

Abstract
Estrogens are considered to play a major role in promoting the proliferation of both the normal and the neoplastic breast epithelium. Their role as breast carcinogens has long been suspected and recently confirmed by epidemiological studies. Three major mechanisms are postulated to be involved in their carcinogenic effects: stimulation of cellular proliferation through their receptor-mediated hormonal activity, direct genotoxic effects by increasing mutation rates through a cytochrome P450-mediated metabolic activation, and induction of aneuploidy. Recently it has been fully demonstrated that estrogens are carcinogenic in the human breast by testing in an experimental system the natural estrogen 17β-estradiol (E2) by itself or its metabolites 2-hydroxy, 4-hydroxy, and 16-a-hydroxy-estradiol (2-OH-E2, 4-OH-E2, and 16-α-OH E2) respectively, by inducing neoplastic transformation of human breast epithelial cells (HBEC) MCF10F in vitro to a degree at least similar to that induced by the chemical carcinogen benz(a)pyrene (BP). Neither TAMOXYFEN (TAM) nor ICI-182,780 abrogated the transforming efficiency of estrogen or its metabolites. The E2 induced expression of anchorage independent growth, loss of ductulogenesis in collagen, invasiveness in Matrigel, is associated with the loss of 9p11-13 and only invasive cells that exhibited a 4p15.3-16 deletion were tumorigenic. Tumors were poorly differentiated ER-α and progesterone receptor negative adenocarcinomas that expressed keratins, EMA and E-cadherin. The E2 induced tumors and tumor-derived cell lines exhibited loss of chromosome 4, deletions in chromosomes 3p12.3-13, 8p11.1-21, 9p21-qter, and 18q, and gains in 1p, and 5q15-qter. The induction of complete transformation of the human breast epithelial cell MCF-10F in vitro confirms the carcinogenicity of E2, supporting the concept that this hormone could act as an initiator of breast cancer in women. This model provides a unique system for understanding the genomic changes that intervene for leading normal cells to tumorigenesis and for testing the functional role of specific genomic events taking place during neoplastic transformation.
Keywords: estrogen, invasiveness, CGH, breast cancer
1.Introduction
Breast cancer is a malignancy whose dependence on ovarian function was first recognized through the regression of both advanced cancer (1) and metastatic disease (2) induced by oophorectomy in premenopausal women. Ulterior correlation of ovarian function with estrogen production (3), and the isolation of the estrogen receptor protein (4,5), combined with the observed greater incidence of estrogen receptor positive tumors in postmenopausal women (6-16), led to the identification of a strong association of estrogen dose and length of exposure with increased breast cancer risk (6,10,13,14). The importance of ovarian steroidogenesis in normal breast development and in the genesis of breast cancer is highlighted by the facts that early menarche and late menopause are associated with greater breast cancer risk, whereas late menarche and early menopause, that occurring before 40 years of age, result in a significant reduction of the same (17-20). Breast development at puberty and during sexual maturity is stimulated by 17β-estradiol (E2), which is the predominant circulating ovarian steroid and the most biologically active hormone in breast tissue (21, 22). At menopause E2 plasma levels decrease by 90% (17-19). In spite of the markedly different circulating levels of estrogens in pre- and postmenopausal women, the concentrations of E2 in breast cancer tissues do not differ between these two groups of women, an indication that its uptake from the circulation might not contribute significantly to the total content of this hormone in breast tumors, but rather that de novo biosynthesis, i.e., peripheral aromatization of ovarian and adrenal androgens, plays a more significant role (23, 24).
Considerable epidemiological and clinical evidence link cumulative and sustained exposure to estrogens with increased risk of developing breast cancer. However, there is no clear understanding of the mechanisms through which estrogens cause cancer. In experimental animal models it has been demonstrated that E2, 11β-methoxyethinylestradiol (Moxestrol), and diethylstilbestrol (DES), as well as their 4-hydroxycatechols, induce kidney cancer in castrated male Syrian golden hamsters (25-27). In rats, continuous administration of supraphysiological doses of estrogens induces a high percentage of mammary adenocarcinomas, whereas low doses given over long periods induce fibroadenomas (28). In both models, however, the tumorigenic effects of estrogens are associated with marked hyperprolactinemia and pituitary hyperplasia resulting from an increase in number of hyperplastic prolactin secreting cells. The dependence on a functional pituitary gland has been further confirmed in hypophysectomized rats in which estrogens are ineffective as carcinogens (29). Nevertheless, the most widely acknowledged mechanism of estrogen carcinogenicity is its binding to its specific nuclear receptor alpha (ER-α) for exerting a potent stimulus on breast cell proliferation through its direct and/or indirect actions on the enhanced production of growth factors (21,22). However, the fact that ER-α knockout mice expressing the Wnt-1 oncogene (ERKO/Wnt-1) develop mammary tumors provides direct evidence that estrogens may cause breast cancer through a genotoxic, non-ER-α-mediated mechanism (30, 31). This postulate is further supported by the observations that when ovariectomized mice are supplemented with E2 they develop a higher tumor incidence with shorter latency time than controls, even in the presence of the pure antiestrogen ICI-182,780. Experimental studies on estrogen metabolism (32,33), formation of DNA adducts (34), carcinogenicity (35-37), mutagenicity (38), and cell transformation (39-42) have supported the hypothesis that reaction of specific estrogen metabolites, namely, catechol estrogen-3,4-quinones (CE-3,4-Q) and to a much lesser extent, CE-2,3-Q, can generate critical DNA mutations that initiate breast, prostate and other cancers (43).
2. Rationale for an in vitro-in vivo model of cell transformation
In order to definitively outline the pathways through which estrogens act as carcinogens in the human breast and for assessing whether one or more of the mechanisms described above are responsible of carcinogenic initiation, it is needed an experimental system in which E2 by itself or its metabolites induce transformation of human breast epithelial cells (HBEC) in a well controlled environment, preferentially in vitro. Recently it has been reported an in vitro-in vivo system of cell transformation that fulfills these requirements (44). Using this model it has been demonstrated that E2 and its metabolite 4-hydroxyestradiol (4-OH-E2) induce transformation of MCF-10F, an ER-αnegative human breast epithelial cell line (39-42, 45). In response to estrogen treatment the cells form colonies in agar methocel, lose the capacity to differentiate by forming three-dimensional structures when grown in a collagen matrix, or their ductulogenic capacity, forming instead spherical and solid masses, and exhibit an increase in cell proliferation and in their invasive capabilities in Matrigel (39-42, 45). More importantly, the expression of these phenotypes indicative of neoplastic transformation was not abrogated by their simultaneous treatment with the anti estrogen ICI-182,780 (ICI), suggesting that the transformation of MCF-10F cells by these compounds did not require the presence of the ER-α(40,41). E2-induced transformation of HBECin vitro increased the invasive potential of the cells. In addition, the selection of the most highly invasive cells in the Matrigel chambers identified transformed cells that express phenotypic and genotypic variations that correlate with their tumorigenic potential in a heterologous host, but still maintained their cell lineage characteristics. It also has been reported that the induced tumors exhibit genomic alterations that are similar to those reported in primary breast cancer, as determined by comparative genomic hybridization (CGH) (44).
