

Abstract
HIV-1 Associated Dementia (HAD) is a significant consequence of HIV infection. Although multiple inflammatory factors contribute to this chronic, progressive dementia, excitotoxic damage appears to be an underlying mechanism in the neurodegenerative process. Excitotoxicity is a cumulative effect of multiple processes occurring in the CNS during HAD. The overstimulation of glutamate receptors, an increased vulnerability of neurons, and disrupted astrocyte support each potentiate excitotoxic damage to neurons. Recent evidence suggests that poorly controlled generation of glutamate by phosphate-activated glutaminase may contribute to the neurotoxic state typical of HAD as well as other neurodegenerative disorders. Glutaminase converts glutamine, a widely available substrate throughout the CNS to glutamate. Inflammatory conditions may precipitate unregulated activity of glutaminase, a potentially important mechanism in HAD pathogenesis.
Keywords: glutaminase, glutamate, excitotoxicity, macrophage, HIV-1 associated dementia, neurodegeneration
Introduction, 2
Human immunodeficiency virus has had a profound impact on global health. With worldwide infections near 40 million and approximately 20 million attributable deaths, HIV continues to be a growing problem (1). HIV notoriously targets the immune system, progressively degrading the host defense system until opportunistic infection or systemic failure results in a fatal outcome. In addition to the attack on immune function, HIV is also linked to a syndrome of cognitive and motor dysfunction termed HIV-associated dementia (HAD) (2). This frank dementia is a result of neuronal cell dysfunction and death in the CNS, yet HIV rarely infects neurons. While much research has investigated this viral induced dementia, a clear mechanism for neuronal damage has yet to be established.
HAD is the clinical consequence of a persistent inflammatory process in the CNS causing neurotoxicity. HIV-1 infection of the brain occurs in the early stages of infection (3), with evidence supporting CNS invasion during the primary viremia that accompanies seroconversion (4). Peripherally infected monocytes cross the blood brain barrier, seeding the brain with virus (5–7). HAD is a diagnosis of exclusion and typically only manifests during the later stages of disease. The dementia includes a spectrum of neurological impairments including forgetfulness, apathy, hallucinations, delirium, coma, and ultimately death (2, 8). The dementia once affected 20–30% of those with advanced HIV, but this number has decreased since the advent of Anti-Retroviral Therapy (ART) in developed countries to around 10% (9, 10). However, ART cannot completely protect from or reverse HAD (9). A more subtle collection of cognitive impairments associated with progressive HIV infection termed minor cognitive/motor disorder (MCMD) exists in 30% of infected individuals (11, 12). Both the number of newly infected individuals and life expectancies of those with HIV continues to rise, and thus the prevalence of HAD has increased as well, making this dementia the most common type for those under the age of 40 (13). Accordingly, HAD is an increasingly significant effect of HIV infection.
The pathologic correlate to HAD, HIV encephalitis (HIVE), is characterized by activated macrophage and microglia, damage of neuronal dendrites and axons, and apoptotic neurons. Productive viral infection is found primarily in mononuclear phagocytes (MP; perivascular and parenchymal macrophages and microglia) (5). In HIVE, infected and activated MP are characterized by the formation of multinucleated giant cells, microglial nodules, and macrophage infiltration into the CNS (14). Because viral products are produced and released in the CNS, neuronal death can be induced through two paths, either directly by viral proteins such as gp120, or indirectly through the inflammatory process of HAD. Macrophage derived neurotoxic products are amplified within the brain as a consequence of productive viral infection and persistent immune activation (15). The congregating immune cells fail to eliminate the immunologic insult but continue to produce additional toxins, inflammatory factors, and virus establishing a chronic state of inflammation leading to neurodegeneration.
The release of various factors by MP including cytokines, viral proteins, excitotoxic amino acids and metabolic factors play contributory roles in the development of HAD. Excitotoxic amino acids such as glutamate have been suggested to contribute to neurotoxicity and the neurodegenerative process (16–20). The predominant excitatory neurotransmitter expressed within the mammalian CNS, glutamate mediates numerous physiological functions through activation of multiple receptors (21–23). However, high concentrations of extracellular glutamate are known to induce neuronal damage (24–27). HIV-1-infected patients have been reported to have significantly higher plasma concentrations of glutamate as compared to uninfected controls (28, 29). HIV-1 infected macrophages appear to be an important source of extracellular glutamate (20), this glutamate production has recently been linked to the activity of phosphate-activated mitochondrial glutaminase (30). Glutaminase catalyzes the conversion of glutamine to glutamate, is the primary enzyme for the production of glutamate in the CNS (31–34) and is also the predominant glutamine-utilizing enzyme of the brain (35, 36). Improper regulation of this enzyme through changes in enzyme activity, expression level, or localization may lead to excitotoxic damage of neuron populations (Fig. 1). This review will discuss HAD and the priming of neuron populations for excitotoxic damage. In addition, the current understanding of glutaminase activity and its potential contributions to neurodegeneration will be presented.
