.
Nuclear receptors such as LXR and PPARγ are able to transport to the nucleus and bind their cognate hexamer halfsites in the presence or absence of ligand. In the absence of ligand, the receptors become associated with corepressors, such as nuclear receptor corepressor (N-CoR) or silencing mediator of retinoid and thyroid receptors (SMRT), which are part of multiprotein complexes including histone deacetylases, which antagonize the basal transcriptional machinery [40]. Ligand-binding causes a conformational change in the receptors that releases the corepressors, allowing binding of coactivators, which induce target gene expression. In the present study, we observed ligand-dependent repression of MPO with LXR ligand T090, and with PPARγ or PPARα ligands in GMCSF- Mφ. This finding is unusual in that ligand-dependent activation is usually observed for receptors bound to two core recognition motifs. One possible explanation is that the inhibition may be due to transrepression, in which LXR or PPAR α/γ bind to NF-κB or AP1, rather than acting throught the AluRRE. An alternative explanation is that these receptors are able to repress the target gene in the specific context of the AluRRE, or the MPO basal promoter, or in GMCSF- treated macrophages, The current models of receptor/coregulator interations were developed through analysis of a small set of target genes, and the outcome (agonist or antagonist) might differ in vivo in a complex inflammatory state with multiple competing receptors, and might change depending on the specific cell type or the specific promoter context [40]. Macrophages derived by treatment with MCSF or GMCSF may have different levels of coactivators and corepressors, resulting in MPO induction by PPARα/γ in MCSF, and repression in GMCSF. Some LXR ligands induce receptor conformations that enhance associations with both corepressors and coactivators, such that the net effect of the ligand-bound receptor on target gene expression may depend on the prevalence of coactivators or corepressors in a particular cell type[42]. Other studies suggest that minor differences in DNA sequence, spacing, or orientation of the hexamer motifs can alter the interactions between receptors and corepressors/coactivators [43–45], determining whether ligand binding has positive or negative effects on target gene expression [46]. The unusual arrangement of the several hexamer halfsites in the AluRRE, some with noncanonical spacing or DNA sequences[12, 39], could influence receptor conformations and coregulator interactions. For example, the PPAR γ/α site is DR-2, while the canonical binding site is DR-1. Also, the canonical binding motif for ER homodimers is an inverted repeat, while an inverted repeat motif is not apparent in the AluRRE. Non-canonical binding sites have previously been reported for PPAR and ER [47], and these sequence variants could alter receptor conformation and thereby coregulator interactions. Thus, it remains unclear at present whether the repression of MPO by LXR ligand, or the activation/repression by PPARγ/α ligands is mediated through the AluRRE halfsites, or through transrepression. One possibility is that activation in MCSF is mediated by PPARγ/α binding to the AluRRE, while the inhibition in GMCSF is due by transrepression. Although beyond the scope of this study, it will be interesting to further decipher the interactions of receptors through the AluRRE: There are approximately one million Alu elements in the human genome, and most had the several hexamer binding sites at the time of insertion [39], therefore many genes are likely to be regulated by nuclear receptors through AluRRE.
In summary, these findings identify human MPO as a novel LXR and PPARα target gene, consistent with the role of these receptors in regulation of inflammatory and atherogenic genes in macrophages.
