

Abstract
Objective
We compared the rate of emergence of thymidine analogue mutations (TAMs) and major protease inhibitor mutations in adherent patients who remained on stable treatment with a thymidine analogue and/or protease inhibitor after the onset of virologic failure.
Design
Follow-up genotypic resistance testing was done using archived plasma obtained from patients having 0 or 1 TAM and/or 0 or 1 major protease inhibitor resistance mutation at the onset of virologic failure.
Results
The median duration of observed failure was 691 days. There were 41 thymidine analogue regimens and 34 protease inhibitor regimens; concomitant ritonavir was used 4 times. New major protease inhibitor mutations emerged more rapidly than did new TAMs (P = 0.0019); new TAMs emerged more rapidly in thymidine analogue regimens that did not include lamivudine (P = 0.0073). The emergence of TAMs and major protease inhibitor mutations did not differ if lamivudine was not part of the thymidine analogue regimen. The evolution of CD4+ cell counts and plasma viral loads (pVLs) during virologic failure was similar regardless of whether or not a new TAM or major protease inhibitor mutations emerged or, for thymidine analogue–containing regimens, whether lamivudine was or was not used.
Conclusions
Major protease inhibitor mutations arose more frequently and rapidly than did TAMs in patients with sustained virologic failure who received lamivudine.
Keywords: HIV resistance, protease inhibitors, resistance mutations, reverse transcriptase inhibitors
Notwithstanding the continued development of new antiretroviral agents with unique profiles of resistance mutations and simpler dosing schedules,1–6 the development of multiclass antiretroviral resistance remains a serious problem for HIV-infected patients.7–12 With the failure of each regimen, the number of remaining effective choices decreases and the virologic threshold for changing antiretroviral therapy necessarily increases.13,14
For persons with clinical and virologic treatment failure, the benefits of changing therapy are evident. Many patients continue to demonstrate prolonged immunologic benefit from treatment, although achieving only partial viral suppression.9,15–21 For these patients, one option is to continue on a stable failing regimen and accept the risk of emergence of further resistance mutations, treatment failure, and the consequent reduction in future treatment choices.13,14 The primary alternative is to change therapy using new agents that are predicted to be effective. If the number or potency of new agents is small, however, application of the latter strategy may fail to suppress viral replication, and thus again lead to the emergence of new resistance mutations and yet further reductions in the number of subsequent viable treatment opportunities. Other options such as complete or partial structured treatment interruptions have failed to show consistent benefit or not undergone careful scrutiny in clinical trials.22–24 Complicating these decisions is the relative paucity of data regarding the rate at which anti-retroviral resistance develops after the onset of virologic treatment failure in patients receiving highly active anti-retroviral therapy (HAART).25–27
The purpose of this study was to define the time to accrual of genotypic resistance mutations, particularly thymidine analogue mutations (TAMs) and major protease inhibitor mutations, in persons with a persistently detectable plasma viral load (pVL) despite being on a stable antiretroviral regimen. We also sought to evaluate the impact of co-administration of lamivudine on the emergence of TAMs given the detrimental effect of the 184V mutation on HIV replication capacity, especially in combination with TAMs.28,29
METHODS
The comprehensive electronic medical records of the Veterans Affairs (VA) Greater Los Angeles Healthcare System were reviewed to identify HIV-infected patients who had a prolonged period of virologic failure while adherent to treatment with HAART, which was defined by the administration of 2 nucleoside reverse transcriptase inhibitors and a nonnucleoside reverse transcriptase inhibitor or a protease inhibitor. Adherence was evaluated per retrospective review of the medication refill records, clinic appearances, and progress notes. All assessments of adherence were performed before performance of resistance genotypes on archived plasma specimens. All treatment decisions had been independently made by the patients’ physicians without intervention by the study team.
To be included in the analysis, patients were required to be receiving at least 3 antiretroviral agents, including a thymidine analogue or a protease inhibitor, despite the onset of virologic treatment failure between August 1996 and February 2002. In addition, at least 2 plasma samples, separated by at least 120 days, had to have been obtained during virologic failure and to be available for analysis. The time of virologic failure was the earliest date that one of the following criteria were satisfied: failure to achieve a 1.0 log10 decrease in the pVL after 1 month of treatment with HAART or, after 3 months of treatment, the first of 2 consecutive pVLs > 1000 copies/mL or the first pVL > 5000 copies/mL. Patients who did not have sufficient medication supplies to allow for 75% adherence during the study period were excluded, as were patients for whom progress notes indicated < 75% of their medications or nonadherence to an unspecified degree. Subjects were allowed to change their antiretroviral therapy while the pVL was < 400 copies/mL, to substitute stavudine for zidovudine (or vice versa), or to add low-dose ritonavir. Courses of therapy were otherwise censored to further follow-up when the antiretroviral regimen was altered.
To avoid a ceiling effect on the potential emergence of mutations, we limited our analyses to patients who lacked evidence of multidrug resistance or multiple TAMs.30 Therefore, analysis for the emergence of TAMs was limited to persons with < 2 TAMs who did not have the Q151M or the T69 insertion complex at the time of the first resistance genotype obtained after virologic failure. Analysis of the emergence of major protease inhibitor mutations was limited to persons with < 2 major protease inhibitor mutations at the time of the first resistance genotype obtained after virologic failure. Resistance mutations were classified as per the November 2004 version of the International AIDS Society (IAS)–USA guidelines.31 Thus, TAMS were defined as the specified mutations at amino acid positions 41, 67, 70, 210, 215, and 219. Major protease inhibitor resistance mutations were defined as those at amino acid positions 30, 46, 48, 50, 82, 84, and 90, and minor resistance mutations were defined as those at amino acid positions 10, 20, 24, 32, 33, 36, 47, 53, 54, 63, 71, 73, 77, and 88.
This study used plasma specimens obtained from patients who had undergone HIV pVL testing at the VA Greater Los Angeles Healthcare System between August 1996 and August 2004. Since August 1996, aliquots of plasma from all patients undergoing pVL analyses at this facility have been routinely stored at − 70° C. All genotyping was done using the TruGeneassay (Bayer, Berkeley, CA). All pVLs were determined by reverse transcriptase polymerase chain reaction (PCR; Amplicor v1.0 or Amplicor v1.5; Roche Diagnostics, Nutley, NJ) with a lower limit of either 50 or 400 copies/mL. pVLs of < 50 or < 400 copies/mL were arbitrarily assigned values of 25 and 200 copies/mL, respectively. All pVL results were log-transformed before statistical manipulation. All data are presented as mean ± SD or median (25%–75% inter-quartile range [IQR]) as appropriate. Statistical analyses utilized the χ 2 test with Yates correction, Student t test, or Cox-Mantel test as appropriate.
This study was approved by the Institutional Review Boards of the VA Greater Los Angeles Healthcare System, Los Angeles, CA, and the University of Texas Medical Branch, Galveston, TX.
RESULTS
Among the 582 individuals who had received at least 1 month of HAART as of February 2002, there were 396 episodes of virologic failure for which there were 120 days of subsequent follow-up. After 276 such episodes, antiretroviral therapy was promptly changed, whereas for the other 123 episodes of virologic failure, patients remained on a stable HAART regimen, and thus were eligible to be considered for inclusion in this study. Subsequently, 40 episodes were excluded because of poor adherence to the treatment regimen, 27 episodes were excluded because of the presence of multiple resistance mutations at the time of virologic failure, 2 episodes were excluded because the failing regimen did not include a thymidine analogue or protease inhibitor, and 8 episodes were excluded because of lack of adequate plasma samples.
