

Abstract
Elevated glycogen synthase kinase-3 (GSK-3) activity is associated with Alzheimer disease. We have found that collapsin response mediator proteins (CRMP) 2 and 4 are physiological substrates of GSK-3. The amino acids targeted by GSK-3 comprise a hyperphosphorylated epitope first identified in plaques isolated from Alzheimer brain. Expression of wild type CRMP2 in primary hippocampal neurons or SH-SY5Y neuroblastoma cells promotes axon elongation. However, a GSK-3-insensitive CRMP2 mutant has dramatically reduced ability to promote axon elongation, a similar effect to pharmacological inhibition of GSK-3. Hence, we propose that phosphorylation of CRMP proteins by GSK-3 regulates axon elongation. This work provides a direct connection between hyperphosphorylation of these residues and elevated GSK-3 activity, both of which are observed in Alzheimer brain.
Glycogen synthase kinase-3 (GSK-3)1 is a Ser/Thr kinase evolutionarily conserved from yeast to humans. In mammals, there are two closely related isoforms expressed from separate genes, namely GSK-3α and GSK-3β (1). Both are ubiquitously expressed, although highest expression is found in the brain (1). GSK-3 has been shown to phosphorylate more than 30 proteins involved in various cellular functions, including glycogen metabolism, signal transduction, apoptosis, gene transcription, microtubule dynamics, embryonic development, ubiquitin-mediated degradation, and nuclear/cytoplasmic trafficking, although only a handful of these proteins have been confirmed as physiological targets for GSK-3 (for reviews, see Refs. 2 and 3). Most GSK-3 substrates require prior phosphorylation by another kinase at a serine or threonine located 4 residues C-terminal to the site phosphorylated by GSK-3 (i.e. S/TXXXS/T(P), where X is any amino acid) (2). This downstream residue is termed a “priming” site and phosphorylation of the substrate by GSK-3 can be regulated indirectly by altering the activity of the “priming” kinase. Meanwhile, GSK-3 activity per se is regulated directly by phosphorylation at two distinct sites, Ser21/9 and Tyr279/216 in GSK-3α/β (4, 5). In addition, GSK-3 activity is regulated by interaction with inhibitory proteins. For example, activation of the Wnt signaling pathway results in the inhibition of GSK-3 activity, possibly by interaction with FRAT/GBP (6).
In the brain, GSK-3 is expressed throughout the cell body and processes of post-mitotic neurons (7, 8). Its expression is widespread throughout all regions of the developing and adult brain, although it is highest in the hippocampus, thalamus, cortex, and Purkinje cells of the cerebellum in the adult, which are regions of greatest neuronal plasticity (7-9). Elevated expression is also observed at the late embryonic/early post-natal period (7, 8, 10). Overexpression of GSK-3β activity in transgenic adult mice causes a relative reduction in the size of neuronal cell bodies (11, 12). Conversely, inhibition of GSK-3 in neuronal cell lines reduces axon elongation rates but increases the size of axon growth cones (13). GSK-3 is also implicated in nerve growth factor control of axon growth (14). These observations suggest that GSK-3 is an important regulator of neuronal process extension and synapse formation, although little is known about the mechanisms regulating the downstream effectors of GSK-3 in the brain.
Our data show that phosphorylation of specific residues on collapsin response mediator protein (CRMP) 2 by GSK-3 is a key part of the mechanism by which these molecules combine to induce axon growth and identifies GSK-3 as the modulator of an Alzheimer-related epitope.
EXPERIMENTAL PROCEDURES
Cloning, Mutagenesis, and Protein Expression
The cDNA encoding full-length hCRMP2 was amplified by PCR from Image clone #6177866 using the primers 5′-GGATCCGCCACCATGGACTACAAGGACGACG-ATGACAAGTCTTATCAGGGGAAGAAAAATATTCCACGC-3′ and 5′-GAATTCTTAGCCCAGGCTGGTGATGTTGGC-3′. The cDNA encoding full-length hCRMP4 was amplified by PCR from Image clone #5725550 using the primers 5′-GAATTCGCCACCATGGACTACAAGGACGACG-ATGACAAGTCCTACCAAGGCAAGAAGAACATCCCG-3′ and 5′-GAA-TTCTTAACTCAGAGATGTGATATTAGAACGGCCG-3′. The PCR products were subcloned into pRK5 for mammalian or pGEX-6 for bacterial expression. The mutants hCRMP2/4[S522A/D] were generated using the QuikChange mutagenesis kit (Stratagene). Recombinant DYRK2 were expressed in Escherichia coli BL21 cells as GST-tagged proteins, while GSK-3β was expressed as a His6-tagged protein in Sf21 cells as described previously (15).