3. The experimental model of transformation of MCF-10F cells by 17-β estradiol treatment
Treatment of the spontaneously immortalized ER-α and progesterone receptor (PgR) negative human breast epithelial cell line MCF-10F (Figure 1) with 70nM E2 twice a week for two weeks formed colonies in agar methocel (Figure 2) and the colony efficiency increased from 0 in controls to 12.0±1 in the treated cells. The positive control cells BP1-Tras and MDA-MB231 cells had a moderately (p<0.02) and significantly (p<0.001) higher colony efficiency than E2-transformed cells, respectively (44). This treatment also affected the ductulogenic pattern of cells grown in collagen gel (Figure 3), which was quantitatively evaluated by counting the total number of ductules and spherical masses formed by 10,000 cells plated in collagen. Control MCF-10F cells formed an average of 110 ductular structures, but did not form solid masses. After treatment with E2, MCF-10F cells almost completely lost their ductulogenic capacity, while acquiring the ability to form spherical solid masses (Figure 3). BP1-Tras and MDA-MB 231 exhibited a complete absence of ductule formation, forming instead solid masses in collagen gel whose values were not significantly different from those formed by E2-treated cells. The differences were highly significant (p<0.0001) (44).
Figure 1.
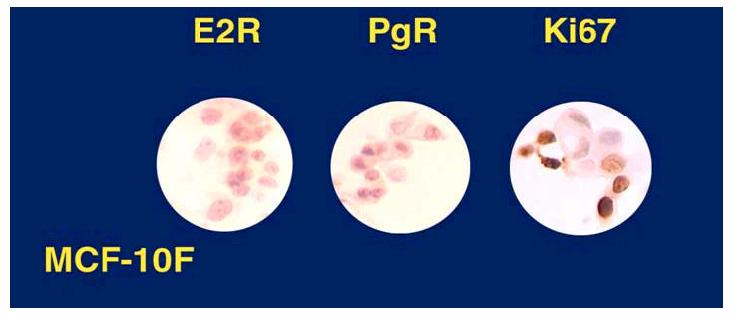
MCF-10F cells are proliferating cells (Ki67 positive), E2R (estrogen receptor) and PgR (progesterone receptor) negative.
Figure 2.
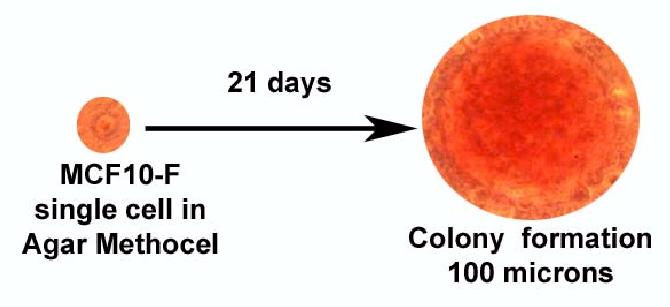
Anchorage independent growth is only observed in MCF-10F cells transformed with estradiol forming colonies of 100 micros in diameter.
Figure 3.
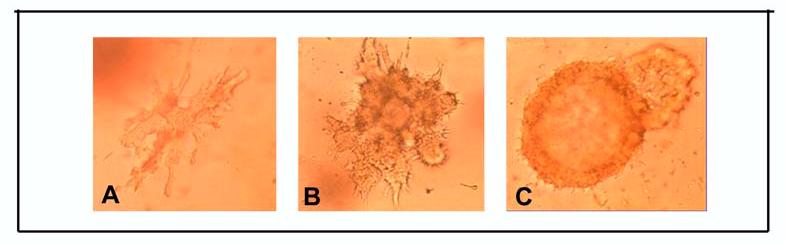
A) MCF-10F cells in collagen matrix form ductules, B) MCF-10F cells transformed with 70nM of E2 have lost the ability to form ductules and C) form solid masses. Phase contrast micrographs, X20)
The ability of cells to invade a Matrigel membrane in vitro is a widely accepted criterion of cell transformation. Control MCF-10F cells exhibited a low invasive capacity (Figure 4A), whereas the invasive capacity of E2-transformed cells at their 9th passage was significantly higher (Figure 4B) (44). BP1-Tras and MDA-MB231 cells had an invasive index that was significantly higher than that of MCF-10F control and E2 transformed cells (44)
Figure 4.
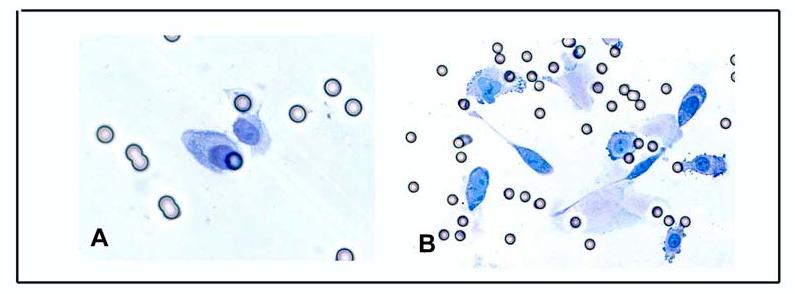
A)Invasion of f MCF-10F Control cells and B) E270nM transformed cells.
MCF-10F cells between passages 130-132 and E2-treated cells between their passages 7 and 9 were injected to 10 SCID mice each for testing their tumorigenic capabilities. Neither control nor E2-treated cells formed tumors after a six-month follow up period. Instead, BP1-Tras and MDA-MB231 cells were highly tumorigenic with a short latency period (44). Because the tumorigenic response of these two cell lines was associated with a highly invasive phenotype, it was tested whether selection of more invasive cells among E2-transformed MCF-10F cells would allow them to express the tumorigenic phenotype, and further to determine whether this phenotype was exclusively induced by estrogen, and not the result of the selection of more invasive control cells. For this purpose, MCF-10F cells in their 133rd passage and E2-treated MCF-10F cells in their 10th passage were trypsinized and seeded in the upper chamber of seven and four matrigel invasion chambers, respectively (44). Those cells that at 22 hours post-seeding had crossed the Matrigel membrane were cultured, giving origin to seven MCF-10F cell lines that were labeled A1 to A-7. From the E2-treated cells four lines were obtained, and were designated B2, C3, C4, and C5 (44). Injection of A1 to A7 cells to SCID mice did not induce a tumorigenic response even after six months of follow up. After injection of the E2-transformed cells B2, C3, C4, and C5 to SCID mice, only C3 and C5 were tumorigenic in 2/12 and 9/10 animals injected, respectively. The clone C5 produced tumors that were larger than the ones produced by C3 (Figure 5). From the 9 tumors obtained from C5 cells, four tumoral cell lines, designated C5-A1-T1, C5-A4-T4, C5-A6-T6 and C5-A8-T8 were derived. These cells were subsequently injected to another set of five SCID mice per cell line for testing their tumorigenic capabilities. All these cell lines formed palpable tumors, being C5-A8-T8 the fastest growing tumor. Cell lines B2 and C4 did not induce tumors even after a nine-month follow up (44).