The remaining 46 episodes of failing antiretroviral therapy that were eligible for analysis occurred in 41 patients; for 5 patients, the emergence of resistance was assessed during 2 separate episodes of failing therapy. There were 29 episodes that satisfied criteria for evaluation of the emergence of TAMs and major protease inhibitor mutations, 12 episodes that were assessed for the emergence of TAMs only, and 5 episodes that were assessed for the emergence of major protease inhibitor mutations only. Twenty-three episodes evaluated for the emergence of TAMs were in patients who had previously received a thymidine analogue (usually before HAART), and 6 of the episodes evaluated for the emergence of major protease inhibitor mutations were in patients who had previously been treated with a protease inhibitor (3 with indinavir and 1 each with nelfinavir, ritonavir, and saquinavir). Among the thymidine analogue regimens, 17 included zidovudine and 24 included stavudine. The protease inhibitor regimens used nelfinavir (18 regimens), indinavir (12 regimens), saquinavir (3 regimens, 2 with 400 mg of ritonavir administered twice daily), and amprenavir (1 regimen). During the course of virologic failure, ritonavir boosting was initiated in 1 patient receiving indinavir and in 1 patient receiving amprenavir.
Figure 1 provides the duration of successful and failing antiretroviral therapy and the CD4+ cell count and pVL values before virologic failure, at the onset of virologic failure, and at the end of follow-up. Episodes monitored for the emergence of TAMs or major protease inhibitor mutations did not differ in regard to prior antiretroviral therapy, duration of successful or failing treatment, CD4+ cell counts, or pVL values. Data regarding the status of individual patients at the time of virologic failure are provided in Table 1. In 30 episodes, a pVL of < 400 copies/mL had been achieved before treatment failure. At virologic failure, no TAMs were present in 35 of 41 episodes evaluated for the emergence of TAMs, whereas no major protease inhibitor mutations were present in 13 of 34 episodes evaluated for the emergence of major protease inhibitor mutations (P = 0.019).
FIGURE 1.
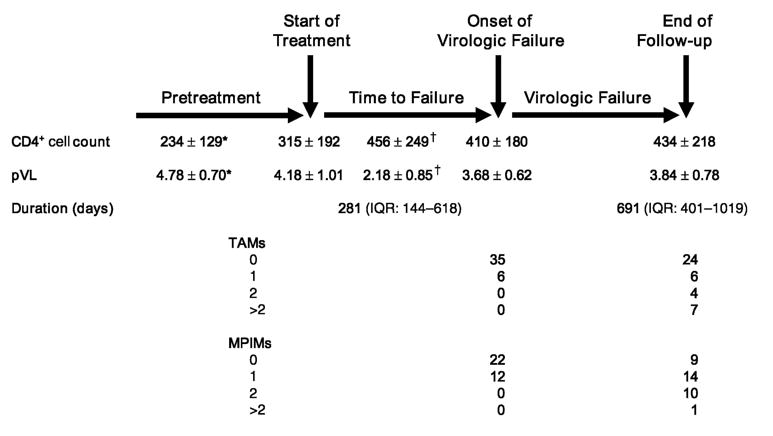
Relation between treatment, virologic failure, CD4+ cell count, pVL, and detection of TAMS and major protease inhibitor mutations (MPIMs). TAMS and MPIMs are shown at the first detection of virologic failure and at the end of follow-up. All data are presented as mean ± SD except for the duration of observation, which is presented as the mean and IQR. *CD4+ cell count is the minimum known value before treatment, whereas pVL is the maximum known value. †CD4+ cell count is the maximum value achieved on the treatment regimen of interest before virologic failure, whereas pVL is the minimum value.
TABLE 1.
Status at Time of Initial Virologic Failure
| Current
|
Mutations
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Patient (Episode) | Course | Prior Therapy | Current Therapy | Days of Therapy | CD4 | pVL | NRTI | PI |
| 2 (1) | 1 | ZDV, 3TC, ddI, ddC | d4T/SQV/RTV | 854 | 592 | 3.10 | 215Y | 63P |
| 5 (1) | 1 | ddI | ddI/3TC/IDV | 126 | 349 | 3.12 | WT | 63P |
| 5 (2) | 2 | 3TC, ddI, IDV | ZDV/3TC/APV | 161 | 343 | 3.19 | WT | 63P, 77I |
| 6 (1) | 1 | None | ZDV/3TC/IDV | 658 | 559 | 3.70 | 184V | 46I |
| 7 (1) | 1 | ZDV, 3TC, d4T, ddI | ZDV/3TC/IDV | 112 | 371 | 3.75 | 184V | 10L/R, 63P, 71A/T, 82T |
| 11 (1) | 1 | None | d4T/3TC/NFV | 397 | 330 | 3.12 | WT | WT |
| 13 (1) | 1 | ZDV, ddC | d4T/3TC/IDV | 160 | 350 | 3.71 | 62V, 184V, 215Y | 10I, 46I, 63P, 71V, 73S, 84V, 90M* |
| 15 (1) | 1 | None | d4T/3TC/NVP | 489 | 387 | 3.74 | 184V | 63P, 71T |
| 16 (1) | 1 | ZDV, d4T, ddI | d4T/3TC/IDV | 112 | 169 | 3.94 | 184V | 71T |
| 21 (1) | 1 | ZDV, 3TC, d4T | ZDV/3TC/IDV | 525 | 355 | 3.03 | 184V | 63P, 71T, 77IV, 82AV |
| 30 (1) | 1 | ZDV, 3TC, ddI | d4T/3TC/IDV | 90 | 219 | 3.00 | 184V | 10I, 71T |
| 30 (2) | 2 | ZDV, d4T, 3TC, ddI, IDV | d4T/NVP/NFV | 133 | 288 | 4.24 | WT | 10I, 24L/I, 46L/M |
| 32 (1) | 1 | None | d4T/ddI/NVP | 581 | 571 | 3.43 | 41L | 63P |
| 105 (1) | 1 | ZDV, d4T, ddI | ZDV/3TC/IDV | 329 | 322 | 3.90 | 184V | WT |
| 106 (1) | 1 | None | ZDV/3TC/NFV | 442 | 729 | 3.48 | 62V, 184V | WT |
| 107 (1) | 1 | None | ZDV/3TC/IDV | 559 | 264 | 4.31 | WT | 46L, 54V, 82A* |
| 108 (1) | 1 | ddI | ZDV/3TC/IDV | 261 | 423 | 3.97 | WT | 63P |
| 109 (1) | 1 | None | ZDV/3TC/NFV | 273 | 498 | 4.69 | 184V | 30N, 63P, 88D |
| 110 (1) | 1 | ZDV, 3TC, d4T | ZDV/3TC/NFV | 315 | 772 | 3.63 | 67N, 70R, 184V† | 30N, 63HP |
| 111 (1) | 1 | None | ddI/3TC/IDV | 821 | 362 | 3.00 | 184V | 46I, 63P |
| 112 (1) | 1 | d4T, 3TC, ddI, NVP, IDV | ZDV/3TC/NFV | 121 | 229 | 4.00 | WT | 20M, 63P |
| 115 (1) | 1 | ZDV, 3TC, d4T | d4T/3TC/IDV | 711 | 215 | 4.89 | 184V | 82A |