Purification of GSK-3 Substrates from Rat Brain
The identification of novel GSK-3 substrates from brain (50 male Sprague-Dawley rats) by KESTREL was performed as described previously (16).
Mass Spectrometry
Tryptic peptides were analyzed using an Applied Biosystems 4700 Proteomics Analyser MALDI-TOF-TOF mass spectrometer with 5 mg/ml α-cyanocinnamic acid as the matrix. Mass spectra were acquired in the reflector mode and peptide sequences were confirmed by high energy tandem MS/MS of selected precursors. The precursor ion masses of tryptic peptides and the daughter ions from MS/MS experiments were scanned against non-redundant protein databases (Celera) using Mascot search software (Matrix Science). Phosphorylated tryptic peptides were enriched by incubating the tryptic digests for 20 min with 2 μl of PHOS-select™ metal chelate beads (Sigma) in 0.25 M acetic acid/30% (v/v) acetonitrile. The beads were then packed into a pipette tip, washed with the same buffer, and the peptides eluted with 20 μl of 0.4 M ammonium hydroxide. Solid phase sequencing of 32P-labeled peptides was performed as described previously (17).
Phosphorylation Assays
Purified rCRMP2/4 was incubated with 1 milliunit or 5 milliunits of His6-GSK-3β, 10 mM magnesium acetate, and 0.1 mM [γ-32P]ATP in buffer containing 50 mM Tris-HCl, pH 7.5, 0.03% (v/v) Brij 35, and 0.1% (v/v) 2-mercaptoethanol at the concentrations and times indicated. Reactions were subjected to SDS-PAGE, transferred to nitrocellulose, and autoradiographed. 32P-Labeled rCRMP bands were analyzed by Cerenkov counting. rCRMP2/4 was incubated with 10 milliunits of the phosphatase PP1, with or without microcystin (1 μM), for 30 min at 30 °C. Microcystin was then added to the tubes that did not already contain it, followed by addition of 5 milliunits of His6-GSK-3β, 10 mM magnesium acetate, and 0.1 mM [γ-32P]ATP to all tubes (1 h, 30 °C). Relative amounts of phosphate incorporated into rCRMP2/4 were determined as described above. Recombinant GST-hCRMP4 (1 μM) was phosphorylated using 25 milliunits of His6-GSK-3β or GST-DYRK2, 10 mM magnesium acetate, and 0.1 mM [γ-32P]ATP at 30 °C for the times indicated. For priming experiments, GST-hCRMP4 (1 μM) was phosphorylated using 2.5 milliunits/μl GST-DYRK2, 10 mM magnesium acetate, and 0.1 mM unlabeled ATP for 30 min at 30 °C. GST-DYRK2 was removed from the reaction mixture by addition of Ni2+-agarose and the supernatant incubated with His6-GSK-3β (2.5 milliunits/μl), Mg-[γ-32P]ATP (final concentration: 10 mM magnesium acetate, 0.1 mM ATP) at 30 °C for the times indicated.
Transfection of HEK293 and SH-SY5Y Cells
HEK293 cells were transfected with wild type and mutant hCRMP2/4 expression vectors using the calcium phosphate method. DNA was incubated with the cells for 4 h, then the medium changed and either Me2SO or the GSK-3β inhibitor CT-99021 (2 μM) was added for 16 h. Cell lysates containing 0.5 mg of protein were immunoprecipitated with anti-FLAG-agarose beads overnight at 4 °C and then subjected to SDS-PAGE. Gels were stained with ProQ Diamond phospho-specific stain (Molecular Probes), followed by CBR-250. SH-SY5Y neuroblastoma cells were transfected using LipofectAMINE 2000 (Invitrogen). Following incubation at 37 °C for 60 h, the cells were fixed in 4% (w/v) paraformaldehyde in phosphate-buffered saline (PBS), permeabilized with 0.1% (v/v) Triton X-100 in PBS, blocked in 10% (v/v) fetal bovine serum, 0.5% (w/v) bovine serum albumin, and 0.1% (v/v) Triton X-100 in PBS, then incubated with an anti-FLAG monoclonal antibody (Sigma), followed by an antimouse secondary antibody pre-conjugated to the Alexa488 fluorophore.