Figure 5.
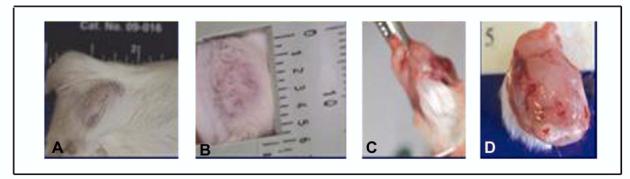
A-D) Palpable tumor formed by E2 70nM-C5 cells.
Histopathological analysis revealed that all the E2 70nM-C5 cells formed tumors and those tumors formed by their derived cells were poorly differentiated adenocarcinomas. They invaded the mammary fat pad and the skeletal muscle of the abdominal wall (Figure 6). The immunocytochemical reactivity of the E2-induced tumors in SCID mice were positive for AE1 and AE3, human low and high molecular weight cytokeratins were expressed in the cytoplasm of the neoplastic cells in all E2 induced tumors in a pattern similar to those observed in normal breast tissues, in primary invasive ductal carcinomas of the breast and in MCF-10Fcells. The cytokeratin peptide 7 and 8 (CAM5.2) diffusely stained the cytoplasm of neoplastic cells with greater variations in the degree of intensity than in the invasive ductal carcinoma of the breast used as positive control. E-cadherin was positive in all E2-induced carcinomas, exhibiting a diffuse and moderate reactivity, which was less intense than that observed in the invasive ductal carcinoma used as a positive control. Epithelial membrane antigen (EMA) had similar level of reactivity in E2-induced tumors than in primary breast cancer, in normal breast tissues and in MCF-10F cells. Estrogen receptor alpha (ER-α), which was positive in normal breast tissues and in primary breast cancer, was negative in MCF-10F cells and in all E2-induced tumors in SCID mice. The same pattern of reactivity was observed for progesterone receptor (44).
Figure 6.
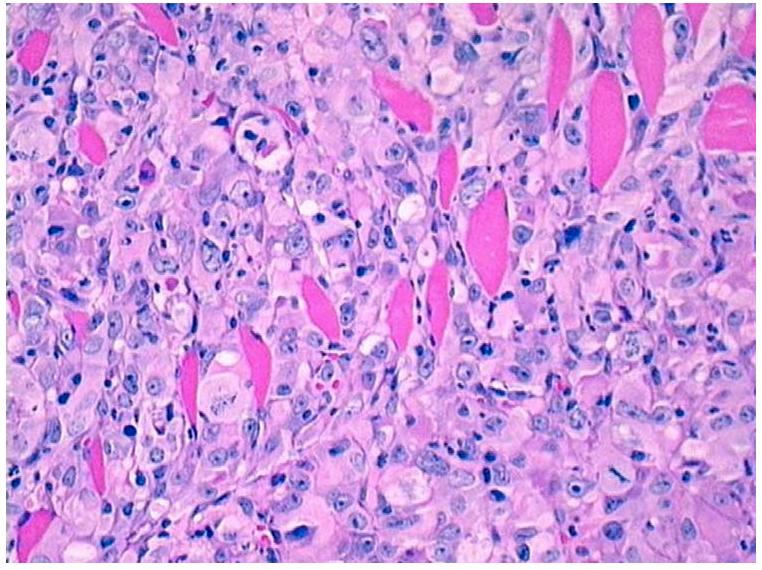
Histological section of an invasive adenocarcinoma growing in the fat pad of a SCID mouse (H&E, 10X).
In summary treatment of the immortalized estrogen receptor alpha (ER-α) negative human breast epithelial cell line MCF-10F with E2 and its metabolites 2- and 4-hydroxyestradiol induce anchorage independent growth, loss of ductulogenic pattern, and invasiveness in a Matrigel basement membrane (2,44, 46). The transforming capabilities of estrogens have been confirmed in MCF-10A, another ER-α negative immortalized human breast epithelial cell line in which E2 and estrogenic substances, such as Zeranol (Ralgro), a nonsteroidal agent with estrogenic activity that is used as a growth promoter in the U.S. beef and veal industry (47) and 4-hydroxyequilenin (48) induce anchorage independent growth. These observations support the concept that estrogens induce neoplastic transformation through non-receptor alpha mediated mechanisms, exerting direct genotoxic effects, as previously suggested (32-38). Our findings of specific mutations in p53, and loss of heterozygosity (LOH) in chromosomes 11 and 13 further support this concept (40, 49).
4. Genomic pathway of 17 beta estradiol induced neoplastic transformation
Using Comparative Genomic Hybridization (CGH) that is a molecular cytogenetic method for screening gains and losses at chromosomal and subchromosomal levels, it has been detected that MCF-10F cells transformed by E2 had lost 9p11-13, a loss that persisted in the invasive cell line E2-70 nM-C5 (Figure 7). This locus contains the serine protease family member PRSS3 (trypsinogen-IV), a putative tumor suppressor gene (50) in which an allelic imbalance has been reported in hepatocellular carcinoma (51), carcinoma in situ of the bladder (52) and renal cell carcinoma (53). The loss of 9p11-13 was not detected by CGH technique in the tumors and tumor-derived cell lines, probably because the change did not reach the threshold for detection or because the cell population was heterogeneous. However, losses in 9p21-pter were clearly evident in the tumor and tumor cell lines. Losses of chromosome 9 regions are frequently reported in bladder carcinoma, especially in premalignant lesions such as hyperplasia and carcinoma in situ (CIS). Simultaneous losses in 9p11-q12 and in 9p21 have been reported in CIS of the bladder (54). Losses in this locus have also been reported in peripheral T cell lymphoma (55), melanoma cell lines (56), malignant fibrous histiocytoma (57) and parathyroid adenomas (58). The 9p21-pter, region includes both the p16 and p15 genes. These observations indicate that loss of these tumor suppressor genes on 9p contribute to the progression of the invasive to the tumorigenic phenotypes in the natural progression of the disease.
Figure 7.