| 116 (1) | 1 | ZDV, 3TC, d4T | d4T/3TC/SQV | 133 | 366 | 3.51 | 184V | No data* |
| 116 (2) | 2 | ZDV, 3TC, d4T, NVP, SQV | d4T/NVP/NFV | 49 | 246 | 3.94 | 184V | 10I, 63P, 71T, 90M |
| 133 (1) | 1 | ZDV, d4T | d4T/3TC/IDV | 505 | 166 | 4.82 | 184V/M | WT |
| 135 (1) | 1 | None | d4T/SQV/RTV | 1170 | 437 | 3.34 | WT | 71T |
| 142 (1) | 1 | None | d4T/3TC/NVP | 216 | 316 | 2.91 | WT | 77I |
| 143 (1) | 1 | d4T, 3TC, ddC | d4T/3TC/EFV/NFV | 979 | 715 | 3.20 | 184V | No data |
| 145 (1) | 1 | ZDV, d4T, ddI | d4T/3TC/NFV | 196 | 405 | 4.08 | 184V | 10I, 36I |
| 146 (1) | 1 | ZDV | d4T/3TC/NFV | 214 | 278 | 3.27 | 184V | 10R, 63P, 77I |
| 152 (1) | 1 | ZDV, 3TC, d4T, ddI | ddI/3TC/NFV | 651 | 228 | 3.11 | 184V | 36I, 88S |
| 153 (1) | 1 | ddI | ddI/3TC/IDV | 812 | 517 | 3.84 | 184V, 215Y | 82A |
| 154 (1) | 1 | None | ZDV/3TC/NFV | 123 | 276 | 4.53 | 184V | 63P |
| 157 (1) | 1 | ddI | ZDV/3TC/NFV | 630 | 444 | 3.52 | 184V | 30N, 63P, 88D |
| 158 (1) | 1 | ddI | ZDV/3TC/IDV | 274 | 203 | 3.16 | 184V | 63P, 77I |
| 159 (1) | 1 | None | ZDV/3TC/NFV | 95 | 128 | 5.78 | 184V | 20R, 36I, 63P |
| 161 (1) | 1 | ZDV, d4T | d4T/3TC/IDV | 1599 | 620 | 4.23 | 41L, 184V | 46I, 50I/V, 63P, 771/V |
| 162 (1) | 1 | ZDV | ZDV/ddC/NFV | 1231 | 578 | 3.22 | WT | 36I |
| 162 (2) | 2 | ZDV, ddC, 3TC, NFV | ZDV/3TC/NFV | 288 | 662 | 3.01 | 184V | 10V, 36I, 90M |
| 164 (1) | 1 | None | d4T/3TC/SQV | 139 | 336 | 4.28 | 184V | 36I, 90M |
| 164 (2) | 2 | d4T, 3TC, SQV | d4T/ddI/APV | 68 | 499 | 3.37 | 184V | 20R/T/K, 36I, 63P, 84V, 90M* |
| 166 (1) | 1 | ZDV, ddI | d4T/3TC/NFV | 1302 | 905 | 3.05 | 184V | WT |
| 167 (1) | 1 | No data | d4T/3TC/NVP | 258 | 373 | 3.50 | 70K/R, 184V | 63P, 77I |
| 169 (1) | 1 | ZDV, 3TC, d4T, IDV, DLV, SQV | d4T/ddI /APV | 174 | 428 | 3.31 | 62V, 215F | 10F, 20M, 24I, 36I, 46I, 54V, 63P, 82A* |
| 170 (1) | 1 | ZDV, 3TC, d4T, ddI, RTV, SQV | d4T/NVP/NFV | 160 | 281 | 3.00 | WT | 10I, 63P, 71V, G73S, 90M |
| 171 (1) | 1 | None | d4T/3TC/NVP | 508 | 768 | 4.35 | 184V | WT |
In 5 patients, the emergence of resistance was assessed in 2 separate courses of therapy.
Emergence of major protease inhibitor mutations while on protease inhibitor therapy was not evaluated because of the inability to sequence HIV protease [patient 116(1)] or the presence of 2 or more major protease inhibitor mutations at the time of virologic failure [patients 13(1), 107(1), 164(2), and 169(1)].
Emergence of TAMS while on thymidine analogue therapy was not evaluated because of the presence of 2 or more TAMS at the time of virologic failure [patient 110(1)].
APV indicates amprenavir; CD4, CD4 cell count; ddI, didanosine; d4T, stavudine; EFV, efavirenz; IDV, indinavir; NFV, nelfinavir; NRTI, nucleoside reverse transcriptase inhibitor; NVP, nevirapine; PI, protease inhibitor; RTV, ritonavir; SQV, saquinavir; 3TC, lamivudine; WT, wild type; ZDV, zidovudine.
The median time between virologic failure and the first subsequent genotype was 218 (IQR: 126–370) days; a median of 5 (IQR: 3–6) genotypes was performed per course of virologic failure. Kaplan-Meier analyses indicated that new major protease inhibitor mutations subsequently emerged more rapidly than did new TAMs. This relation held true when all regimens involving thymidine analogues or protease inhibitors were evaluated (Fig. 2; P = 0.026) and when evaluation was limited to the 29 regimens in which there was concurrent use of a thymidine analogue and a protease inhibitor (Fig. 3; P = 0.0019). Finally, the emergence of TAMs was substantially slower in the 32 thymidine analogue–containing regimens that included lamivudine than in the 9 regimens that did not (Fig. 4; P = 0.0073). When regimens not including lamivudine were excluded, the rate of emergence of TAMs and major protease inhibitor mutations did not differ. To compensate for potential bias introduced by including a second course of therapy, we also assessed the time to onset of new TAMS and major protease inhibitor mutations after limiting the data set to each patient’s first course of therapy. In these analyses, new TAMS were again shown to emerge more slowly than new major protease inhibitor mutations (P = 0.0072 for all treatment regimens and P = 0.0024 for patients receiving current therapy with a thymidine analogue and a protease inhibitor). The emergence of TAMS was not shown to differ in patients who received thymidine analogue regimens with or without concurrent use of lamivudine (P = 0.203), however, possibly because of the small residual sample size (30 patients with receipt of lamivudine and 6 patients without receipt of lamivudine).
FIGURE 2.
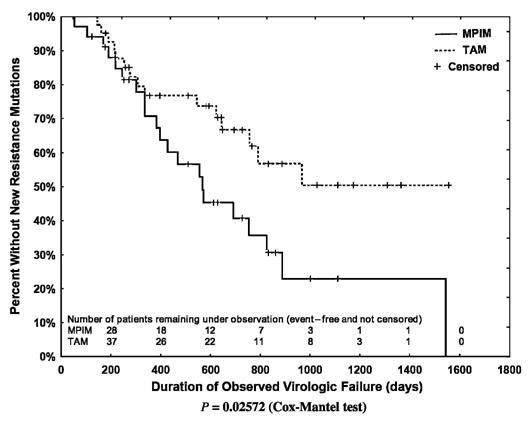
Kaplan-Meier plot of the emergence of a new TAM among the 41 thymidine analogue regimens and of a new major protease inhibitor mutation (MPIM) among the 34 protease inhibitor regimens (Cox-Mantel test, P = 0.026).