Hippocampal Neuron Cell Culture, Transfection, and Immunofluorescence Analysis
Hippocampi isolated from 2-3-day-old Sprague-Dawley rats were isolated as described previously (18). Following plating, cells were incubated in neurobasal medium containing 2% (v/v) B27 serum replacement (Sigma) and 2 mM L-glutamine for 3-4 h and then transfected using LipofectAMINE 2000. After 36 h, the cells were fixed, permeabilized, and blocked as described above. The cells were sequentially incubated with anti-FLAG and anti-MAP2 (rabbit polyclonal; Chemicon) antibodies and then incubated simultaneously with antimouse and anti-rabbit secondary antibodies, conjugated to Alexa488 and Cy5 fluorophores, respectively. A laser scanning confocal imaging system (LSM 510 Carl Zeiss, Oberkochen, Germany) and accompanying software was used for image acquisition and analysis (cells analyzed; n = 35 GFP, n > 80 hCRMP2 and n > 80 hCRMP2[S522A]).
RESULTS AND DISCUSSION
Identification of a Novel Substrate of GSK-3
The KESTREL technique (16) was used to identify novel GSK-3 substrates in rat brain lysate. Rat brains were homogenized and fractionated by heparin chromatography. An aliquot from each fraction was incubated with recombinant His6-GSK-3β and radiolabeled ATP. In addition to the GSK-3β autophosphorylation band, several 32P-labeled proteins (potential GSK-3 substrates) of varying molecular weight were detected. Of particular interest were a group of 32P-labeled bands present at Mr 60,000-70,000. The fractions containing these proteins were further purified by ion exchange and gel filtration chromatography. These substrates (62,000-64,000 Mr) were excised from a gel, digested with trypsin, and identified by peptide mass fingerprinting. The masses of 28 peptides were matched with rat collapsin response mediator protein (rCRMP) 2 (accession number gi 1351260), covering 62.8% of the protein, while the masses of 25 peptides were matched with rCRMP4 (accession number gi 25742568), covering 51.6% of the protein. Some of the peptides detected were common to both isoforms, although many were specific for either isoform. Both isoforms contained consensus sequences for phosphorylation by GSK-3. No other proteins were detectable in the tryptic digest mixture. CRMPs are a family of five microtubule-associated proteins primarily expressed during neuronal development (19-21). CRMP2 was first identified in a screen for proteins that mediate Semaphorin 3A activity (22). It is key to axon formation during neuronal polarization and is enriched in elongating axons of the hippocampus (23). CRMP2 binds to tubulin heterodimers in preference to polymerized tubulin and is thought to play a regulatory role in polymerization of microtubules (24).
CRMP Phosphorylation by GSK-3 in Vitro
The mixture of purified rCRMP2 and rCRMP4 was incubated with GSK-3β and γ-32P]ATP to label the proteins and two 32P-labeled bands of roughly 62 and 64 kDa were observed by SDS-PAGE. The bands were individually digested and phosphorylated tryptic peptides enriched using immobilized metal ion affinity chromatography (IMAC) before being subjected to MS analysis. Three phosphopeptides were detected in each band (T1-T3 in Fig. 1A), while four phosphorylation sites were determined by analyzing the MS/MS sequence data using Mascot software (data for the lower band is shown in Fig. 1B). They represented equivalent residues in each isoform; namely Ser522, Ser518, Thr514, and Thr509 (Fig. 1B). Phosphorylation of Thr509 is the first example of a GSK-3 phosphorylation site located 5 residues N-terminal to a Ser or Thr residue. Thus, the consensus sequence for phosphorylation by GSK-3 will have to be modified to include this variation. The IMAC-enriched phosphopeptides (T1-T3) were also subjected to N-terminal solid phase sequence analysis. γ-32P was detected in cycles corresponding to phosphorylation of Thr514, Ser518, and Thr509, respectively (data not shown). γ-32P was not detected in Ser522, but a phosphopeptide containing phospho-Ser522 was detected by MS (T2 in Fig. 1B), suggesting that Ser522 was already phosphorylated when purified from rat brain. Therefore, Ser522 is phosphorylated in intact brain and acts as a priming site for subsequent GSK-3 phosphorylation.
FIG.1.