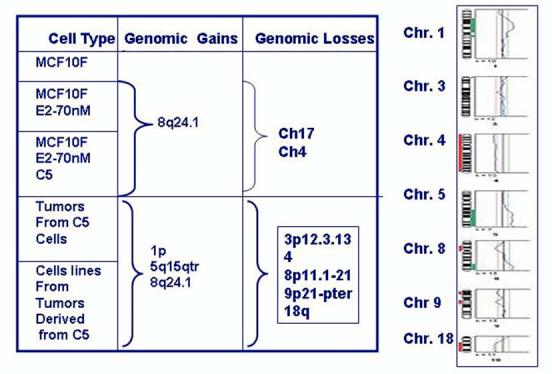
Comparative Genomic Hybridization (CGH) that is a molecular cytogenetic method for screening gains and losses at chromosomal and sub chromosomal levels.
5. Genomic changes during the tumorigenic stage of malignant transformation
E2 induces, in addition to the expression of early phenotypes of neoplastic transformation, tumorigenesis in a heterologous host (Figure 8). This phenomenon became possible only after the selection of invasive cells that exhibit specific changes, such as the deletion of chromosome 4p15.3-16, which was the first one, detected (Figures 7 and 8). Interestingly enough, injection of these cells to SCID mice resulted in the formation of tumors in which the entire chromosome 4 was deleted, a change that became a permanent feature of all tumors and tumor-derived cell lines. Allelic losses at one or both arms of chromosome 4 have been frequently reported in several tumor types, including breast cancers, either sporadic or occurring in BRCA1 and BRCA2 germline mutation carriers (59, 60). Regions that have been frequently reported to be deleted are 4p16.3 (50 %), 4p15.1-15.3 (57%), 4q25-26 (63%), and 4q33-34 (76%) (61). The tumors induced by E2-transformed cells in SCID mice are fast growing and ER negative, being similar in these aspects to the tumors exhibiting similar deletions and that are diagnosed in young women, in whom tumors are large at the time of diagnosis, having a high percentage of cells in S-phase and being negative for estrogen receptors (59, 60). Chromosome 4 contains numerous genes of potential interest in cancer development, among them is Slit2, a gene located at 4p15.2 that encodes a protein that inhibits leukocyte chemotaxis and is a putative ligand for the ROBO receptors gene (62). SLIT2 is primarily a secreted protein that in conditioned medium suppresses the growth of several breast cancer lines (62). Therefore the loss of the 4p15.3-16 region in E2-70 nM C5 cells could be the event that triggers a cascade that select tumorigenic cell population.
Figure 8.
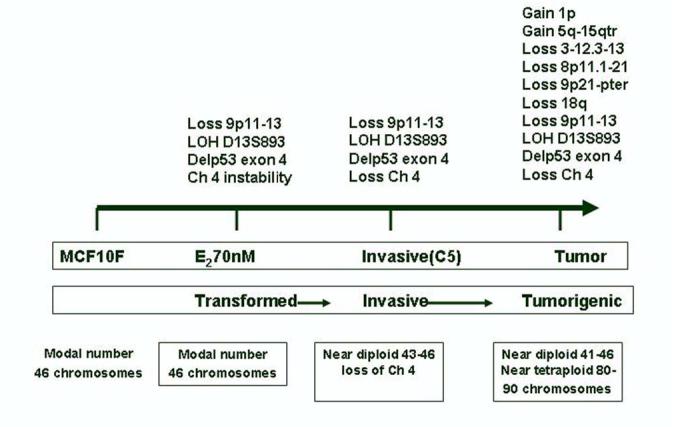
Schematic representation of the cumulative genomic changes observed in the MCF10F cells transformed with 17 beta estradiol.
Additional losses that were initially detected in the tumors and that were maintained in the tumor-derived cell lines were in chromosomes, 3 p12.3-13, 8 p11-21 and 18q. The region lost in chromosome 3 (p12.3-13) has been reported to exhibit imbalances in MCF-7 cells developing resistance to tamoxifen (63); the region 8p11-21 encodes the frizzled-related gene FRP1/FRZB, that is turned off in 78% of breast carcinomas (64), and associated with androgen in prostate cancer (65); the loss of chromosome arm 18q is a common event in primary breast cancers (60-70), ductal hyperplasia (71), and in breast cancer cell lines (72), and it is often interpreted as representing loss of one or more tumor-suppressor genes. The relevance of these losses in estrogen-induced cell transformation is that among the genes located in the q arm of chromosome 18 are two independent tumor-suppressor loci in segment 18q21.1, one at SMAD4 and the other potentially at an enhancer of DCC or an unrelated novel gene (66,70).
Treatment of MCF-10F cells with E2 induced genomic gains in 1p and 5q15-qter, both of which became evident in tumors and remained at the same level of expression in all tumor-derived cells. Amplification of 1p has already been reported in primary breast cancer (73-78) and in established breast cancer cell lines (79). Gain in 5q15-qter has not been frequently found in breast cancer (80), but it has been reported in previously immortalized human ovarian surface epithelial (HOSE) cells using HPV16E6E7 ORFs (81) and in the cell lines SW480 and SW620, derived from different stages of colon carcinoma in the same patient (82). Although at the present time the role played by these gains in 1p and 5q15-qter in the process of estrogen-induced tumorigenesis is not known, a likely explanation is that the gains resulted from amplifications of smaller chromosomal segments that probably arose through real DNA amplification processes, suggesting that many genes present in these chromosomal loci are potential targets for the carcinogenic effect of 17-β-estradiol (83).
6. Conclusions
17-β-estradiol is able to induce complete neoplastic transformation of human breast epithelial cells, as proven by the formation of tumors in SCID mice. This model demonstrates a sequence of chromosomal changes that correlates with specific stages of neoplastic progression. The data also support the concept that 17-β-estradiol can act as a carcinogenic agent without the need of the ERα, although we cannot rule out thus far the possibility that other receptors such as ERβ, or other mechanisms could play a role in the transformation of human breast epithelial cells. These are areas of active research in our laboratory. The knowledge that breast cancer in women is associated with prolonged exposure to high levels of estrogens gives relevance to this model of estrogen induced carcinogenesis (6,8-10,15,16). For this reason this model is extremely valuable for furthering our understanding of estrogen induced carcinogenicity.
Acknowledgements
This work was supported by grants DMAD17-00-1-0247, DMAD17-03-1-0229.