FIGURE 3.
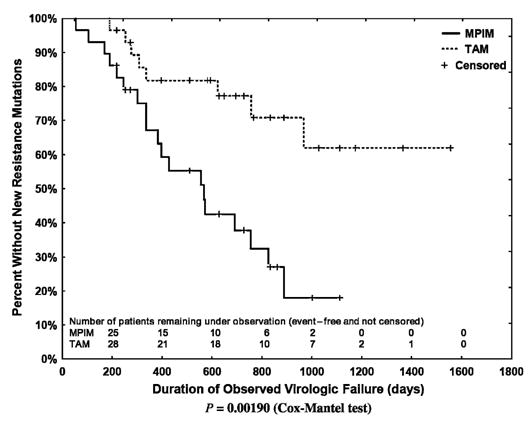
Kaplan-Meier plot of the emergence of a new TAM among the 29 regimens that included a thymidine analogue and a protease inhibitor (Cox-Mantel test, P = 0.0019).
FIGURE 4.
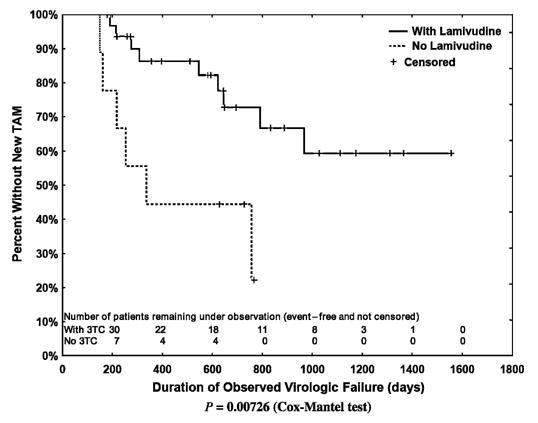
Kaplan-Meier plot of the emergence of a new TAM among the thymidine analogue–containing regimens that did (n = 32) or did not (n = 9) include lamivudine (Cox-Mantel test, P = 0.0073). 3TC indicates lamivudine.
The median follow-up was similar for episodes of failure evaluated for emergent TAMs and for major protease inhibitor mutations, 695 (IQR: 414–1014) days and 745 (IQR: 529– 1018) days, respectively. During virologic failure, new TAMs were detected in 15 of 41 eligible episodes and new major protease inhibitor mutations were detected in 21 of 34 eligible episodes (P = 0.20). In the subset of courses with no detectable TAMs (n = 35) or major protease inhibitor mutations (n = 21) at the onset of virologic failure, new TAMS emerged 11 times and new major protease inhibitor mutations emerged 13 times (P = 0.026).
Among the 41 thymidine analogue–containing regimens, a single TAM was detected at virologic failure in 3 of 9 courses that did not include lamivudine versus 3 of 32 courses that did include lamivudine (P = 0.072). New TAMs later emerged in 6 of the 9 thymidine analogue regimens with lamivudine versus 9 of the 32 thymidine analogue regimens without lamivudine (P = 0.034). Of the 9 thymidine analogue regimens that did not include lamivudine, 3 included didanosine, 1 included zalcitabine, and 5 included a nonnucleoside reverse transcriptase inhibitor or full-dose ritonavir plus a protease inhibitor. Among the 3 courses in which stavudine was combined with didanosine, new TAMS were detected after 150, 161, and 218 days; for the 6 courses in which stavudine (n = 5) or zidovudine (n = 1) was the sole nucleoside reverse transcriptase inhibitor, new TAMS were detected after 253, 336, and 757 days in 3 instances and were not detected at final follow-up (274, 728 and 767 days) in 3 instances. The frequency of protease inhibitors versus nonnucleoside reverse transcriptase inhibitors did not differ in thymidine analogue–containing courses that did or did not include lamivudine.
For 11 of the 15 episodes in which a new TAM emerged, further follow-up on the same course of therapy was available for a median of 553 (IQR: 262–762) days. During 8 of these 11 periods, additional TAMs emerged. Similarly, there was a median of 403 (IQR: 258–586) days of follow-up for 18 of the 21 episodes in which a major protease inhibitor mutation emerged; additional major protease inhibitor mutations emerged in 7 of these 18 periods. Figure 1 shows the final distribution of TAMs and major protease inhibitor mutations among the thymidine analogue– and protease inhibitor–containing regimens.
The 62V, 118I, and 184V mutations were the only non-TAM nucleoside reverse transcriptase inhibitor resistance mutations present at virologic failure or that arose during the subsequent period (Table 2). The 2 instances of 118I emergence were in persons receiving didanosine and stavudine, both of whom had previously accumulated 3 TAMS. The 151M, 44A/D, and T69 insertion mutations were not found in any patients. Although the 62V mutation can be found in association with the 69S insertion complex (with 41L, 70R, 210W, 215Y/F, and 219Q/E) or the 151 complex (with 75I, 77L, and 116Y),31 neither of these associations was observed in any of the 6 studied treatment courses in which this mutation was detected.
TABLE 2.
Status at Final Follow-Up
| Days to First New
|
Mutations
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Patient (Episode) | Final Therapy | Failure Days | TAM | MPIM | CD4 | pVL | NRTI | PI |
| 2 (1) | d4T/SQV/RTV | 728 | > 728* | > 728 | 600 | 3.69 | 215Y | 36I, 63P |
| 5 (1) | ddI/3TC/IDV | 613 | N/A | > 613 | 264 | 3.05 | WT | 63P, 77I |
| 5 (2) | ZDV/3TC/APV/r | 511 | > 511 | > 511 | 137 | 3.44 | WT | 63P, 77IV |
| 6 (1) | ZDV/3TC/IDV | 648 | > 648 | 556 | 570 | 3.03 | 184V | 46I, 63P, 71V, 82A, 90M |
| 7 (1) | ZDV/3TC/NFV | 1988 | 645 | N/A | 534 | 3.31 | 67N, 70R, 184V, 219Q | 10VI, 36I, 54V, 63P, 71T, 82TA, 88S |
| 11 (1) | d4T/3TC/NFV | 1896 | 967 | 56 | 386 | 3.09 | 41L, 184V, 215Y | 36I, 46I, 63P, 71V, 90M |
| 13 (1) | d4T/3TC/NFV | 1311 | > 1311 | N/A | 524 | 3.23 | 62V, 184V, 215YC | 10I, 46I, 63P, 71V, 84V, 90M |
| 15 (1) | d4T/3TC/NFV | 1556 | > 1556 | 567 | 469 | 3.50 | 184V | 10I, 46I, 54L, 63P, 71T, 90M |
| 16 (1) | d4T/ddI/NFV | 693 | > 693 | 301 | 261 | 3.04 | 184V | 30N, 46L, 63P, 71T, 88D |
| 21 (1) | ZDV/3TC/IDV | 763 | 623 | 336 | 556 | 4.45 | 41L, 67N, 70R, 184V, 219E | 54V, 77I, 84V |
| 30 (1) | d4T/3TC/IDV | 218 | > 218 | > 218 | 273 | 3.06 | 184V | 10I, 24I, 46L |
| 30 (2) | d4T/NVP/NFV | 253 | 253 | > 253 | 229 | 4.43 | 70R, 215SYTN | 10I, 24I, 36I |
| 32 (1) | d4T/ddI/NVP | 430 | 150 | N/A | 718 | 3.31 | 41L, 210W, 215Y | 63P |
| 105 (1) | d4T/3TC/IDV | 1366 | > 1366 | 826 | 385 | 3.50 | 184V | 46L, 82A |
| 106 (1) | ZDV/3TC/NFV | 300 | 190 | 190 | 805 | 4.75 | 41L, 62V, 184V | 46I, 77I |