Figure 1.
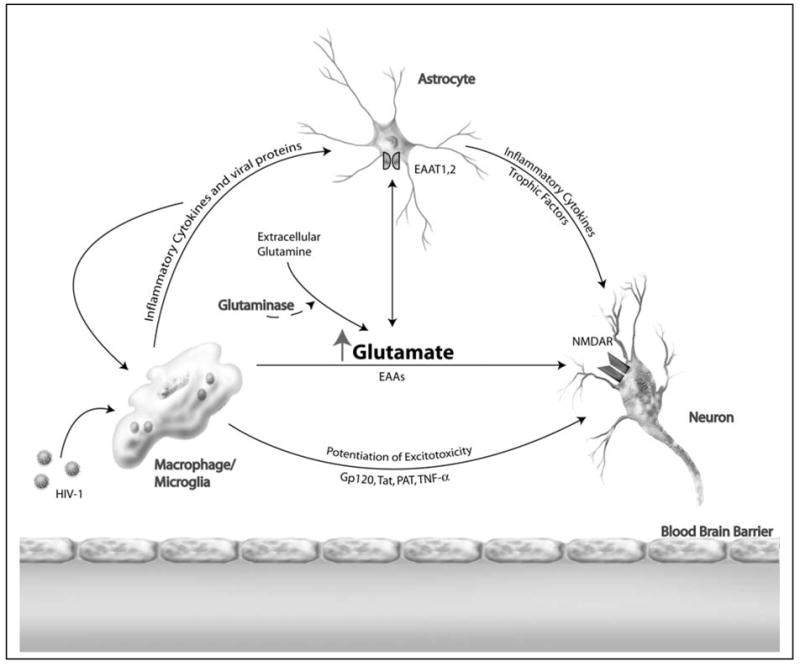
HAD Potentiation of Excitotoxicity. HAD causes an increase in extracellular glutamate and enhances the susceptibility of neurons to damage by excitotoxins. Infected mononuclear phagocytes (MP) produce soluble factors including inflammatory cytokines and viral proteins, inducing functional changes in the astrocyte population and causing damage to neurons via excitotoxic insult. Astrocytes can also express inflammatory cytokines and trophic support of neurons may be altered. In addition, changes in glutamate uptake leads to altered glutamate homeostasis. Extracellular glutamine, a widely available amino acid, may be converted to glutamate by glutaminase, leading to further glutamate increase and neuronal damage.
Macrophage Driven Pathogenesis, 3
Macrophage and microglia are the resident CNS cell population productively infected by HIV-1 (37) and the primary route of viral entry through the blood brain barrier, as such, MP are crucial to HAD pathogenesis. However, the number and localization of infected cells does not explain the extensive and diffuse pathology found in HAD. Apoptotic neurons do not strictly co-localize with infected cells, supporting an indirect or soluble factor mechanism of toxicity (38). Transient exposure of otherwise healthy cells to viral particles can be sufficient to trigger cascades resulting in production of inflammatory factors that ultimately lead to neuronal damage (39). Various viral proteins such as gp120, Tat, Nef, Vpr, and gp41 induce neuronal injury as has been well-established in various culture systems and animal models, but to what extent and in what conditions the viral proteins induce toxicity in vivo is still unclear (40). Viral particles, however, are not the only mediators of CNS dysfunction and neuron toxicity. HIV-induced encephalitis is associated with immune activation of glial cells that results in alterations of normal cell biology, notably secretory functions (41–45). Improper regulation of these factors can lead to impaired cellular functioning, modified neurotransmitter action, amplified inflammation and neuronal damage. Various potential neurotoxins are generated and released during the inflammatory process including reactive oxygen species, tumor necrosis factor-α (TNF-α), arachidonic acid, platelet-activating factor (PAF), nitric oxide, quinolinic acid, and glutamate (20, 30, 46–52).