Footnotes
Grant Numbers and Sources of support: This work was supported by the U.S. Army Medical and Research Materiel Command under grants DAMD17-00-1-0247 and DAMD17-03-1-0229.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beatson G. On the treatment of inoperable cases of carcinoma of the mammary. Suggestions for a new method of treatment with illustrative cases. Lancet. 1896;2:104–107. [PMC free article] [PubMed] [Google Scholar]
- 2.Boyd S. An oophorectomy in cancer of the breast. Br. Med. J. 1900;2:1161–1167. [Google Scholar]
- 3.Block G. Estrogen excretion following operative and irradiation castration in cases of mammary cancer. Surgery. 1958;43:415–422. [PubMed] [Google Scholar]
- 4.Toft D, Shyamala G, Gorski J. A receptor molecule for estrogens: Studies using a cell free system. Proc. Natl. Acad. Sci. USA. 1967;57:1740–1743. doi: 10.1073/pnas.57.6.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Green S, Walter P, Greene G, Krust A, Goffin C, Jensen E, Scrace G, Waterfield M, Chambon P. Cloning of the human estrogen receptor cDNA. J. Steroid Biochem. 1986;24:77–83. doi: 10.1016/0022-4731(86)90035-x. [DOI] [PubMed] [Google Scholar]
- 6.Dorgan JF, Longcope C, Stephenson HE, Jr., Falk RT, Miller R, Franz C, Kahle L, Campbell WS, Tangrea JA, Schatzkin A. Relation of prediagnostic serum estrogen and androgen levels to breast cancer risk. Cancer Epidemiol. Biomarkers Prev. 1996;5:533–539. [PubMed] [Google Scholar]
- 7.Dorgan JF, Stanczyk FZ, Longcope C, Stephenson HE, Jr., Chang L, Miller R, Franz C, Falk RT, Kahle L. Relationship of serum dehydroepiandrosterone (DHEA), DHEA sulfate, and 5-androstate-3β, 17β-diol to risk of breast cancer in postmenopausal women. Cancer Epidemiol. Biomarkers Prev. 1997;6:177–181. [PubMed] [Google Scholar]
- 8.Thomas HV, Key TJ, Allen DS, Moore JW, Dowsett M, Fentiman IS, Wang DY. A prospective study of endogenous serum hormone concentrations and breast cancer risk in postmenopausal women on the island of Guernsey. Br. J. Cancer. 1997;76:401–405. doi: 10.1038/bjc.1997.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hankinson SE, Willett WC, Manson JE, Colditz GA, Hunter DJ, Spiegelman D, Barbieri RL, Speizer FE. Plasma sex steroid hormone levels and risk of breast cancer in postmenopausal women. J. Natl. Cancer Inst. 1998;90:1292–1299. doi: 10.1093/jnci/90.17.1292. [DOI] [PubMed] [Google Scholar]
- 10.Toniolo PG, Levitz M, Zeleniuch-Jacquotte A, Banerjee S, Koenig KL, Shore RE, Strax P, Pasternack BS. A prospective study of endogenous estrogens and breast cancer in postmenopausal women. J. Natl. Cancer Inst. 1995;87:190–197. doi: 10.1093/jnci/87.3.190. [DOI] [PubMed] [Google Scholar]
- 11.Berrino F, Muti P, Micheli A, Bolelli G, Krogh V, Sciajno R, Pisani P, Panico S, Secreto G. Serum sex hormone levels after menopause and subsequent breast cancer. J. Natl. Cancer Inst. 1996;88:291–296. doi: 10.1093/jnci/88.5.291. [DOI] [PubMed] [Google Scholar]
- 12.Garland CF, Friedlander NJ, Barrett-Connor E, Khaw K-T. Sex hormones and postmenopausal breast cancer: a prospective study in an adult community. Am. J. Epidemiol. 1992;135:1220–1230. doi: 10.1093/oxfordjournals.aje.a116228. [DOI] [PubMed] [Google Scholar]
- 13.Kabuto M, Akiba S, Stevens RG, Neriiski K, Land CE. A prospective study of estradiol and breast cancer in Japanese women. Cancer Epidemiol. Biomarkers Prev. 2000;9:575–579. [PubMed] [Google Scholar]
- 14.Cauley JA, Lucas FL, Kuller LH, Stone K, Browner W, Cummings SR, for the Study of Osteoporotic Fractures Research Group Elevated serum estradiol and testosterone concentrations are associated with a high risk for breast cancer. Ann. Intern Med. 1999;130:270–277. doi: 10.7326/0003-4819-130-4_part_1-199902160-00004. [DOI] [PubMed] [Google Scholar]
- 15.Helzlsouer KJ, Alberg AJ, Bush TL, Longcope C, Gordon GB, Comstock GW. A prospective study of endogenous hormones and breast cancer. Cancer Detect. Prev. 1994;18:79–85. [PubMed] [Google Scholar]
- 16.Endogenous Hormones and Breast Cancer Collaborative Group Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J. Natl. Cancer Inst. 2002;94:606–616. doi: 10.1093/jnci/94.8.606. [DOI] [PubMed] [Google Scholar]
- 17.Henderson BE, Ross R, Bernstein L. Estrogens as a cause of human cancer: the Richard and Hinda Rosenthal Foundation award lecture. Cancer Res. 1988;48:246–253. [PubMed] [Google Scholar]
- 18.Pike MC, Spicer DV, Dahmoush L, Press MF. Estrogens, progestagens, normal breast cell proliferation, and breast cancer risk. Epidemiol. Rev. 1993;15:17–35. doi: 10.1093/oxfordjournals.epirev.a036102. [DOI] [PubMed] [Google Scholar]
- 19.Kelsey JL, Gammon MD, John EM. Reproductive factors and breast cancer. Epidemiol. Rev. 1993;15:36–47. doi: 10.1093/oxfordjournals.epirev.a036115. [DOI] [PubMed] [Google Scholar]
- 20.Bernstein L, Ross RK. Endogenous hormones and breast cancer risk. Epidemiol. Rev. 1993;15:48–65. doi: 10.1093/oxfordjournals.epirev.a036116. [DOI] [PubMed] [Google Scholar]
- 21.Russo J, Ao X, Grill C, Russo IH. Pattern of distribution of cells positive for estrogen receptor alpha and progesterone receptor in relation to proliferating cells in the mammary gland. Breast Cancer Res. Treat. 1999;53:217–227. doi: 10.1023/a:1006186719322. [DOI] [PubMed] [Google Scholar]
- 22.Russo IH, Russo J. The role of estrogen in breast cancer. In: Russo J, Russo IH, editors. Molecular Basis of Breast Cancer Prevention and Treatment. Springer-Verlag; Heidelberg: 2005. pp. 89–136. [Google Scholar]
- 23.Miller WR, O’Neill J. The importance of local synthesis of estrogen within the breast. Steroids. 1987;50:537–548. doi: 10.1016/0039-128x(87)90037-7. [DOI] [PubMed] [Google Scholar]