| 107 (1) | ZDV/3TC/IDV | 179 | > 179 | N/A | 295 | 4.68 | 184V | 46L, 54V, 63P, 71V, 82A |
| 108 (1) | ZDV/3TC/IDV | 397 | > 397 | 397 | 398 | 4.27 | 184V | 82A |
| 109 (1) | ZDV/3TC/NFV | 833 | > 833 | > 833 | 543 | 4.70 | 184V | 30N, 63P, 88D |
| 110 (1) | ZDV/3TC/NFV | 1218 | N/A | 469 | 472 | 4.33 | 67N, 70R, 184V, 219Q | 30N, 63QHP, 71IV, 90M |
| 111 (1) | ddI/3TC/IDV/r | 1545 | N/A | 1545 | 349 | 3.53 | 184V | 10I, 36I, 46I, 63P, 90M |
| 112 (1) | ZDV/3TC/NFV | 1020 | > 1020 | 691 | 352 | 4.18 | 184V | 71V, 90M |
| 115 (1) | d4T/3TC/IDV/r | 888 | > 888 | 888 | 172 | 6.44 | 184V | 10I, 24I, 46I, 63P, 82A |
| 116 (1) | d4T/3TC/NVP/NFV | 644 | > 644 | N/A | 234 | 4.31 | 184V | 10I, 63P, 71T, 90M |
| 116 (2) | d4T/NVP/NFV | 778 | 336 | 572 | 188 | 3.89 | 67N, 70R, 219E | 30N, 36I, 63P, 71V, 88D, 90M |
| 133 (1) | d4T/3TC/IDV | 1112 | > 1112 | > 1112 | 166 | 4.03 | 184V | WT |
| 135 (1) | d4T/SQV/RTV | 767 | > 767 | 383 | 210 | 4.25 | WT | 10I, 48V, 54V, 71T, 82A |
| 142 (1) | d4T/3TC/NVP | 357 | > 357 | N/A | 474 | 4.32 | WT | 77I |
| 143 (1) | d4T/3TC/EFV/NFV | 259 | > 259 | N/A | 1000 | 3.16 | 184V | 30N, 36I, 46I, 77I, 88D |
| 145 (1) | d4T/3TC/NFV | 1029 | > 1029 | 427 | 405 | 3.70 | 184V | 10I, 20R, 30N, 36I, 88D |
| 146 (1) | d4T/3TC/NFV | 584 | > 584 | 169 | 405 | 4.66 | 184V | 30N, 63P, 71T, 77I, 88D |
| 152 (1) | ddI/3TC/NFV | 126 | N/A | > 126 | 372 | 3.86 | 184V | 36I, 88S |
| 153 (1) | ddI/3TC/IDV | 177 | N/A | > 177 | 676 | 4.06 | 184V, 215Y | 82A |
| 154 (1) | ZDV/3TC/NFV | 695 | > 695 | 219 | 363 | 3.85 | 184V | 30N, 63P, 71V, 77I, 88D |
| 157 (1) | ZDV/3TC/NFV | 861 | 308 | > 861 | 364 | 3.40 | 41L, 184V, 215Y | 30N, 36I, 63P, 88D |
| 158 (1) | d4T/3TC/IDV/r | 1175 | > 1175 | 244 | 109 | 3.14 | WT | 10I, 63P, 73S, 77I |
| 159 (1) | ZDV/3TC/NFV | 276 | 276 | 105 | 124 | 5.17 | 70R, 184V | 20R, 30N, 36I, 63P |
| 161 (1) | d4T/3TC/IDV | 214 | 214 | N/A | 318 | 4.18 | 184V, 210W, 215Y | 10IV, 46I, 63P, 71V |
| 162 (1) | ZDV/ddC/NFV | 628 | > 628 | > 628 | 625 | 2.43 | WT | 10V, 36I |
| 162 (2) | ZDV/3TC/NFV | 274 | > 274 | > 274 | 478 | 3.54 | 184V | 36I, 90M |
| 164 (1) | d4T/3TC/SQV/NVP | 1014 | > 1014 | 755 | 534 | 2.18 | 184V | 20RT, 36I, 63P, 84V, 90M |
| 164 (2) | d4T/DDI/APV/r | 816 | 161 | N/A | 617 | 4.01 | 41L, 67N, 118I, 210W, 215FY | 10F, 20R, 32I, 33F, 361, 54L, 63P, 84V, 90M |
| 166 (1) | d4T/3TC/NFV | 595 | > 595 | 337 | 826 | 3.11 | 184V | 10F, 88S |
| 167 (1) | d4T/3TC/NVP | 545 | 545 | N/A | 265 | 3.36 | 67N, 70R, 184V, 219E | 63P, 77I |
| 169 (1) | d4T/DDI/APV | 414 | 218 | N/A | 483 | 3.70 | 41L, 118I, 210W, 215F | 20M, 24I, 36I, 46I, 54V, 63P, 82A |
| 170 (1) | d4T/NVP/NFV | 1002 | 757 | > 1002 | 329 | 3.77 | 215FY | 10I, 63P, 71V, 73S, 77I, 90M |
| 171 (1) | d4T/3TC/NVP | 1620 | 791 | N/A | 1100 | 5.58 | 70R, 184V | WT |
No mutation was observed at the end of the follow-up period.
APV indicates amprenavir; CD4, CD4 cell count; ddI, didanosine; d4T, stavudine; EFV, efavirenz; IDV, indinavir; ; MPIM, major protease inhibitor mutation; NFV, nelfinavir; NRTI, nucleoside reverse transcriptase inhibitor; NVP, nevirapine; PI, protease inhibitor; r, ritonavir; RTV, ritonavir; SQV, saquinavir; 3TC, lamivudine; WT, wild type; ZDV, zidovudine.
Minor protease inhibitor resistance mutations were present at virologic failure in 25 regimens, and new minor protease inhibitor resistance mutations were detected during the period of observation in 22 of the 34 regimens. In 7 of the 8 patients receiving nonnucleoside reverse transcriptase inhibitors, genotypic evidence of resistance to these agents was present at virologic failure.
Compared with the time of virologic failure, at final follow-up, there was an increase of 24 ± 137 CD4 cells/μL and an increase of 0.16 ± 0.72 log10 copies/mL in the pVL; neither of these changes were statistically significant. There were no differences in the change of the CD4+ cell count or the pVL from the time of virologic failure to last follow-up regardless of the emergence of a new TAM or major protease inhibitor mutation or for thymidine analogue–containing regimens in which lamivudine was or was not used.
DISCUSSION
This study utilized an unusual opportunity to evaluate the rate of emergence of genotypic resistance to thymidine analogues and protease inhibitors retrospectively in patients who remained on stable HAART after the onset of virologic treatment failure. In these 46 studied episodes of prolonged virologic failure (mean duration of 110 weeks), there was a significantly greater rate of emergence of major protease inhibitor mutations than of TAMs. This difference was observed only when thymidine analogues were used in combination with lamivudine; importantly, dual–protease inhibitor therapy with ritonavir or ritonavir boosting was used in only 4 of the 34 evaluable courses of protease inhibitor therapy. In addition, the emergence of TAMs was substantially slower in lamivudine-containing regimens. Overall, the mean time to detection of a new TAM in treatment courses that included lamivudine was > 300 days longer than the mean time until the detection of a new major protease inhibitor mutation.