A great deal of research has been conducted on HAD pathogenesis, and a multitude of factors have been identified with the potential to cause neuronal harm (53). Disease pathogenesis undoubtedly involves many of these different factors and processes; however, there may be fundamental or initiating cascades that drive the degenerative process of HAD as well as other CNS disorders. Characterizing the mechanism of neuronal demise is challenging in a slow progressive disorder with profound variations between individuals whom experience unique courses of infection. While multiple factors play important roles in MP mediated neuronal injury, accumulated evidence in various neurodegenerative disorders seems to indicate glutamate excitotoxicity as a prominent mechanism of neuronal damage. Excitotoxicity is the end product in a complex system that involves a confluence of factors leading to a state of heightened vulnerability of neurons to excitation in conditions with elevated excitatory toxins. Through the priming of neuron populations, the disruption of astrocyte support, and the excess generation of glutamate and other excitatory amino acids, excitotoxicity is a critical process in HAD.
A number of toxic factors released from mononuclear phagocytes in HAD affect neurons through excitotoxicity. Supernatants of HIV infected monocytes are known to cause NMDA receptor dependent cell death (20, 54–57). Excitotoxic amino acids (EAA) notably glutamate, as well as related substances including quinolate, cysteine and Ntox, a neurotoxic amine, target glutamate receptors potentially leading to Ca2+ dysregulation and cell death. (18, 20, 58). The inflammatory process of HAD potentiates glutamate excitotoxicity through multiple factors including PAF, TNF- α, IL-1β, and various HIV proteins (59–66). Glutamate excitotoxicity may be the common pathway to cognitive dysfunction (67). In conjunction with the release of a variety of EAAs, extracellular glutamate levels are known to be increased in a variety of CNS inflammatory disorders. There are multiple potential sources of this excess glutamate including release from dying cells, alterations in homeostasis, and the generation of excess glutamate through enzymatic activity.
Glutamate Function and Homeostasis, 4
Glutamate Homeostasis
Glutamate regulation is highly energy dependent and many brain functions revolve around the production, release, and consequent removal of glutamate from the extracellular space. Intracellular glutamate is abundant, and provides an important metabolic product and fuel that is integral to the TCA cycle yielding energy or to be used in intracellular signaling, protein synthesis, and ammonia fixation. Glutamate in plasma is relatively high, but the blood-brain barrier efficiently prevents entry into the CNS, requiring the brain to produce nearly all of the present glutamate. Glutamate is produced by various cells in the brain and is released by neurons as a transmitter, but extracellular concentrations are kept low. While diffusion allows some glutamate to clear synapses, there is no enzymatic consumption of glutamate, and consequently, most must be actively removed. This removal is generally the role of the supporting astrocytes.
The primary mechanism to remove extracellular glutamate is uptake through excitatory amino acid transporters (EAATs). Five subtypes have been cloned: EAAT1/GLAST (glutamate-aspartate transporter, EAAT2/GLT-1 (glutamate transporter), EAAT3/EAAC1 (excitatory amino acid carrier), EAAT4, and EAAT5, with EAAT1 and EAAT2 expressed predominantly in astrocytes. These transporters utilize Na+ gradients to drive glutamate and aspartate into the cell against their concentration gradients. The GLAST and GLT expression at specific parts of the astrocyte membrane depends on the surrounding cells. Regions of the astrocyte membrane facing neurons have high concentrations of GLAST and GLT, while regions facing capillary epithelium have low concentrations (68). Another piece of evidence that glutamate transporters are localized to specific regions is that EAAT5 has a protein-binding domain on its C-terminal similar to the PSD-95 binding domain of NMDA receptors, which would localize EAAT5 to synaptic areas (68, 69).
Returning glutamate to neurons through the extracellular space poses the danger of interfering with concentration dependent signaling, yet neurons cannot sustain the production of glutamate without a supply of TCA intermediates from supporting astrocytes (70). The biological response is the conversion of glutamate to glutamine in astrocytes, which is then made available to neurons once released back to the extracellular space. This sets up an exchange called the glutamate-glutamine cycle where there is glutamate release, uptake, conversion and then subsequent release. This process, however, is not simply a one for one exchange, where each glutamate removed from a synapse is converted over to glutamine. The efficiency of the glutamate-glutamine cycle is dependent upon the compartmentalization of key enzymes to neurons or astrocytes. Glutamate is a valuable metabolic fuel that can also be oxidatively degraded by astrocytes. Synaptic glutamate is cleared by supporting astrocytes, where it is either metabolized or converted to glutamine by the astrocyte specific glutamine synthetase, an enzyme absent in neurons (71). This glutamine is then released back to the extracellular space where glutamine is widely available, and does not possess the transmitter potential of glutamate. Neuron populations efficiently take up glutamine, where it is then hydrolyzed by phosphate-activated glutaminase (PAG) to glutamate. PAG is known to be present in both neurons and astrocytes, thus neurons cannot generate glutamine, but both neurons and astrocytes can form glutamate from glutamine (Fig. 2).
Figure 2.