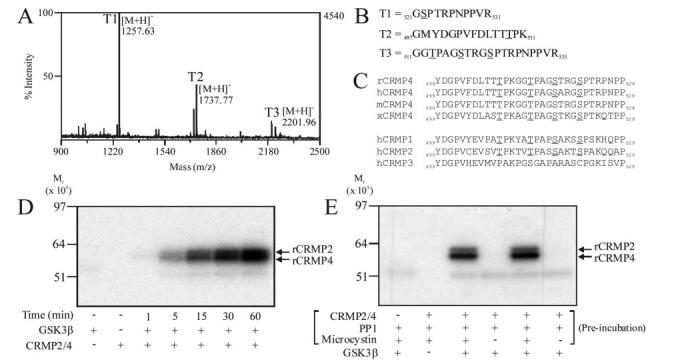
Identification of the residues in rCRMP phosphorylated by GSK-3β. Purified rCRMP was phosphorylated using GSK-3β and then subjected to SDS-PAGE. Phosphorylated tryptic peptides were isolated using IMAC chromatography and identified using MALDI-TOF mass spectrometry. A, MS of tryptic phospho-peptides from rCRMP4. B, peptide identification of T1, T2, and T3. C, sequence alignment of residues Tyr499 to Pro529 of rCRMP4 with other species (rat, human, mouse, Xenopus), as well as other CRMP isoforms. Phosphorylated residues are underlined. D, purified rat brain CRMP2/4 was labeled for the times indicated using GSK-3β and the reactions subjected to SDS-PAGE, electrotransferred to nitrocellulose membrane, and autoradiographed. E, purified rCRMP2/4 was preincubated with the protein phosphatase PP1, with or without the phosphatase inhibitor microcystin as indicated. Microcystin was then added to the tubes that did not already contain it followed by 5 milliunits of GSK-3β and [γ-32P]ATP. Phosphate incorporated in each reaction is presented.
To compare the efficiency of phosphorylation by GSK-3β, the purified rCRMP isoforms were incubated with GSK-3β and [γ-32P]ATP for up to 1 h (Fig. 1D). The amount of γ-32P incorporated into rCRMP2 and rCRMP4 increased proportionally with time (Fig. 1D). Similar rates of incorporation of γ-32P into each isoform were observed. Preincubation of purified rCRMP2 and rCRMP4 with the protein phosphatase PP1 (to remove endogenous phosphorylation) ablated the ability of GSK-3β to phosphorylate rCRMP2/4 in a subsequent reaction (Fig. 1E). This confirms that at least a portion of the purified rCRMP is phosphorylated at Ser522, priming for subsequent phosphorylation by GSK-3. Similarly, GSK-3β did not phosphorylate bacterially expressed GST-hCRMP4 to any appreciable extent (Fig. 2), demonstrating that Ser522 is not a target for GSK-3. Examination of the CRMP4 primary sequence surrounding Ser522 identified dual tyrosine regulated kinase (DYRK) as a likely Ser522 kinase. Indeed, purified DYRK2 phosphorylated GST-hCRMP4 almost stoichiometrically (Fig. 2A). Meanwhile, MS analysis identified Ser522 as the major (and possibly only) residue phosphorylated by DYRK2 (data not shown). Therefore, GST-hCRMP4 was preincubated with DYRK2 to promote phosphorylation of Ser522, prior to incubation with GSK-3β. Nearly 1.2 mol of phosphate/mol of GST-hCRMP4 was incorporated into hCRMP4 by GSK-3β but only following preincubation of hCRMP4 with DYRK2 (Fig. 2B). The residues in GST-hCRMP4 phosphorylated by GSK-3 were identified by MS as Thr509, Thr514, and Ser518 (data not shown). Therefore, Ser522 phoshorylation is sufficient to prime CRMP4 for subsequent phosphorylation by GSK-3β in vitro.
FIG.2.