Our results extend and generally corroborate the observations of previous studies regarding the rate of emergence of TAMs and protease mutations after virologic failure but provide a substantially longer period of follow-up. Kempf et al26 reported that genotypic evidence of resistance to lamivudine, stavudine, and nelfinavir emerged in 100%, 15%, and 74%, respectively, of 80 previously treatment-naive individuals with up to 48 weeks of ongoing virologic failure. Similarly, Aleman et al25 reported that among 10 patients with fewer than 2 TAMs at the time of initial virologic failure who continued to receive a thymidine analogue and protease inhibitors, 2 developed 1 or more TAMs and 8 developed new major protease inhibitor resistance mutations over a mean follow-up period of 103 weeks. In contrast, Napravnik et al32 found that the incidence of new major protease inhibitor mutations was lower than the incidence of nucleoside reverse transcriptase inhibitor–associated mutations in persons who remained on stable combination antiretroviral therapy with detectable viremia for a median of 9.3 months. Importantly, the rate of development of new resistance mutations was greater among persons who maintained an average viral load of 3 to 4 HIV RNA log10 copies/mL than in persons whose average viral load was lower or higher. These authors also found that the adjusted incidence rate of new mutations was lowest among persons who had 1 to 3 resistance mutations (including minor protease inhibitor mutations) at baseline or whose viral load was stable during follow-up (slopes ≤0.2 per month). Unlike our study, however, Napravnik et al32 did not specifically assess the emergence of TAMs. In addition, the proportion of patients receiving thymidine analogues (with or without lamivudine) or ritonavir-boosted protease inhibitor regimens was not specified. Other analyses of the emergence of resistance mutations during virologic failure fail to describe the specific emergence of TAMs or major protease inhibitor resistance mutations, include patients with high levels of preexistent resistance, or do not provide separate analyses of patients with few mutations at baseline.30,33–37
Our observations regarding the slower rate of emergence of TAMs after treatment failure in persons receiving lamivudine are consistent with and extend previous observations. In particular, Kuritzkes et al38 previously reported that in 49 treatment-naive patients given 52 to 72 weeks of nonsuppressive therapy, TAMs emerged more often with zidovudine monotherapy than with zidovudine plus lamivudine. Other studies have shown that 184V is usually the first nucleoside reverse transcriptase inhibitor mutation to arise in failing zidovudine plus lamivudine regimens but have not examined the long-term evolution of TAMs after initial treatment failure.39–44
The relatively gradual rate of accrual of TAMs in patients receiving lamivudine may be related to the observation that in the presence of the 184V mutation, the emergence of TAMs decreases viral fitness as measured by replication capacity.29 Thus, in the presence of M184V, mutations that diminish nucleoside reverse transcriptase inhibitor activity (TAMs) may not be favored. In contrast, the emergence of multiple minor protease inhibitor mutations may improve replication capacity and allow for the development of further major protease inhibitor mutations.45 Of note, the emergence of TAMs in patients not receiving lamivudine was similar to the rate of emergence of major protease inhibitor mutations. We were not otherwise able to determine predictors of the emergence of antiretroviral resistance. In particular, the CD4+ lymphocyte count tended to remain stable and the magnitude of pVL changes before or after treatment failure did not correlate with the presence or absence of antiretroviral resistance mutations at the time of first failure or the subsequent emergence of these mutations. As previously shown, there was no difference in the emergence of TAMs in subjects who received zidovudine versus stavudine.46
The strengths of this study are the prolonged period of observation during virologic failure on a stable antiretroviral regimen and the frequency with which resistance genotypes were obtained. We believe that the differential rate of emergence of TAMs and major protease inhibitor mutations is not likely to be attributable to variances in adherence to these medications, especially because there was no correlation between the emergence of resistance mutations and the observed virologic responses.46,47 Furthermore, at the time of final follow-up, in all but 5 patients, the pVL was less than the pre-HAART level, and in all but 2 patients, the CD4+ lymphocyte count was greater than the pre-HAART value (data not shown). Although other studies have examined differences in the presence of TAMs and major protease inhibitor mutations at the time of initial treatment failure, few studies have evaluated the subsequent course of patients who remain on stable nonsuppressive regimens25–27 and none, to our knowledge, have followed as large a cohort of failing patients with low rates of baseline resistance who continue to receive a stable regimen for as long a period.
The principal limitation of this study is the retrospective and potentially biased case selection. In particular, persons with virologic failure that is progressive or accompanied by immunologic and/or clinical deterioration are much more likely to have had their antiretroviral therapy rapidly changed, and thus would not have been eligible for inclusion in this study. Such individuals may have more rapidly developed resistance than did the individuals we studied. Furthermore, exclusion of persons with 2 or more TAMS or major protease inhibitor mutations at the time of virologic failure may bias the analysis by eliminating patients in whom resistance emerges particularly quickly. Our results may also be relevant only for the specific regimens received by our subjects, particularly because few patients received ritonavir-boosted protease inhibitor regimens,26,27 and for the resistance profiles observed at the time of virologic failure. Previous studies have shown that the rate of emergence of major protease inhibitor mutations is less in persons receiving a ritonavir-boosted pro-tease inhibitor than when ritonavir boosting is not used.26,27 In addition, our ability to assess the time of onset of anti-retroviral resistance was limited by our reliance on having access to sufficient stored plasma that had been obtained for clinical reasons; thus, we were not able to assess the emergence of resistance at regular fixed intervals. Our results should also be interpreted in the context of our definition of virologic failure. This relied principally on reproducibly demonstrating a pVL > 1000 copies/mL. This was based on treatment recommendations for changing antiretroviral therapy13,14 and on the practical consideration of the pVL threshold for reliably performing genotypic assays for anti-retroviral resistance.
The relatively slow rate of accrual of TAMs in our subjects provides support for further studies to understand better the determinants of the pattern of emergence of anti-retroviral resistance mutations and of strategies to delay the emergence of resistance in the context of virologic failure. We strongly caution that these results should not be used to make clinical management decisions in persons with virologic failure while receiving HAART, however, especially for those persons who have access to remaining effective antiretroviral agents. Although TAMs emerged slowly in most patients, our data clearly indicate that continued failing therapy is likely to result in the ultimate emergence of TAMs and major protease inhibitor mutations, with resultant cross-resistance that limits future treatment options and leads to poor clinical outcomes. Finally, regardless of whether there is initial immunologic benefit, patients with virologic treatment failure ultimately develop immunologic failure and, where excellent treatment options exist, have been shown to have worse clinical outcomes than do persons who achieve full virologic suppression.17,48
Footnotes
Supported by a Collaborative Research grant from GlaxoSmithKline.