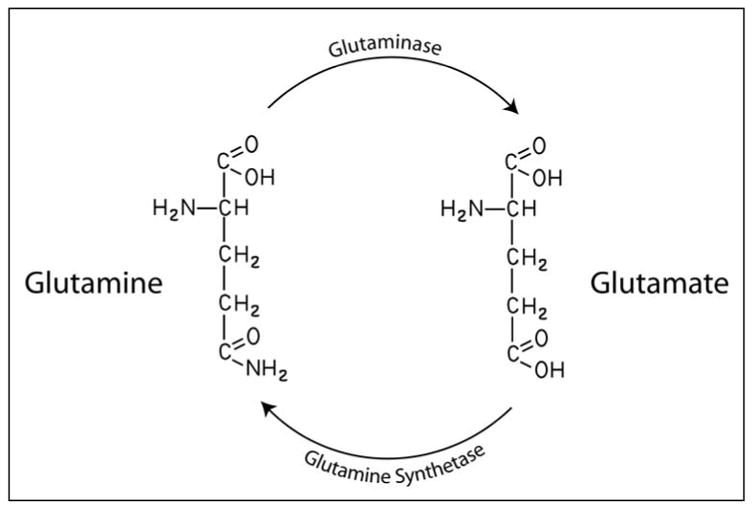
Glutamine-Glutamate Interconversion. Glutamine is deaminated to glutamate in an energy free process mediated by the enzyme phosphate-activated glutaminase. The reverse reaction is an energy dependent process carried out by the enzyme glutamine synthetase.
Glutamate Receptors
Glutamate is the major mediator of excitatory synaptic transmission in the mammalian CNS and is critical to cognition, memory, learning, as well as playing vital roles in development, migration, and differentiation. Glutamate acts on several receptor types, which are classified into three ionotropic classes: N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors, and kainate; and three metabotropic classes (72–74). Ionotropic receptors are ion channels that open upon the binding of glutamate, leading to the influx of sodium and/or calcium and the efflux of potassium, while metabotropic receptor activation leads to G-protein coupled release of Ca2+ from intracellular stores (73, 75).
NMDA receptors are tetra-heteromeric structures permeable to sodium, potassium, zinc and calcium (72). At normal physiological resting membrane potential, magnesium blocks the channel pore (76); when removed, the ligand activated NMDA receptor allows an influx of calcium, leading to postsynaptic depolarization and action potential in the postsynaptic neuron (77). NMDA receptor antagonists can block most excitotoxic effects of glutamate (78). Calcium entering through over-activated NMDA receptors results in more cell death as opposed to calcium entering through non-NMDA glutamate receptors or voltage-gated calcium channels, suggesting NMDA receptor-mediated neurotoxicity occurs through distinct calcium signaling pathways that may involve the NMDA receptor-specific interaction with PSD, a family of postsynaptic scaffold proteins (75, 79).
AMPA receptors are permeable to sodium, potassium, zinc and occasionally calcium. The efficiency of calcium permeability through AMPA receptors is highly dependent upon the combination of subunits making up the heteromeric receptor (80) (81). The pre-mRNA editing of one subunit, GluR2, causes the replacement of a neutral glutamine with a positively charged arginine residue in the channel-forming membrane loop segment (82). Presence of an edited GluR2, as is the case in an overwhelming majority of cells expressing AMPA, renders the heteromeric receptor mostly impermeable to calcium (80, 81). Calcium-impermeable AMPA receptors can still cause excitotoxicity by allowing sodium influx to slightly depolarize the cell membrane, leading to the subsequent activation of NMDA receptors (83, 84), as has been demonstrated by multiple investigators (84–88).
Kainate receptors are heteromeric receptors permeable to sodium, potassium, and sometimes calcium (89). Excitotoxicity resulting from kainate receptor stimulation may proceed by apoptotic pathways rather than the necrotic pathway sometimes observed with NMDA receptor mediated cell death (90). Excitotoxicity enhanced by kainate receptor activation may be due to release of glutamate and sodium influx to depolarize the membrane and release the magnesium blockade of NMDA, leading to the subsequent activation of NMDA receptors (83, 84, 91).
Metabotropic glutamate receptors have been grouped into three categories (Group I–III) based on pharmacological properties, signal transduction mechanisms, and sequence similarities. Group I mGlu receptors play a role in regulating multiple calcium, potassium, and non-selective cationic channels as well as NMDA, and AMPA receptors, which may influence the firing patterns of neurons (92, 93). Group I mGlu receptors potentiate NMDA receptor activation, thus effecting transmission, synaptic plasticity, and the generation of long-term potentiation (93, 94). Group II and III mGlu receptors inhibit various calcium channels and may inhibit presynaptic release of neurotransmitters (92, 93, 95). Group I mGluR activation may enhance neurodegeneration through excitotoxic mechanisms, while Group II and III mGluR stimulation may be neuroprotective (96).