- 24.Russo J, Hu YF, Russo IH. Estrogens and breast cancer in humans. In: Metzler M, editor. Endocrine disrupters of the Environment. Springer-Verlag; Heidelberg: 2000. pp. 1–26. [Google Scholar]
- 25.Li JJ, Li SA, Klicka JK, Parsons JA, Lam LK. Relative carcinogenic activity of various synthetic and natural estrogens in the Syrian hamster kidney. Cancer Res. 1983;43:5200–5204. [PubMed] [Google Scholar]
- 26.Li JJ, Li SA, Oberley TD, Parsons JA. Carcinogenic activities of various steroidal and nonsteroidal estrogens in the hamster kidney: Relation to hormonal activity and cell proliferation. Cancer Res. 1995;55:4347–4351. [PubMed] [Google Scholar]
- 27.Yager JD. Endogenous estrogens as carcinogens through metabolic activation. J. Natl. Cancer Institute Monogr. 2000;27:67–73. doi: 10.1093/oxfordjournals.jncimonographs.a024245. [DOI] [PubMed] [Google Scholar]
- 28.Russo IH, Russo J. Mammary Gland Neoplasia in Long-Term Rodent Studies. Environ. Health Perspect. 1996;104:938–967. doi: 10.1289/ehp.96104938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meites J. Relation of prolactin and estrogen to mammary tumorigenesis in the rat. J Natl. Cancer Inst. 1972;48:1217–1223. [PubMed] [Google Scholar]
- 30.Bocchinfuso WP, Korach KS. Mammary gland development and tumorigenesis in estrogen receptor knockout mice. J. Mammary Gland Biol. Neoplasia. 1997;2:323–334. doi: 10.1023/a:1026339111278. [DOI] [PubMed] [Google Scholar]
- 31.Bocchinfuso WP, Hively WP, Couse JF, Varmus HE, Korach KS. A mouse mammary tumor virus-Wnt1 transgene induces mammary gland hyperplasia and tumorigenesis in mice lacking estrogen receptor-alpha. Cancer Res. 1999;59:1869–1876. [PubMed] [Google Scholar]
- 32.Cavalieri EL, Kumar S, Todorovic R, Higginbotham S, Badawi AF, Rogan EG. Imbalance of estrogen homeostasis in kidney and liver of hamsters treated with estradiol: Implications for estrogen-induced initiation of renal tumors. Chem. Res. Toxicol. 2001;14:1041–1050. doi: 10.1021/tx010042g. [DOI] [PubMed] [Google Scholar]
- 33.Rogan EG, Badawi AF, Devanesan PD, Meza JL, Edney JA, West WW, Higginbotham SM, Cavalieri EL. Relative imbalances in estrogen metabolism and conjugation in breast tissue of women with carcinoma: Potential biomarkers of susceptibility to cancer. Carcinogenesis. 2003;24:697–702. doi: 10.1093/carcin/bgg004. [DOI] [PubMed] [Google Scholar]
- 34.Cavalieri EL, Stack DE, Devanesan PD, Todorovic R, Dwivedy I, Higginbotham S, Johansson SL, Patil KD, Gross ML, Gooden JK, Ramanathan R, Cerny RL, Rogan EG. Molecular origin of cancer: Catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc. Natl. Acad. 1997;99:10937–10942. doi: 10.1073/pnas.94.20.10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liehr JG, Fang WF, Sirbasku DA, Ari-Ulubelen A. Carcinogenicity of catecholestrogens in Syrian hamsters. J. Steroid Biochem. 1986;24:353–356. doi: 10.1016/0022-4731(86)90080-4. [DOI] [PubMed] [Google Scholar]
- 36.Li JJ, Li SA. Estrogen carcinogenesis in Syrian hamster tissue: Role of metabolism. Fed. Proc. 1987;46:1858–1863. [PubMed] [Google Scholar]
- 37.Newbold RR, Liehr JG. Induction of uterine adenocarcinoma in CD-1 mice by catechol estrogens. Cancer Res. 2000;60:235–237. [PubMed] [Google Scholar]
- 38.Chakravarti D, Mailander P, Li K-M, Higginbotham S, Zhang HL, Gross ML, Meza JL, Cavalieri EL, Rogan EG. Evidence that a burst of DNA depurination in SENCAR mouse skin induces error-prone repair and forms mutations in the H-ras gene. Oncogene. 2001;20:7945–7953. doi: 10.1038/sj.onc.1204969. [DOI] [PubMed] [Google Scholar]
- 39.Russo J, Lareef MH, Tahin Q, Hu YF, Slater C, Ao X, Russo IH. 17β-Estradiol is carcinogenic in human breast epithelial cells. J Steroid Biochem Mol Biol. 2002;80:149–162. doi: 10.1016/s0960-0760(01)00183-2. [DOI] [PubMed] [Google Scholar]
- 40.Russo J, Lareef MH, Balogh G, Guo S, Russo IH. Estrogen and its metabolites are carcinogenic agents in human breast epithelial cells. J. Steroid Biochem. Mol. Biol. 2003;87:1–25. doi: 10.1016/s0960-0760(03)00390-x. [DOI] [PubMed] [Google Scholar]
- 41.Lareef MH, Garber J, Russo PA, Russo IH, Heulings R, Russo J. The estrogen antagonist ICI-182,780 does not inhibit the transformation phenotypes induced by 17-beta-estradiol and 4-OH estradiol in human breast epithelial cells. Int. J. Oncol. 2005;26:423–429. [PubMed] [Google Scholar]
- 42.Fernandez SV, Russo IH, Lareef MH, Balsara B, Russo J. Comparative genomic hybridization of human breast epithelial cells transformed by estrogen and its metabolites. Int. J. Oncol. 2005;26:691–695. [PubMed] [Google Scholar]
- 43.Cavalieri E, Rogan E, Chakravarti D. The role of endogenous catechol quinones in the initiation of cancer and neurodegenerative diseases. In: Sies H, Packer L, editors. Methods in Enzymology, Quinones and Quinone Enzymes. Vol. 382. Elsevier, Duesseldorf; Germany: 2004. pp. 293–319. Part B. [DOI] [PubMed] [Google Scholar]
- 44.Russo J, Fernandez SV, Russo PA, Fernbaugh R, Sheriff FS, Lareef HM, Garber J, Russo IH. 17 beta estradiol induces transformation and tumorigenesis in human breast epithelial cells. FASEB J. 2006;20:1–13. doi: 10.1096/fj.05-5399com. [DOI] [PubMed] [Google Scholar]
- 45.Russo J, Hu YF, Tahin Q, Mihaila D, Slater C, Lareef MH, Russo IH. Carcinogenicity of estrogens in human breast epithelial cells. APMIS. 2001;109:39–52. doi: 10.1111/j.1600-0463.2001.tb00013.x. [DOI] [PubMed] [Google Scholar]
- 46.Russo J. Estrogens as carcinogens in the human breast. Proc. Am. Assoc. Cancer Res. 2005;46:SY14–1. [Google Scholar]