References
- 1.Walmsley S, Bernstein B, King M, et al. Lopinavirritonavir versus nelfinavir for the initial treatment of HIV infection. N Engl J Med. 2002;346:2039–2046. doi: 10.1056/NEJMoa012354. [DOI] [PubMed] [Google Scholar]
- 2.Gallant JE, Staszewski S, Pozniak AL, et al. Efficacy and safety of tenofovir DF vs stavudine in combination therapy in antiretroviral-naive patients: a 3-year randomized trial. JAMA. 2004;292:191–201. doi: 10.1001/jama.292.2.191. [DOI] [PubMed] [Google Scholar]
- 3.Johnson M, Grinsztejn B, Rodriguez C, et al. Atazanavir plus ritonavir or saquinavir, and lopinavir/ritonavir in patients experiencing multiple virological failures. AIDS. 2005;19:685–694. doi: 10.1097/01.aids.0000166091.39317.99. [DOI] [PubMed] [Google Scholar]
- 4.Lalezari JP, Henry K, O’Hearn M, et al. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N Engl J Med. 2003;348:2175–2185. doi: 10.1056/NEJMoa035026. [DOI] [PubMed] [Google Scholar]
- 5.Lazzarin A, Clotet B, Cooper D, et al. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N Engl J Med. 2003;348:2186–2195. doi: 10.1056/NEJMoa035211. [DOI] [PubMed] [Google Scholar]
- 6.Cooper D, Hicks C, Cahn P, et al. 24-Week RESIST study analyses: the efficacy of tipranavir/ritonavir is superior to lopinavir/ritonavir, and the TPV/r treatment response is enhanced by inclusion of genotypically active antiretrovirals in the optimized background regimen [abstract 560]; Presented at: 12th Conference on Retroviruses and Opportunistic Infections; Boston, MA. 2005. [Google Scholar]
- 7.Sabin CA, Hill T, Lampe F, et al. Emergence of antiretroviral resistance in HIV-positive women receiving combination antiretroviral therapy in pregnancy. AIDS. 2005;19:63–67. doi: 10.1097/00002030-200501030-00007. [DOI] [PubMed] [Google Scholar]
- 8.Phillips AN, Dunn D, Sabin C, et al. Long term probability of detection of HIV-1 drug resistance after starting antiretroviral therapy in routine clinical practice. AIDS. 2005;19:487–494. doi: 10.1097/01.aids.0000162337.58557.3d. [DOI] [PubMed] [Google Scholar]
- 9.Ledergerber B, Lundgren JD, Walker AS, et al. Predictors of trend in CD4-positive T-cell count and mortality among HIV-1-infected individuals with virological failure to all three antiretroviral-drug classes. Lancet. 2004;364:51–62. doi: 10.1016/S0140-6736(04)16589-6. [DOI] [PubMed] [Google Scholar]
- 10.Mocroft A, Ledergerber B, Viard JP, et al. Time to virological failure of 3 classes of antiretrovirals after initiation of highly active antiretroviral therapy: results from the EuroSIDA study group. J Infect Dis. 2004;190:1947–1956. doi: 10.1086/425424. [DOI] [PubMed] [Google Scholar]
- 11.Lohse N, Obel N, Kronborg G, et al. Declining risk of triple-class antiretroviral drug failure in Danish HIV-infected individuals. AIDS. 2005;19:815–822. doi: 10.1097/01.aids.0000168976.51843.9f. [DOI] [PubMed] [Google Scholar]
- 12.Sabin CA, Hill T, Lampe F, et al. Treatment exhaustion of highly active antiretroviral therapy (HAART) among individuals infected with HIV in the United Kingdom: multicentre cohort study. BMJ. 2005;330:695–699. doi: 10.1136/bmj.38369.669850.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Panel on Clinical Practices for Treatment of HIV Infection. [Accessed January 15, 2006.];Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Available at: http://www.aidsinfo.nih.gov/guidelines/adult/AA_100605.pdf.
- 14.Yeni PG, Hammer SM, Hirsch MS, et al. Treatment for adult HIV infection: 2004 recommendations of the International AIDS Society-USA Panel. JAMA. 2004;292:251–265. doi: 10.1001/jama.292.2.251. [DOI] [PubMed] [Google Scholar]
- 15.Kaufmann D, Pantaleo G, Sudre P, et al. CD4-cell count in HIV-1-infected individuals remaining viraemic with highly active antiretroviral therapy (HAART) Lancet. 1998;351:723–724. doi: 10.1016/s0140-6736(98)24010-4. [DOI] [PubMed] [Google Scholar]
- 16.Deeks SG, Barbour JD, Martin JN, et al. Sustained CD4+ T cell response after virologic failure of protease inhibitor-based regimens in patients with human immunodeficiency virus infection. J Infect Dis. 2000;181:946–953. doi: 10.1086/315334. [DOI] [PubMed] [Google Scholar]
- 17.Deeks SG, Barbour JD, Grant RM, et al. Duration and predictors of CD4 T-cell gains in patients who continue combination therapy despite detectable plasma viremia. AIDS. 2002;16:201–207. doi: 10.1097/00002030-200201250-00009. [DOI] [PubMed] [Google Scholar]
- 18.Cozzi-Lepri A, Phillips AN, Miller V, et al. Changes in viral load in people with virological failure who remain on the same HAART regimen. Antivir Ther. 2003;8:127–136. [PubMed] [Google Scholar]
- 19.Grabar S, Le MV, Goujard C, et al. Clinical outcome of patients with HIV-1 infection according to immunologic and virologic response after 6 months of highly active antiretroviral therapy. Ann Intern Med. 2000;133:401–410. doi: 10.7326/0003-4819-133-6-200009190-00007. [DOI] [PubMed] [Google Scholar]
- 20.Piketty C, Weiss L, Thomas F, et al. Long-term clinical outcome of human immunodeficiency virus-infected patients with discordant immunologic and virologic responses to a protease inhibitor-containing regimen. J Infect Dis. 2001;183:1328–1335. doi: 10.1086/319861. [DOI] [PubMed] [Google Scholar]
- 21.Raffanti SP, Fusco JS, Sherrill BH, et al. Effect of persistent moderate viremia on disease progression during HIV therapy. J Acquir Immune Defic Syndr. 2004;37:1147–1154. doi: 10.1097/01.qai.0000136738.24090.d0. [DOI] [PubMed] [Google Scholar]
- 22.Lawrence J, Mayers DL, Hullsiek KH, et al. Structured treatment interruption in patients with multidrug-resistant human immunodeficiency virus. N Engl J Med. 2003;349:837–846. doi: 10.1056/NEJMoa035103. [DOI] [PubMed] [Google Scholar]
- 23.Katlama C, Dominguez S, Gourlain K, et al. Benefit of treatment interruption in HIV-infected patients with multiple therapeutic failures: a randomized controlled trial (ANRS 097) AIDS. 2004;18:217–226. doi: 10.1097/00002030-200401230-00011. [DOI] [PubMed] [Google Scholar]
- 24.Deeks SG, Hoh R, Neilands TB, et al. Interruption of treatment with individual therapeutic drug classes in adults with multidrug-resistant HIV-1 infection. J Infect Dis. 2005;192:1537–1544. doi: 10.1086/496892. [DOI] [PubMed] [Google Scholar]
- 25.Aleman S, Soderbarg K, Visco-Comandini U, et al. Drug resistance at low viraemia in HIV-1-infected patients with antiretroviral combination therapy. AIDS. 2002;16:1039–1044. doi: 10.1097/00002030-200205030-00010. [DOI] [PubMed] [Google Scholar]