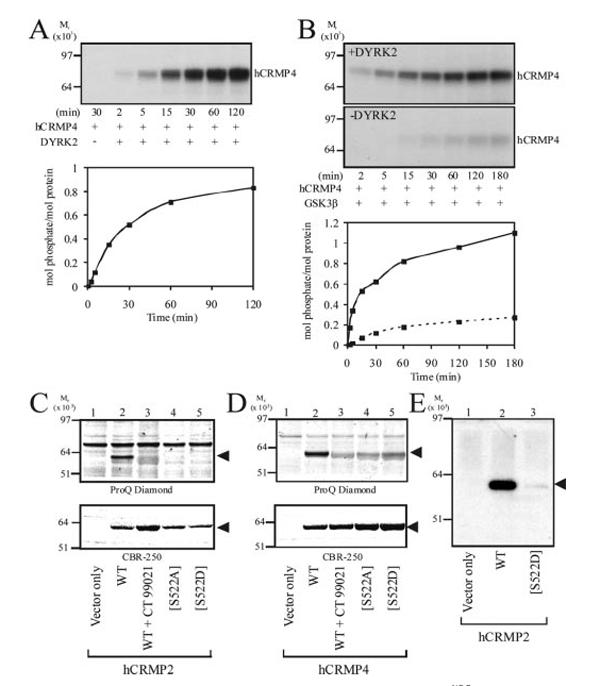
GST-hCRMP4 requires priming at Ser522 for subsequent phosphorylation by GSK-3β. A, GST-hCRMP4 was incubated with DYRK2 and [γ-32P]ATP for the times indicated, prior to SDS-PAGE and autoradiography. Stoichiometry of phosphorylation at each time point is presented below the autoradiograph. B, GST-hCRMP4 was incubated with (upper panel) or without (lower panel) DYRK2 and non-radioactive ATP for 30 min. DYRK2 was removed using Ni2+-agarose and the supernatant incubated with His6-GSK-3β and [γ-32P]ATP. Aliquots were removed and analyzed as described for A. The stiochiometry of phosphorylation is presented below the autoradiograph (dashed line in the absence of DYRK2 preincubation). C, HEK293 cells were transfected with expression vectors as indicated and then incubated with the GSK-3 inhibitor CT-99021 (lane 3) or Me2SO. FLAG-tagged hCRMP2 was immunoprecipitated, subjected to SDS-PAGE, and phosphorylated proteins detected using ProQ Diamond phospho-specific stain (upper panel). Gels were subsequently stained with CBR-250 to detect protein content (lower panel). D, HEK293 cells were transfected with the equivalent constructs for hCRMP4 and analyzed as described for C. E, HEK293 cells transfected with the indicated expression vectors were lysed and hCRMP2 proteins immunoprecipitated subjected to Western blotting using an antibody that specifically recognizes hCRMP2 only when phosphorylated at both Thr509 and Thr514. hCRMP proteins in C, D, and E are indicated by arrows.
CRMP Phosphorylation by GSK-3 in Cells
It is important to demonstrate that phosphorylation observed in vitro also occurs in vivo. Therefore, HEK293 cells were transfected with wild type FLAG-tagged hCRMP2 or hCRMP4 in the presence or absence of the GSK-3 inhibitor CT-99021 (Fig. 2C). This compound is the most selective GSK-3 inhibitor reported to date (25). Following cell lysis, hCRMP2 and hCRMP4 were immunopurified using anti-FLAG-agarose beads and subjected to SDS-PAGE. The gels were stained with ProQ Diamond, which specifically detects phospho-proteins and then CBR-250 to quantify loading. Both hCRMP2 (Fig. 2C) and hCRMP4 (Fig. 2D) were phosphorylated; however, this was completely inhibited by co-incubation with CT-99021. In addition, expression of the priming site mutants Ser522 → Ala (S522A) and Ser522 → Asp (S522D) prevented phosphorylation of either isoform (Fig. 2, C and D). Phosphorylation of wild type hCRMP2 was confirmed using a phospho-specific antibody raised against a peptide representing hCRMP2-(505-517) phosphorylated at Thr509 and Thr514 (Fig. 2E). This antibody recognizes dual phosphorylated peptide (509 and 514) but not peptide phosphorylated at only 509 or 514. This antibody did not detect hCRMP2[S522D]. Together, these data demonstrate that hCRMP2 and hCRMP4 are phosphorylated at three residues by endogenous GSK-3 in intact cells and that phosphorylation of Ser522 is critical for priming in vivo. In addition, mutation of Ser522 to the negatively charged residue aspartate (S522D) does not substitute for priming, while the non-consensus phosphorylation of Thr509 by GSK-3 occurs in intact cells.