Excitotoxicity
Glutamate regulation is critical because improper management of glutamate levels may impair not only its signaling properties, but can lead to cell death via excitotoxicity (78, 97). The concept of excitotoxicity was first proposed by Olney in 1969 as a toxic effect of excessive or prolonged activation of receptors by excitatory amino acids (EAA) (98). Excitatory amino acids refer principally to glutamate (glutamic acid), but also include various metabolites that act via glutamate receptors including endogenous molecules such as aspartic acid, quinolinic acid, homocysteic acid, and exogenous molecules such as NMDA and kainate (reviewed in (84)). Excessive or persistent activation of glutamate-gated ion channels, resulting in inappropriate regulation of glutamatergic neurotransmission has been implicated in a wide range of neuronal degenerative processes (99). The prolonged activation of glutamate receptors or alteration in proper glutamate receptor function results in a pathological increase in free intracellular Ca2+.
The EAA induced neurotoxic response is classically comprised of two stages: an initial influx of extracellular Na+ and Cl− and a secondary influx of Ca2+ through the opening of NMDA receptor channels and or voltage gated Ca2+ channels. Excitotoxic cell death can occur directly via two mechanisms, one rapid and necrotic while the second is delayed and through apoptosis. The fast mechanism is instigated by the massive influx of Na+ ions following overstimulation of glutamate receptors. This influx disrupts cell homeostasis via hypertonic-induced swelling, ATP depletion and ultimately membrane failure and necrotic cell death. The delayed mechanism is independent of cell swelling, and involves Ca2+ influx that triggers multiple neurotoxic cascades including p38 and JNK MAP kinase activation, release of cytochrome c, caspase activation, lipid peroxidation, and chromatin condensation (40, 100–102). The massive influx of Ca2+ triggers release of Ca2+ from intracellular stores, further flooding the intracellular space with free Ca2+ (Fig. 3).
Figure 3.
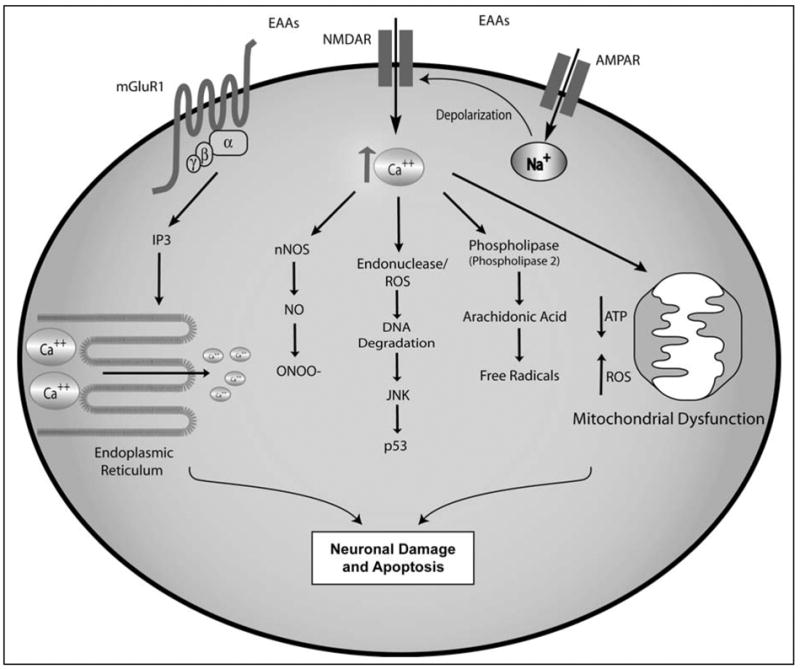
Pathways to Excitotoxicity. Over-stimulation of glutamate receptors initiates multiple cascades with the potential to induce cellular damage and death. NMDAR activation by agonists, notably glutamate, leads to the influx of Ca2+ and Na+ ions, an effect potentiated by AMPA receptors. MGluR activation, particularly mGluR1, leads to additional Ca2+ release. The excess Ca2+ triggers multiple pathways including nNOS, JNK, and Phospholipase, as well as leading to mitochondrial stress and dysfunction.