- 47.Liu S, Lin YC. Transformation of MCF-10A human breast epithelial cells by zeranol and estradiol-17beta. Breast J. 2004;10:514–521. doi: 10.1111/j.1075-122X.2004.21410.x. [DOI] [PubMed] [Google Scholar]
- 48.Cuendet M, Liu X, Pisha E, Li Y, Yao J, Yu L, Bolton JL. Equine estrogen metabolite 4-hydroxyequilenin induces anchorage-independent growth of human mammary epithelial MCF-10A cells: differential gene expression. Mutat. Res. 2004;550:109–121. doi: 10.1016/j.mrfmmm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 49.Fernandez SV, Russo IH, Russo J. Estradiol and its metabolites 4-hydroxyestradiol and 2-hydroxyestradiol induce mutations in human breast epithelial cells. Int. J. Cancer. 2006;118:1862–1868. doi: 10.1002/ijc.21590. [DOI] [PubMed] [Google Scholar]
- 50.Marsit CJ, Okpukpara C, Danaee H, Kelsey KT. Epigenetic silencing of the PRSS3 putative tumor suppressor gene in non-small cell lung cancer. Mol. Carcinog. 2005;44:146–150. doi: 10.1002/mc.20125. [DOI] [PubMed] [Google Scholar]
- 51.Zhang LH, Qin LX, Ma ZC, Ye SL, Liu YK, Ye QH, Wu X, Huang W, Tang ZY. Allelic imbalance regions on chromosomes 8p, 17p and 19p related to metastasis of hepatocellular carcinoma: comparison between matched primary and metastatic lesions in 22 patients by genome-wide microsatellite analysis. J. Cancer Res. Clin. Oncol. 2003;129:279–286. doi: 10.1007/s00432-002-0407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hopman AH, Kamps MA, Speel EJ, Schapers RF, Sauter G, Ramaekers FC. Identification of chromosome 9 alterations and p53 accumulation in isolated carcinoma in situ of the urinary bladder versus carcinoma in situ associated with carcinoma. Am. J. Pathol. 2002;161:1119–1125. doi: 10.1016/S0002-9440(10)64388-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Verdorfer I, Hobisch A, Hittmair A, Duba HC, Bartsch G, Utermann G, Erdel M. Cytogenetic characterization of 22 human renal cell tumors in relation to a histopathological classification. Cancer Genet. Cytogenet. 1999;111:61–70. doi: 10.1016/s0165-4608(98)00217-9. [DOI] [PubMed] [Google Scholar]
- 54.Sarkar S, Roy BC, Hatano N, Aoyagi T, Gohji K, Kiyama R. A novel ankyrin repeat-containing gene (Kank) located at 9p24 is a growth suppressor of renal cell carcinoma. J. Biol. Chem. 2002;277:36585–36591. doi: 10.1074/jbc.M204244200. [DOI] [PubMed] [Google Scholar]
- 55.Zettl A, Rudiger T, Konrad MA, Chott A, Simonitsch-Klupp I, Sonnen R, Muller-Hermelink HK, Ott G. Genomic profiling of peripheral T-cell lymphoma, unspecified, and anaplastic large T-cell lymphoma delineates novel recurrent chromosomal alterations. Am. J. Pathol. 2004;164:1837–1848. doi: 10.1016/S0002-9440(10)63742-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adam Z, Adany R, Ladanyi A, Timar J, Balazs M. Liver metastatic ability of human melanoma cell line is associated with losses of chromosomes 4, 9p21-pter and 10p. Clin. Exp. Metastasis. 2000;18:295–302. doi: 10.1023/a:1011043412634. [DOI] [PubMed] [Google Scholar]
- 57.Simons A, Schepens M, Jeuken J, Sprenger S, van de Zande G, Bjerkehagen B, Forus A, Weibolt V, Molenaar I, van den Berg E, Myklebost O, Bridge J, van Kessel AG, Suijkerbuijk R. Frequent loss of 9p21 (p16(INK4A)) and other genomic imbalances in human malignant fibrous histiocytoma. Cancer Genet. Cytogenet. 2000;118:89–98. doi: 10.1016/s0165-4608(99)00178-8. [DOI] [PubMed] [Google Scholar]
- 58.Tahara H, Smith AP, Gaz RD, Arnold A. Loss of chromosome arm 9p DNA and analysis of the p16 and p15 cyclin-dependent kinase inhibitor genes in human parathyroid adenomas. J. Clin. Endocrinol. Metab. 1996;81:3663–3667. doi: 10.1210/jcem.81.10.8855819. [DOI] [PubMed] [Google Scholar]
- 59.Johannsdottir HK, Johannesdottir G, Agnarsson BA, Eerola H, Arason A, Johannsson OT, Heikkila P, Egilsson V, Olsson H, Borg A, Nevanlinna H, Barkardottir RB. Deletions on chromosome 4 in sporadic and BRCA mutated tumors and association with pathological variables. Anticancer Res. 2004;24:2681–2687. [PubMed] [Google Scholar]
- 60.Zinn-Justin A, Ziegler A, Abel L. Multipoint development of the weighted pairwise correlation (WPC) linkage method for pedigrees of arbitrary size and application to the analysis of breast cancer and alcoholism familial data. Genet. Epidemiol. 2001;21:40–52. doi: 10.1002/gepi.1017. [DOI] [PubMed] [Google Scholar]
- 61.Shivapurkar N, Sood S, Wistuba II, Virmani AK, Maitra A, Milchgrub S, Minna JD, Gazdar AF. Multiple regions of chromosome 4 demonstrating allelic losses in breast carcinomas. Cancer Res. 1999;59:3576–3580. [PubMed] [Google Scholar]
- 62.Dallol A, Da Silva NF, Viacava P, Minna JD, Bieche I, Maher ER, Latif F. SLIT2, a human homologue of the Drosophila Slit2 gene, has tumor suppressor activity and is frequently inactivated in lung and breast cancers. Cancer Res. 2002;62:5874–5880. [PubMed] [Google Scholar]
- 63.Achuthan R, Bell SM, Roberts P, Leek JP, Horgan K, Markham AF, MacLennan KA, Speirs V. Genetic events during the transformation of a tamoxifen-sensitive human breast cancer cell line into a drug-resistant clone. Cancer Genet. Cytogenet. 2001;130:166–172. doi: 10.1016/s0165-4608(01)00475-7. [DOI] [PubMed] [Google Scholar]
- 64.Ugolini F, Adelaide J, Charafe-Jauffret E, Nguyen C, Jacquemier J, Jordan B, Birnbaum D, Pebusque MJ. Differential expression assay of chromosome arm 8p genes identifies Frizzled-related (FRP1/FRZB) and Fibroblast Growth Factor Receptor 1 (FGFR1) as candidate breast cancer genes. Oncogene. 1999;18:1903–1910. doi: 10.1038/sj.onc.1202739. [DOI] [PubMed] [Google Scholar]