Acknowledgments
We thank Magnus Pfahl and Maxocore for providing PPARalpha ligand GW9578, and we thank our administrative assistant Carrie Baum. This research was supported by grants from the National Institutes of Health, AG-017879 and AG-026539, to W.F.R.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stocker R, Keaney JF., Jr Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 3.Libby P. Atherosclerosis: the new view. Sci Am. 2002;286:46–55. doi: 10.1038/scientificamerican0502-46. [DOI] [PubMed] [Google Scholar]
- 4.Eiserich JP, Baldus S, Brennan ML, Ma W, Zhang C, Tousson A, Castro L, Lusis AJ, Nauseef WM, White CR, Freeman BA. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science. 2002;296:2391–2394. doi: 10.1126/science.1106830. [DOI] [PubMed] [Google Scholar]
- 5.Baldus S, Eiserich JP, Brennan ML, Jackson RM, Alexander CB, Freeman BA. Spatial mapping of pulmonary and vascular nitrotyrosine reveals the pivotal role of myeloperoxidase as a catalyst for tyrosine nitration in inflammatory diseases. Free Radic Biol Med. 2002;33:1010. doi: 10.1016/s0891-5849(02)00993-0. [DOI] [PubMed] [Google Scholar]
- 6.Daugherty A, Dunn JL, Rateri DL, Heinecke JW. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J Clin Invest. 1994;94:437–444. doi: 10.1172/JCI117342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hazen SL, Crowley JR, Mueller DM, Heinecke JW. Mass spectrometric quantification of 3-chlorotyrosine in human tissues with attomole sensitivity: a sensitive and specific marker for myeloperoxidase-catalyzed chlorination at sites of inflammation. Free Radic Biol Med. 1997;23:909–916. doi: 10.1016/s0891-5849(97)00084-1. [DOI] [PubMed] [Google Scholar]
- 8.Sugiyama S, Okada Y, Sukhova GK, Virmani R, Heinecke JW, Libby P. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol. 2001;158:879–891. doi: 10.1016/S0002-9440(10)64036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergt C, Pennathur S, Fu X, Byun J, O'Brien K, McDonald TO, Singh P, Anantharamaiah GM, Chait A, Brunzell J, Geary RL, Oram JF, Heinecke JW. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc Natl Acad Sci U S A. 2004;101:13032–13037. doi: 10.1073/pnas.0405292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng L, Nukuna B, Brennan ML, Sun M, Goormastic M, Settle M, Schmitt D, Fu X, Thomson L, Fox PL, Ischiropoulos H, Smith JD, Kinter M, Hazen SL. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest. 2004;114:529–541. doi: 10.1172/JCI21109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar AP, Reynolds WF. Statins downregulate myeloperoxidase gene expression in macrophages. Biochem Biophys Res Commun. 2005;331:442–451. doi: 10.1016/j.bbrc.2005.03.204. [DOI] [PubMed] [Google Scholar]
- 12.Piedrafita FJ, Molander RB, Vansant G, Orlova EA, Pfahl M, Reynolds WF. An Alu element in the myeloperoxidase promoter contains a composite SP1-thyroid hormone-retinoic acid response element. J Biol Chem. 1996;271:14412–14420. doi: 10.1074/jbc.271.24.14412. [DOI] [PubMed] [Google Scholar]
- 13.Kumar AP, Piedrafita FJ, Reynolds WF. Peroxisome proliferator-activated receptor gamma ligands regulate myeloperoxidase expression in macrophages by an estrogen-dependent mechanism involving the −463GA promoter polymorphism. J Biol Chem. 2004;279:8300–8315. doi: 10.1074/jbc.M311625200. [DOI] [PubMed] [Google Scholar]
- 14.Asselbergs FW, Reynolds WF, Cohen-Tervaert JW, Jessurun GA, Tio RA. Myeloperoxidase polymorphism related to cardiovascular events in coronary artery disease. Am J Med. 2004;116:429–430. doi: 10.1016/j.amjmed.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 15.Nikpoor B, Turecki G, Fournier C, Theroux P, Rouleau GA. A functional myeloperoxidase polymorphic variant is associated with coronary artery disease in French-Canadians. Am Heart J. 2001;142:336–339. doi: 10.1067/mhj.2001.116769. [DOI] [PubMed] [Google Scholar]