- 26.Kempf DJ, King MS, Bernstein B, et al. Incidence of resistance in a double-blind study comparing lopinavir/ritonavir plus stavudine and lamivudine to nelfinavir plus stavudine and lamivudine. J Infect Dis. 2004;189:51–60. doi: 10.1086/380509. [DOI] [PubMed] [Google Scholar]
- 27.Macmanus S, Yates P, White S, et al. GW433908 in ART-naive subjects: absence of resistance at 48 weeks with boosted regimen and APV-like resistance profile with unboosted regimen [abstract 598]; Presented at: 10th Conference on Retroviruses and Opportunistic Infections; Boston, MA. 2003. [Google Scholar]
- 28.Wei X, Liang C, Gotte M, et al. The M184V mutation in HIV-1 reverse transcriptase reduces the restoration of wild-type replication by attenuated viruses. AIDS. 2002;16:2391–2398. doi: 10.1097/00002030-200212060-00003. [DOI] [PubMed] [Google Scholar]
- 29.Miller MD, White KL, Petropoulos CJ, et al. Decreased replication capacity of HIV-1 clinical isolates containing K65R or M184V RT mutations [abstract 616]; Presented at: 10th Conference on Retroviruses and Opportunistic Infections; Boston, MA. 2003. [Google Scholar]
- 30.Kantor R, Shafer RW, Follansbee S, et al. Evolution of resistance to drugs in HIV-1-infected patients failing antiretroviral therapy. AIDS. 2004;18:1503–1511. doi: 10.1097/01.aids.0000131358.29586.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson VA, Brun-Vezinet F, Clotet B, et al. Update of the drug resistance mutations in HIV-1: 2004. Top HIV Med. 2004;12:119–124. [PubMed] [Google Scholar]
- 32.Napravnik S, Edwards D, Stewart P, et al. HIV-1 drug resistance evolution among patients on potent combination antiretroviral therapy with detectable viremia. J Acquir Immune Defic Syndr. 2005;40:34–40. doi: 10.1097/01.qai.0000174929.87015.d6. [DOI] [PubMed] [Google Scholar]
- 33.Maggiolo F, Callegaro A, Quinzan GP, et al. [abstract H- 2057] Evolution of HIV reverse transcriptase and protease mutations in patients on antiretroviral therapy; Presented at: 42nd Interscience Conference on Antimicrobial Agents and Chemotherapy; 2002; San Diego. [Google Scholar]
- 34.Fitzgibbon JE, Gaur S, Walsman SM, et al. Emergence of drug resistance mutations in a group of HIV-infected children taking nelfinavir-containing regimens. AIDS Res Hum Retroviruses. 2001;20:1321–1328. doi: 10.1089/08892220152596579. [DOI] [PubMed] [Google Scholar]
- 35.Hatano H, Hunt P, Weidler J, et al. Rate of viral evolution and risk of losing future drug options in heavily pre-treated patients remaining on a stable partially suppressive regimen [abstract 615]; Presented at: 13th Conference on Retroviruses and Opportunistic Infection; Denver, CO. 2006. [Google Scholar]
- 36.Riddler S, Jiang H, Deeks S, et al. A randomized pilot study of delayed ART switch in subjects with detectable viremia on HAART [abstract 522]; Presented at: 13th Conference on Retroviruses and Opportunistic Infection; Denver, CO. 2006. [Google Scholar]
- 37.Nasta P, Matti A, Cocca G, et al. Early vs deferred HAART switch in heavily pre-treated HIV patients with low viral load level and stable CD4 cell count [abstract 523]; Presented at: 13th Conference on Retroviruses and Opportunistic Infection; Denver, CO. 2006. [Google Scholar]
- 38.Kuritzkes DR, Shugarts D, Bakhtiari M, et al. Emergence of dual resistance to zidovudine and lamivudine in HIV-1-infected patients treated with zidovudine plus lamivudine as initial therapy. J Acquir Immune Defic Syndr. 2000;23:26–34. doi: 10.1097/00126334-200001010-00004. [DOI] [PubMed] [Google Scholar]
- 39.Kuritzkes DR, Quinn JB, Benoit SL, et al. Drug resistance and virologic response in NUCA 3001, a randomized trial of lamivudine (3TC) versus zidovudine (ZDV) versus ZDV plus 3TC in previously untreated patients. AIDS. 1996;10:975–981. doi: 10.1097/00002030-199610090-00007. [DOI] [PubMed] [Google Scholar]
- 40.Maguire M, Gartland M, Moore S, et al. Absence of zidovudine resistance in antiretroviral-naive patients following zidovudine/lamivudine/protease inhibitor combination therapy: virological evaluation of the AVANTI 2 and AVANTI 3 studies. AIDS. 2000;14:1195–1201. doi: 10.1097/00002030-200006160-00017. [DOI] [PubMed] [Google Scholar]
- 41.Havlir DV, Hellmann NS, Petropoulos CJ, et al. Drug susceptibility in HIV infection after viral rebound in patients receiving indinavir-containing regimens. JAMA. 2000;283:229–234. doi: 10.1001/jama.283.2.229. [DOI] [PubMed] [Google Scholar]
- 42.Mouroux M, Descamps D, Izopet J, et al. Low-rate emergence of thymidine analogue mutations and multi-drug resistance mutations in the HIV-1 reverse transcriptase gene in therapy-naive patients receiving stavudine plus lamivudine combination therapy. Antivir Ther. 2001;6:179–183. [PubMed] [Google Scholar]
- 43.Ferrer E, Podzamczer D, Arnedo M, et al. Genotype and phenotype at baseline and at failure in human immunodeficiency virus-infected antiretroviral-naive patients in a randomized trial comparing zidovudine and lamivudine plus nelfinavir or nevirapine. J Infect Dis. 2003;187:687–690. doi: 10.1086/367987. [DOI] [PubMed] [Google Scholar]
- 44.Ait-Khaled M, Stone C, Amphlett G, et al. M184V is associated with a low incidence of thymidine analogue mutations and low phenotypic resistance to zidovudine and stavudine. AIDS. 2002;16:1686–1689. doi: 10.1097/00002030-200208160-00017. [DOI] [PubMed] [Google Scholar]
- 45.Barbour JD, Wrin T, Grant RM, et al. Evolution of phenotypic drug susceptibility and viral replication capacity during long-term virologic failure of protease inhibitor therapy in human immunodeficiency virus-infected adults. J Virol. 2002;76:11104–11112. doi: 10.1128/JVI.76.21.11104-11112.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuritzkes DR, Bassett RL, Hazelwood JD, et al. Rate of thymidine analogue resistance mutation accumulation with zidovudine- or stavudine-based regimens. J Acquir Immune Defic Syndr. 2004;36:600–603. doi: 10.1097/00126334-200405010-00008. [DOI] [PubMed] [Google Scholar]
- 47.King MS, Brun SC, Kempf DJ. Relationship between adherence and the development of resistance in antiretroviral-naive, HIV-1-infected patients receiving lopinavir/ritonavir or nelfinavir. J Infect Dis. 2005;191:2046–2052. doi: 10.1086/430387. [DOI] [PubMed] [Google Scholar]
- 48.Moore DM, Hogg RS, Yip B, et al. Discordant immunologic and virologic responses to highly active antiretroviral therapy are associated with increased mortality and poor adherence to therapy. J Acquir Immune Defic Syndr. 2005;40:288–293. doi: 10.1097/01.qai.0000182847.38098.d1. [DOI] [PubMed] [Google Scholar]