A critical by-product of over stimulation via glutamate is the generation of reactive oxygen species, ROS (reviewed in (103)). One mechanism of radical generation is through the activation of neuronal nitric oxide synthase (nNOS), and the consequent generation of nitric oxide, NO. PSD-95 specifically interacts with NMDA receptors and nitric oxide synthase. The close proximity of NMDA and nNOS mediated by PSD-95, may account for preferential activation of nNOS and subsequent toxicity caused by calcium influx through NMDA receptors as opposed to non-NMDA receptors (75, 104). NO readily combines with superoxide anion (O2-) and forms highly reactive peroxynitrite (ONOO-) all leading to oxidative stress and further mitochondrial injury (105).
The presence of excess glutamate may cause CNS oxidative stress through glial cells such as macrophage and/or microglia by another mechanism. A glutamate/cystine exchanger brings one cystine into the cell in exchange for one internal glutamate (106). Extracellular glutamate inhibits cystine uptake, and if the concentration of extracellular glutamate is high enough, cystine may be released as glutamate enters through the exchanger. This loss of cystine may facilitate oxidative stress due to the requirement of cysteine for the synthesis of glutathione, a critical antioxidant (68).
Mitochondria are a critical site in the evolution of an excitotoxic event. Mitochondria generate ATP, consume oxygen, produce free radicals, and mobilize intracellular Ca2+ (107). Mitochondria have the ability to sequester large amounts of Ca2+, however, this carries a risk for mitochondrial dysfunction (103). Through the disruption of mitochondrial potential, excess Ca2+ can reduce ATP synthesis, rendering a cell more vulnerable to death. In addition, mitochondrial dysfunction also leads to increased generation of ROS. Mitochondria appear to be the primary mediators of cell death caused by abnormal levels of intracellular Ca2+ during excitotoxicity (108, 109).
The capacity to respond to stress determines the viability of individual cells. The influx of Na+ and Ca2+ ions requires significant energy expenditure to re-establish normal gradients. This energetic burden is even greater in cells with limited mitochondrial capacity due to oxidative damage or Ca2+ overload. In addition, the capacity of surrounding glial support cells to remove extracellular glutamate, as well as provide trophic factors also contributes to cellular outcome. Cell death is typically not rampant, particularly in chronic disease states. The interpretation of various external and internal factors by individual neurons ultimately determines the outcome of each cell. In a toxic inflammatory environment, the most susceptible regions and cells will be selectively targeted for impairment.
While the benchmark for neurotoxicity has classically been cell death, synaptic dysfunction may be a better measure of neurodegeneration (excellent review by Bellizzi (110)). In a disease of chronic inflammation, incremental stages of increasing stress do not immediately overwhelm a cell’s capacity to respond, the stress required to overwhelm cellular repair and homeostatic mechanisms compromise synaptic function long before the induction of cell death mechanisms. In HAD, damage to dendritic arbors and reduction in synaptic density are important neuropathological signatures of HIVE (111, 112). This type of damage to the neural network is thought to be an early event in the pathway leading to neuronal dropout via apoptosis (55, 113). Although, a direct link between HIV-1 infection of brain macrophages and alterations in the dendritic arbor and synaptic density has been suggested (55, 113), the factors from HIV-1 infected macrophages and the signaling pathways through which HIV-1 mediates damage to the neuronal network has yet to be elucidated. An important note is neuronal damage that spares the cell body is often reversible, with removal of the neurotoxic stimulus allowing recovery of functional synapses (114–117). Excitotoxicity seems to play an important role in this process (113, 118). The clinical manifestations of cognitive dysfunction in neuroinflammatory states may be better ascribed to accumulating synaptic damage.
As is often the case in chronic CNS disorders, insult resulting in disruption of homeostasis has the potential to establish an amplification cycle that is difficult to interrupt. Excitotoxic insult activates glutamate receptors, increasing energy consumption and the production of oxidative radicals. With time, mitochondria may begin to fail, or sufficient cellular damage can result in the induction of death processes. Necrotic or apoptotic death of cells may lead to amplification of inflammation and the targeting of additional cells for stress and perhaps elimination (119). As cell damage increases, released chemotactic factors recruit additional effector cells such as MP, resulting in further amplification of the neurotoxic inflammatory cycle (120). The CNS may be ill-equipped to interrupt this type of excitotoxic positive feedback cycle, a potentially important process to many neurodegenerative disorders.