Functional Consequences of CRMP2 Phosphorylation by GSK-3 in Neuronal Cells
It has previously been reported that transfection of N1E-115 neuroblastoma cells or primary neurons with hCRMP2 enhances neurite formation and growth (24). Therefore, we transfected the human neuroblastoma cell line, SH-SY5Y, with mammalian expression vectors containing either GFP, hCRMP2, or hCRMP2[S522A] and incubated for 60 h (in the absence of other differentiation factors (26)). Transfected cells were detected by immunoflourescence and analyzed using LSM 510 image software. Transfection with hCRMP2 induced a 2-3-fold increase in the number of transfected cells bearing neurites greater than 20 μm in length and a 4-fold increase in cells bearing neurites greater than 50 μm in length when compared with cells transfected with GFP (Fig. 3, A-C). In contrast, transfection with hCRMP2[S522A] only increased the number of cells bearing neurites greater than 20 μm in length ∼1.5-fold over control, while there was no significant difference in the number of cells with neurites greater than 50 μm in length between hCRMP2[S522A] and control cells.
FIG.3.
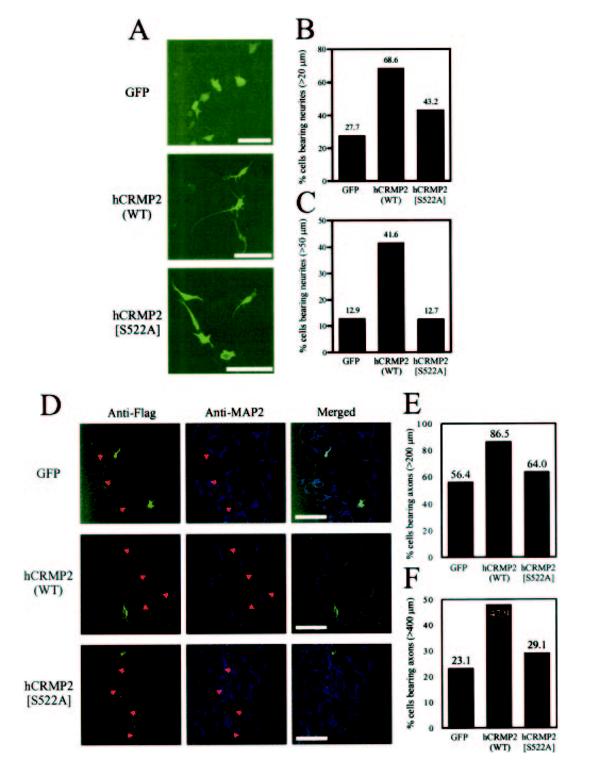
Phosphorylation of hCRMP2 by GSK-3 is required for neurite formation and elongation. A, SH-SY5Y cells were transfected with expression vectors containing GFP, hCRMP2, or hCRMP2[S522A] and incubated at 37 °C for 60 h. Cells transfected with hCRMP2 or hCRMP2[S522A] were detected using an anti-FLAG monoclonal antibody, followed by treatment with an Alexa488-conjugated secondary antibody, and neurite lengths were subsequently analyzed using LSM 510 image analysis software. The scale bar is equivalent to 100 μm. Cells bearing neurites greater than 200 μm (B) or 50 μm (C)in length were determined as a percentage of the total number of cells analyzed for each sample group (n > 100 in each group). D, confocal images of hippocampal neurons transfected with expression vectors containing GFP, hCRMP2, or hCRMP2[S522A]. Cells transfected with hCRMP2 or hCRMP2[S522A] were detected using an anti-FLAG antibody and neurones identified with an antibody that recognizes the somato-dendritic marker protein, MAP2. Axon length was obtained using LSM 510 software. The scale bar is equivalent to 100 μm. The proportion of cells with axons greater than 200 μm (E) or 400 μm (F)in length is presented for each sample group. Average axon length is given in the text.
In a separate experiment, primary hippocampal cultures were transfected with the same constructs (Fig. 3, D and E). Axons and dendrites were differentially identified according to their morphology and staining for the somato-dendritic marker, MAP2. As reported previously (24), cells transfected with wild type hCRMP2 display a significant increase in axon elongation (427.5 μm versus 280.11 μm average axon length, p < 0.0005) when compared with GFP-transfected neurons (Fig. 3, D-F). There was a trend (but no significant difference) toward increased axon elongation in cells transfected with hCRMP2[S522A] (average length: 305.9 μm) compared with control cells. However, the average axon length of transfected neurons containing wild type hCRMP2 was significantly greater (p < 0.0002) than those containing hCRMP2[S522A]. In addition, clear differences in the number of cells with axons over 200 or 400 μm were observed (Fig. 3, D-F). These data demonstrate that CRMP2 expression can promote axon outgrowth in primary neurons and initiate neurite extension in a neuroblastoma, while phosphorylation by GSK-3 greatly enhances these actions.