- 16.Pecoits-Filho R, Stenvinkel P, Marchlewska A, Heimburger O, Barany P, Hoff CM, Holmes CJ, Suliman M, Lindholm B, Schalling M, Nordfors L. A functional variant of the myeloperoxidase gene is associated with cardiovascular disease in end-stage renal disease patients. Kidney Int. 2003;Suppl:S172–176. doi: 10.1046/j.1523-1755.63.s84.32.x. [DOI] [PubMed] [Google Scholar]
- 17.Makela R, Laaksonen R, Janatuinen T, Vesalainen R, Nuutila P, Jaakkola O, Knuuti J, Lehtimaki T. Myeloperoxidase gene variation and coronary flow reserve in young healthy men. J Biomed Sci. 2004;11:59–64. doi: 10.1007/BF02256549. [DOI] [PubMed] [Google Scholar]
- 18.Makela R, Karhunen PJ, Kunnas TA, Ilveskoski E, Kajander OA, Mikkelsson J, Perola M, Penttila A, Lehtimaki T. Myeloperoxidase gene variation as a determinant of atherosclerosis progression in the abdominal and thoracic aorta: an autopsy study. Lab Invest. 2003;83:919–925. doi: 10.1097/01.lab.0000077981.49367.46. [DOI] [PubMed] [Google Scholar]
- 19.Zhang R, Brennan ML, Fu X, Aviles RJ, Pearce GL, Penn MS, Topol EJ, Sprecher DL, Hazen SL. Association between myeloperoxidase levels and risk of coronary artery disease. Jama. 2001;286:2136–2142. doi: 10.1001/jama.286.17.2136. [DOI] [PubMed] [Google Scholar]
- 20.Brennan ML, Penn MS, Van Lente F, Nambi V, Shishehbor MH, Aviles RJ, Goormastic M, Pepoy ML, McErlean ES, Topol EJ, Nissen SE, Hazen SL. Prognostic value of myeloperoxidase in patients with chest pain. N Engl J Med. 2003;349:1595–1604. doi: 10.1056/NEJMoa035003. [DOI] [PubMed] [Google Scholar]
- 21.Kutter D, Devaquet P, Vanderstocken G, Paulus JM, Marchal V, Gothot A. Consequences of total and subtotal myeloperoxidase deficiency: risk or benefit? Acta Haematol. 2000;104:10–15. doi: 10.1159/000041062. [DOI] [PubMed] [Google Scholar]
- 22.Brennan M, Gaur A, Pahuja A, Lusis AJ, Reynolds WF. Mice lacking myeloperoxidase are more susceptible to experimental autoimmune encephalomyelitis. J Neuroimmunol. 2001;112:97–105. doi: 10.1016/s0165-5728(00)00392-1. [DOI] [PubMed] [Google Scholar]
- 23.Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93:229–240. doi: 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- 24.Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93:241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 25.Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, Evans RM, Tontonoz P. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7:161–171. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- 26.Lehrke M, Lazar MA. The many faces of PPARgamma. Cell. 2005;123:993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 27.Li AC, Palinski W. Peroxisome proliferator-activated receptors: how their effects on macrophages can lead to the development of a new drug therapy against atherosclerosis. Annu Rev Pharmacol Toxicol. 2006;46:1–39. doi: 10.1146/annurev.pharmtox.46.120604.141247. [DOI] [PubMed] [Google Scholar]
- 28.Reynolds WF, Hiltunen M, Pirskanen M, Mannermaa A, Helisalmi S, Lehtovirta M, Alafuzoff I, Soininen H. MPO and APOEepsilon4 polymorphisms interact to increase risk for AD in Finnish males. Neurology. 2000;55:1284–1290. doi: 10.1212/wnl.55.9.1284. [DOI] [PubMed] [Google Scholar]
- 29.Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. 2003;9:213–219. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- 30.Tangirala RK, Bischoff ED, Joseph SB, Wagner BL, Walczak R, Laffitte BA, Daige CL, Thomas D, Heyman RA, Mangelsdorf DJ, Wang X, Lusis AJ, Tontonoz P, Schulman IG. Identification of macrophage liver X receptors as inhibitors of atherosclerosis. Proc Natl Acad Sci U S A. 2002;99:11896–11901. doi: 10.1073/pnas.182199799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janowski BA, Grogan MJ, Jones SA, Wisely GB, Kliewer SA, Corey EJ, Mangelsdorf DJ. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc Natl Acad Sci U S A. 1999;96:266–271. doi: 10.1073/pnas.96.1.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li AC, Glass CK. PPAR- and LXR-dependent pathways controlling lipid metabolism and the development of atherosclerosis. J Lipid Res. 2004;45:2161–2173. doi: 10.1194/jlr.R400010-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Zelcer N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest. 2006;116:607–614. doi: 10.1172/JCI27883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu L, Hammer RE, Li-Hawkins J, Von Bergmann K, Lutjohann D, Cohen JC, Hobbs HH. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc Natl Acad Sci U S A. 2002;99:16237–16242. doi: 10.1073/pnas.252582399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levin N, Bischoff ED, Daige CL, Thomas D, Vu CT, Heyman RA, Tangirala RK, Schulman IG. Macrophage liver X receptor is required for antiatherogenic activity of LXR agonists. Arterioscler Thromb Vasc Biol. 2005;25:135–142. doi: 10.1161/01.ATV.0000150044.84012.68. [DOI] [PubMed] [Google Scholar]
- 36.Lefebvre P, Chinetti G, Fruchart JC, Staels B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J Clin Invest. 2006;116:571–580. doi: 10.1172/JCI27989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, Teissier E, Minnich A, Jaye M, Duverger N, Brewer HB, Fruchart JC, Clavey V, Staels B. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med. 2001;7:53–58. doi: 10.1038/83348. [DOI] [PubMed] [Google Scholar]
- 38.Castellani LW, Chang JJ, Wang X, Lusis AJ, Reynolds WF. Transgenic mice express human MPO −463G/A alleles at atherosclerotic lesions, developing hyperlipidemia and obesity in −463G males. J Lipid Res. 2006;47:1366–1377. doi: 10.1194/jlr.M600005-JLR200. [DOI] [PubMed] [Google Scholar]
- 39.Vansant G, Reynolds WF. The consensus sequence of a major Alu subfamily contains a functional retinoic acid response element. Proc Natl Acad Sci U S A. 1995;92:8229–8233. doi: 10.1073/pnas.92.18.8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol. 2006;6:44–55. doi: 10.1038/nri1748. [DOI] [PubMed] [Google Scholar]
- 41.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 42.Albers M, Blume B, Schlueter T, Wright MB, Kober I, Kremoser C, Deuschle U, Koegl M. A novel principle for partial agonism of liver X receptor ligands. Competitive recruitment of activators and repressors. J Biol Chem. 2006;281:4920–4930. doi: 10.1074/jbc.M510101200. [DOI] [PubMed] [Google Scholar]
- 43.Diamond MI, Miner JN, Yoshinaga SK, Yamamoto KR. Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science. 1990;249:1266–1272. doi: 10.1126/science.2119054. [DOI] [PubMed] [Google Scholar]
- 44.Zhu P, Baek SH, Bourk EM, Ohgi KA, Garcia-Bassets I, Sanjo H, Akira S, Kotol PF, Glass CK, Rosenfeld MG, Rose DW. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell. 2006;124:615–629. doi: 10.1016/j.cell.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 45.Scully KM, Jacobson EM, Jepsen K, Lunyak V, Viadiu H, Carriere C, Rose DW, Hooshmand F, Aggarwal AK, Rosenfeld MG. Allosteric effects of Pit-1 DNA sites on long-term repression in cell type specification. Science. 2000;290:1127–1131. doi: 10.1126/science.290.5494.1127. [DOI] [PubMed] [Google Scholar]
- 46.Kurokawa R, DiRenzo J, Boehm M, Sugarman J, Gloss B, Rosenfeld MG, Heyman RA, Glass CK. Regulation of retinoid signalling by receptor polarity and allosteric control of ligand binding. Nature. 1994;371:528–531. doi: 10.1038/371528a0. [DOI] [PubMed] [Google Scholar]
- 47.Norris J, Fan D, Aleman C, Marks JR, Futreal PA, Wiseman RW, Iglehart JD, Deininger PL, McDonnell DP. Identification of a new subclass of Alu DNA repeats which can function as estrogen receptor-dependent transcriptional enhancers. J Biol Chem. 1995;270:22777–22782. doi: 10.1074/jbc.270.39.22777. [DOI] [PubMed] [Google Scholar]