HAD Potentiation of Excitotoxicity, 5
The Priming of Neurons
There is general agreement that HIV does not infect neurons (5, 7), and while there is evidence for direct effects of viral proteins upon neuron viability, the indirect form of neurotoxicity seems to predominate (40, 121, 122). Supernatants from HIV-1 infected MP induce NMDA receptor dependent neurotoxicity (20, 55). This effect is mediated through upregulation of neurotoxins that directly target NMDA receptors, and indirectly through inflammatory mediators produced by MP that injure neurons by increasing vulnerability to Ca2+ dependent glutamate excitotoxicity. This is not a HIV specific phenomenon, beta-amyloid stimulated microglial cultures induce neuronal cell death, dependent upon TNF-α and co-activation of NMDA receptors (123). This type of neurotoxicity is probably aggravated by inflammatory cytokines such as IL-1β, TNF-α, arachidonate, free radicals and viral protein (37). For instance, PAF was recently shown to sensitize neuron synapses to glutamate, causing excitotoxicity from typical synaptic transmission (124); whereas IL-1β was shown to trigger phosphorylation of NMDA receptors, increasing Ca2+ influx (125). In HAD, gp120 was shown to interact with the glycine binding site on NMDA receptors, and Tat protein was shown to synergistically enhance the NMDA specific neurotoxic response (126, 127). Thus, even without change in glutamate levels, neurons in inflammatory states are susceptible to excitotoxic effects.
Impairment of Astrocytes in HAD
In the brain, microglia/macrophage are the primary targets of HIV-1 infection (128), but in vivo infection of astrocytes has been documented in multiple studies (129–136). Astrocytes present an abundant CNS target, but the course of infection is generally a long-term latent process. These infected astrocytes lack the productive infection characteristic of MP cells, and express predominantly viral regulatory genes rather than structural genes (132, 135). However, any lack of infective robustness does not diminish the importance of astrocyte dysfunction in the dementia process. Astrocytes are critical to maintaining homeostasis, preserving the integrity of the blood brain barrier and regulating extracellular glutamate (68, 137). Increasing evidence also supports a role for astrocytes in signal transmission through modulation of synapses and neuronal function (138–141).
Uptake of the excitatory amino acid glutamate from the synaptic cleft is pivotal to glutamatergic neurotransmission and avoiding excitotoxicity (142). The glutamate uptake mediated through the EAAT transporters results in concentration gradients of glutamate 10,000 times greater inside the cell as compared to extracellular space (143). In vivo, mice lacking EAAT2/GLT-1 develop epilepsy and increased susceptibility to glutamate (144). In experimental autoimmune encephalitis (EAE), impaired glutamate clearance and degradation by astrocytes and oligodendrocytes has been observed (145–147). Thus, there is a clear connection between improper uptake capacity and CNS damage. Products of HIV infection, gp120 and TNF-α, inhibit glutamate uptake by astrocytes (148, 149). Compounding these effects, HIV-1 has been shown to downregulate glutamate transporter EAAT2, causing disruptions in glutamate uptake (149). Further, G-protein linked mGlu receptors may cause increased intracellular Ca2+, leading to glutamate release from astrocytes (150). Astrocytic release of glutamate has been shown to be stimulated by arachidonic acid, TNF-α, and CXCL12 (50, 148, 151). In addition, numerous studies have indicated a potential for the reversal of glutamate uptake, causing further glutamate dumping (152, 153).
Excess Generation of Extracellular Glutamate in HAD
Increased extracellular glutamate is a common theme to various neurodegenerative disorders (68). Increased Glu has been found in CSF of acute MS patients (154) and secondary progressive MS (155). Excess glutamate has also been found in HAD patient populations (156). Increases in extracellular glutamate are known to occur through a variety of mechanisms including reversal of glutamate uptake carriers, impairment of membrane integrity, swelling-induced opening of anion channels, compromise of the blood brain barrier and enzymatic conversion of glutamine to glutamate (157). The relative contribution of these mechanisms is still unclear, and other processes may cause substantial glutamate accumulation.
Glutaminase, 6
Compartmentalization of glutamate is critical, however glutamine is abundant in the extracellular space, the highest concentration of any amino acid in brain extracellular fluid (158). Glutamine is freely passed between cells, and does not pose the excitotoxic threat of EAAs. Phosphate-activated glutaminase (PAG, EC 3.5.1.2) is a mitochondrial enzyme that catalyzes the deamination of glutamine to glutamate, a hydrolysis resulting in stoichiometric amounts of glutamate and ammonia. There are two loci in humans that yield structurally related but distinct glutaminase enzymes. Liver type glutaminase (LGA) is primarily expressed in periportal hepatocytes, although transcripts have been found in the brain (159). Kidney type glutaminase (KGA) is abundant not only in the kidney, but also the brain, intestine, liver, lymphocytes and various tumors (34). Although LGA is present in the brain, the ratio of LGA to KGA is very small, with most brain glutaminase being of the kidney type (160). Any isoform specific function has yet to be identified, although some work has identified unique molecular interactions of liver versus kidney type glutaminase in the brain (161). Much of the interest in glutaminase has stemmed from its association with various cancers, where glutaminase activity is hypothesized to be critical in tumor growth and a potential target for treatment (162, 163). Various tumors have been linked to glutaminase overexpression (164–167). These findings indicate the potential for isoform specific regulation of glutaminase in various circumstances.