Neuronal polarity requires a series of complex intracellular processes to be initiated and regulated. For example, manipulation of actin structure allows transport of axon specific proteins that designate this phenotype to the appropriate neurite. Meanwhile, assembly of microtubules is required for axon outgrowth and growth cone positioning (19). Inhibition of GSK-3 is known to reduce axon outgrowth (13) and also protect against neuronal apoptosis (27, 28). Indeed, many microtubule-associated proteins that regulate the rate and direction of axon growth are known to be substrates for GSK-3. For example, GSK-3 phosphorylates tau, promoting dissociation of this protein from microtubules (29). In addition, phosphorylation of microtubule-associated protein 1B (MAP1B) regulates microtubule dynamics, presumably via similar mechanisms to tau (13). In this way, GSK-3 is proposed to regulate microtubule stability. We now propose that phosphorylation of CRMP proteins is another mechanism by which GSK-3 regulates axon growth.
There is considerable genetic and biochemical evidence that GSK-3 influences the progression of Alzheimer disease (AD). Patients with AD develop neuronal plaques that contain insoluble deposits of β-amyloid protein. The production and aggregation of the β-amyloid protein is promoted by GSK-3α while reduced by pharmacological inhibition of GSK-3α (30). In addition, AD tissue commonly contains neurofibrillary tangles, which are formed in part by hyperphosphorylation of the microtubule-associated protein tau, another well characterized substrate of GSK-3. Hence, the precise role(s) of GSK-3 in the development of AD and other related “tau-opathies” remains the subject of much study. Interestingly, abnormal CRMP2 expression has been linked to AD development, while hyperphosphorylated CRMP2 is found in AD tissue, and the residues that exhibit increased phosphorylation match those identified as targets for GSK-3 in this study (31). Of course, whether abnormal phosphorylation of CRMP2 by GSK-3 is a cause or a consequence of AD will require further study. We now show that axon growth will be affected by hyperphosphorylation of this epitope following up-regulation of GSK-3 activity in AD.
In summary, GSK-3 phosphorylates CRMP2 and CRMP4 on a cluster of three residues, and this modification regulates a major function of CRMP2. This interaction will play a role in the observed effect of GSK-3 inhibitors on axon growth and neuronal polarity, while loss of regulation of GSK-3 and/or the Ser522 kinase (and hence CRMP phosphorylation) may explain the association of both GSK-3 and CRMP2 with the development of a number of neurological disorders.
Acknowledgments
We thank James Hastie, Hilary Mclauchlan, Rudolfo Marquez, and Sir Philip Cohen, School of Life Sciences, University of Dundee for reagents and Dervla O’Malley, Dave Finlay, Chris Lipina, Hilary Laidlaw, and Laura Burgess for technical advice and assistance.
Footnotes
This work was supported in part by the Biotechnology and Biological Sciences Research Council (94/C18727). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- GSK-3
- glycogen synthase kinase-3
- CRMP
- collapsin response mediator protein
- rCRMP
- rat CRMP
- hCRMP
- human CRMP
- DYRK
- dual tyrosine-regulated kinase
- MS
- mass spectrometry
- GST
- glutathione S-transferase
- MALDI-TOF
- matrix-assisted laser desorption ionization time-of-flight
- PBS
- phosphate-buffered saline
- GFP
- green fluorescent protein
- IMAC
- immobilized metal ion affinity chromatographyl
- AD
- Alzheimer disease
REFERENCES