- 65.Gelsi-Boyer V, Orsetti B, Cervera N, Finetti P, Sircoulomb F, Rouge C, Lasorsa L, Letessier A, Ginestier C, Monville F, Esteyries S, Adelaide J, Esterni B, Henry C, Ethier SP, Bibeau F, Mozziconacci MJ, Charafe-Jauffret E, Jacquemier J, Bertucci F, Birnbaum D, Theillet C, Chaffanet M. Comprehensive profiling of 8p11-12 amplification in breast cancer. Mol Cancer Res. 2005;3:655–667. doi: 10.1158/1541-7786.MCR-05-0128. [DOI] [PubMed] [Google Scholar]
- 66.Jakob J, Nagase S, Gazdar A, Chien M, Morozova I, Russo JJ, Nandula SV, Murty VV, Li CM, Tycko B, Parsons R. Two somatic biallelic lesions within and near SMAD4 in a human breast cancer cell line. Genes Chromosomes Cancer. 2005;42:372–383. doi: 10.1002/gcc.20142. [DOI] [PubMed] [Google Scholar]
- 67.Weber-Mangal S, Sinn HP, Popp S, Klaes R, Emig R, Bentz M, Mansmann U, Bastert G, Bartram CR, Jauch A. Breast cancer in young women (< or = 35 years): Genomic aberrations detected by comparative genomic hybridization. Int. J. Cancer. 2003;107:583–592. doi: 10.1002/ijc.11460. [DOI] [PubMed] [Google Scholar]
- 68.Richard F, Pacyna-Gengelbach M, Schluns K, Fleige B, Winzer KJ, Szymas J, Dietel M, Petersen I, Schwendel A. Patterns of chromosomal imbalances in invasive breast cancer. Int. J. Cancer. 2000;89:305–310. [PubMed] [Google Scholar]
- 69.Dellas A, Torhorst J, Schultheiss E, Mihatsch MJ, Moch H. DNA sequence losses on chromosomes 11p and 18q are associated with clinical outcome in lymph node-negative ductal breast cancer. Clin. Cancer Res. 2002;8:1210–1216. [PubMed] [Google Scholar]
- 70.Yokota T, Matsumoto S, Yoshimoto M, Kasumi F, Akiyama F, Sakamoto G, Nakamura Y, Emi M. Mapping of a breast cancer tumor suppressor gene locus to a 4-cM interval on chromosome 18q21. Jpn. J. Cancer Res. 1997;88:959–964. doi: 10.1111/j.1349-7006.1997.tb00315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kaneko M, Arihiro K, Takeshima Y, Fujii S, Inai K. Loss of heterozygosity and microsatellite instability in epithelial hyperplasia of the breast. J. Exp. Ther. Oncol. 2002;2:9–18. doi: 10.1046/j.1359-4117.2002.01001.x. [DOI] [PubMed] [Google Scholar]
- 72.Forozan F, Veldman R, Ammerman CA, Parsa NZ, Kallioniemi A, Kallioniemi OP, Ethier SP. Molecular cytogenetic analysis of 11 new breast cancer cell lines. Br. J. Cancer. 1999;81:1328–1334. doi: 10.1038/sj.bjc.6695007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nessling M, Richter K, Schwaenen C, Roerig P, Wrobel G, Wessendorf S, Fritz B, Bentz M, Sinn HP, Radlwimmer B, Lichter P. Candidate genes in breast cancer revealed by microarray-based comparative genomic hybridization of archived tissue. Cancer Res. 2005;65:439–447. [PubMed] [Google Scholar]
- 74.Loveday RL, Greenman J, Simcox DL, Speirs V, Drew PJ, Monson JR, Kerin MJ. Genetic changes in breast cancer detected by comparative genomic hybridisation. Int. J. Cancer. 2000;86:494–500. doi: 10.1002/(sici)1097-0215(20000515)86:4<494::aid-ijc8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 75.Weber-Mangal S, Sinn HP, Popp S, Klaes R, Emig R, Bentz M, Mansmann U, Bastert G, Bartram CR, Jauch A. Breast cancer in young women (< or = 35 years): Genomic aberrations detected by comparative genomic hybridization. Int. J. Cancer. 2003;107:583–592. doi: 10.1002/ijc.11460. [DOI] [PubMed] [Google Scholar]
- 76.Bieche I, Khodja A, Lidereau R. Deletion mapping of chromosomal region 1p32-pter in primary breast cancer. Genes Chromosomes Cancer. 1999;24:255–263. doi: 10.1002/(sici)1098-2264(199903)24:3<255::aid-gcc11>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 77.Ragnarsson G, Eiriksdottir G, Johannsdottir JT, Jonasson JG, Egilsson V, ngvarsson S. Loss of heterozygosity at chromosome 1p in different solid human tumours: association with survival. Br. J. Cancer. 1999;79:1468–1474. doi: 10.1038/sj.bjc.6690234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bieche I, Khodja A, Lidereau R. Deletion mapping in breast tumor cell lines points to two distinct tumor-suppressor genes in the 1p32-pter region, one of deleted regions (1p36.2) being located within the consensus region of LOH in neuroblastoma. Oncol. Rep. 1998;5:267–272. doi: 10.3892/or.5.1.267. [DOI] [PubMed] [Google Scholar]
- 79.Forozan F, Mahlamaki EH, Monni O, Chen Y, Veldman R, Jiang Y, Gooden GC, Ethier SP, Kallioniemi A, Kallioniemi OP. Comparative genomic hybridization analysis of 38 breast cancer cell lines: a basis for interpreting complementary DNA microarray data. Cancer Res. 2000;60:4519–4525. [PubMed] [Google Scholar]
- 80.Loveday RL, Greenman J, Simcox DL, Speirs V, Drew PJ, Monson JR, Kerin MJ. Genetic changes in breast cancer detected by comparative genomic hybridisation. Int. J. Cancer. 2000;86:494–500. doi: 10.1002/(sici)1097-0215(20000515)86:4<494::aid-ijc8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 81.Tsao SW, Wong N, Wang X, Liu Y, Wan TS, Fung LF, Lancaster WD, Gregoire L, Wong YC. Nonrandom chromosomal imbalances in human ovarian surface epithelial cells immortalized by HPV16-E6E7 viral oncogenes. Cancer Genet. Cytogenet. 2001;130:141–149. doi: 10.1016/s0165-4608(01)00473-3. [DOI] [PubMed] [Google Scholar]
- 82.Melcher R, Steinlein C, Feichtinger W, Muller CR, Menzel T, Luhrs H, Scheppach W, Schmid M. Spectral karyotyping of the human colon cancer cell lines SW480 and SW620. Cytogenet. Cell Genet. 2000;88:145–152. doi: 10.1159/000015508. [DOI] [PubMed] [Google Scholar]
- 83.Muleris M, Almeida A, Gerbault-Seureau M, Malfoy B, Dutrillaux B. Detection of DNA amplification in 17 primary breast carcinomas with homogeneously staining regions by a modified comparative genomic hybridization technique. Genes, Chromosomes Cancer. 1994;10:160–170. doi: 10.1002/gcc.2870100303. [DOI] [PubMed] [Google Scholar]