- 1.Woodgett JR. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frame S, Cohen P. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doble BW, Woodgett JR. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sutherland C, Leighton IA, Cohen P. Biochem J. 1993;296:15–19. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cole A, Frame S, Cohen P. Biochem J. 2004;377:249–255. doi: 10.1042/BJ20031259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas GM, Frame S, Goedert M, Nathke I, Polakis P, Cohen P. FEBS Lett. 1999;458:247–251. doi: 10.1016/s0014-5793(99)01161-8. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi M, Tomizawa K, Kato R, Sato K, Uchida T, Fujita SC, Imahori K. J Neurochem. 1994;63:245–255. doi: 10.1046/j.1471-4159.1994.63010245.x. [DOI] [PubMed] [Google Scholar]
- 8.Leroy K, Brion JP. J Chem Neuroanat. 1999;16:279–293. doi: 10.1016/s0891-0618(99)00012-5. [DOI] [PubMed] [Google Scholar]
- 9.Yao HB, Shaw PC, Wong CC, Wan DC. J Chem Neuroanat. 2002;23:291–297. doi: 10.1016/s0891-0618(02)00014-5. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi M, Tomizawa K, Ishiguro K. Brain Res. 2000;857:193–206. doi: 10.1016/s0006-8993(99)02424-5. [DOI] [PubMed] [Google Scholar]
- 11.Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J. EMBO J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spittaels K, Van den HC, Van Dorpe J, Terwel D, Vandezande K, Lasrado R, Bruynseels K, Irizarry M, Verhoye M, Van Lint J, Vandenheede JR, Ashton D, Mercken M, Loos R, Hyman B, Van der LA, Geerts H, Van Leuven F. Neuroscience. 2002;113:797–808. doi: 10.1016/s0306-4522(02)00236-1. [DOI] [PubMed] [Google Scholar]
- 13.Owen R, Gordon-Weeks PR. Mol Cell Neurosci. 2003;23:626–637. doi: 10.1016/s1044-7431(03)00095-2. [DOI] [PubMed] [Google Scholar]
- 14.Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD. Neuron. 2004;42:897–912. doi: 10.1016/j.neuron.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 15.Frame S, Cohen P, Biondi RM. Mol Cell. 2001;7:1321–1327. doi: 10.1016/s1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- 16.Knebel A, Morrice N, Cohen P. EMBO J. 2001;20:4360–4369. doi: 10.1093/emboj/20.16.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campbell DG, Morrice NA. J Biomol Technol. 2002;13:119–130. [PMC free article] [PubMed] [Google Scholar]
- 18.Rae MG, Martin DJ, Collingridge GL, Irving AJ. J Neurosci. 2000;20:8628–8636. doi: 10.1523/JNEUROSCI.20-23-08628.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fukata Y, Kimura T, Kaibuchi K. Neurosci Res. 2002;43:305–315. doi: 10.1016/s0168-0102(02)00062-7. [DOI] [PubMed] [Google Scholar]
- 20.Charrier E, Reibel S, Rogemond V, Aguera M, Thomasset N, Honnorat J. Mol Neurobiol. 2003;28:51–64. doi: 10.1385/MN:28:1:51. [DOI] [PubMed] [Google Scholar]
- 21.Arimura N, Menager C, Fukata Y, Kaibuchi K. J Neurobiol. 2004;58:34–47. doi: 10.1002/neu.10269. [DOI] [PubMed] [Google Scholar]
- 22.Goshima Y, Nakamura F, Strittmatter P, Strittmatter SM. Nature. 1995;376:509–514. doi: 10.1038/376509a0. [DOI] [PubMed] [Google Scholar]
- 23.Inagaki N, Chihara K, Arimura N, Menager C, Kawano Y, Matsuo N, Nishimura T, Amano M, Kaibuchi K. Nat Neurosci. 2001;4:781–782. doi: 10.1038/90476. [DOI] [PubMed] [Google Scholar]
- 24.Fukata Y, Itoh TJ, Kimura T, Menager C, Nishimura T, Shiromizu T, Watanabe H, Inagaki N, Iwamatsu A, Hotani H, Kaibuchi K. Nat Cell Biol. 2002;4:583–591. doi: 10.1038/ncb825. [DOI] [PubMed] [Google Scholar]
- 25.Cohen P, Goedert M. Nat Rev Drug Discov. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- 26.Jamsa A, Hasslund K, Cowburn RF, Backstrom A, Vasange M. Biochem Biophys Res Commun. 2004;319:993–1000. doi: 10.1016/j.bbrc.2004.05.075. [DOI] [PubMed] [Google Scholar]
- 27.Pap M, Cooper GM. J Biol Chem. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- 28.Cross DA, Culbert AA, Chalmers KA, Facci L, Skaper SD, Reith AD. J Neurochem. 2001;77:94–102. doi: 10.1046/j.1471-4159.2001.t01-1-00251.x. [DOI] [PubMed] [Google Scholar]
- 29.Wang JZ, Wu Q, Smith A, Grundke-Iqbal I, Iqbal K. FEBS Lett. 1998;436:28–34. doi: 10.1016/s0014-5793(98)01090-4. [DOI] [PubMed] [Google Scholar]
- 30.Phiel CJ, Wilson CA, Lee VM, Klein PS. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- 31.Gu Y, Hamajima N, Ihara Y. Biochemistry. 2000;39:4267–4275. doi: 10.1021/bi992323h. [DOI] [PubMed] [Google Scholar]