KGA, found on chromosome two in humans (168) is located immediately next to the STAT1 gene (169). KGA has various isoforms generated through tissue specific alternative splicing. The first 16 N-terminal amino acids of KGA encode a mitochondrial targeting sequence (170); after trafficking, KGA is trimmed resulting in a 66kd protein. Elgadi et. al. first described two additional isoforms hGAM and hGAC; hGAM is only found in cardiac and skeletal muscle, while GAC is known to be present in the brain (166). All KGA isoforms, share 5’ sequence regions but vary significantly in both 3’ coding and non-coding regions. The GAM isoform contains a significantly reduced coding region and is presumed nonfunctional. GAC mRNA is produced by alternative splicing of a single exon within the KGA gene (171). The resulting protein shares much of the functional glutaminase regions, but contains a unique 3’ tail. The KGA gene also contains multiple poly-adenylation sites, potentially adding further variability (171). The various forms of KGA make expression analysis somewhat difficult, requiring 3’ specific primers to identify specific isoforms, but begs the question of what purpose the various forms serve (Table 1). For instance, acidosis was shown to lead to mRNA stabilization of KGA (171). This complex gene arrangement facilitates various forms of regulation that may or may not prove to be important to disease.
Table 1.
Human Glutaminase Subtypes
| Type | Genetic Structure | Cellular Location | Tissue Expresson | References |
|---|---|---|---|---|
| hLGA | Chromosome 12 | Mitochondrial, Nuclear | Liver, Pancreas, Brain | (34), (196), (159) |
| hKGA | Chromosome 2, multiple poly-adenylation sites | Mitochondrial | Kidney, Brain, Intestine, Lymphocytes, Fetal Liver | (166), (171) (172) |
| hGAC | KGA isoform: Unique 3’ tail derived from exon 15 | Mitochondrial | Cardiac muscle, Pancreas, Placenta, Kidney, Lung, Brain | (166), (171) |
Glutaminase is the primary enzyme consuming glutamine and generates much of the glutamate used in signal transduction. Glutaminase is generally localized to the inner membrane of the mitochondria (172–174); although additional studies have recognized a nuclear population of glutaminase (159, 167). Increase in amount, activity or release of glutaminase could facilitate uncontrolled generation of glutamate in the CNS extracellular space. Enzymatic conversion of glutamine to glutamate presents two potential problems: improper compartmentalization of glutamate interfering with signal transduction and glutamate induced excitotoxicity. Neuronal cultures depleted of astrocytes were shown to have glutamine-potentiated neurotoxicity, an effect inhibited by NMDA receptor antagonists (175). The neurotoxic effects of glutamate generation were first identified as a glutamine dependent generation of glutamate in neuron cultures (176). The unregulated enzymatic generation of glutamate in inflammatory states may establish a positive feedback system of excitotoxic damage leading to further enzyme release and conversion of glutamine to glutamate causing additional excitoxicity and cellular damage.
In stroke, significant regions of tissue can experience catastrophic damage and cell death. In this type of model, Newcomb and colleagues proposed widespread neuron death as a means of freeing glutaminase, resulting in unregulated glutamate generation, fed by an abundant substrate of glutamine (27). Using an in vivo ischemic model, significant glutamine dependent glutamate generation was observed 24 hours post-lesion (177). The significance of glutaminase is supported by the observation of enhanced extracellular activity of glutaminase upon removal of intracellular feedback inhibition (178). However, this type of widespread neuronal cell death is generally not shared by other neurodegenerative disorders such as HAD, Alzheimer’s, and Multiple Sclerosis (MS).
Chronic neurodegenerative disorders typically lack rampant cell death, and are better characterized by prolonged inflammation, or heightened immune activation, where neuron death, while present, is not a dominant feature. However, these disorders do typically have noticeable lymphocyte and monocyte infiltration and accumulation. In these circumstances, there is increasing evidence for an important contribution of glutamate generation by enzymatic conversion of glutamine. Using human brain, immunohistochemistry identified enhanced glutaminase expression in MS lesions as compared to control specimens. This increased glutaminase correlated with axonal damage (179). Additional experiments identified increased glutaminase in both tropical spastic paraparesis and subacute sclerosing panencephalitis. Although lacking the focal increases of glutaminase found in the MS lesions, this diffuse increase in enzyme also seems to indicate an enzymatic role in excitotoxic damage. Another study in rats measured the conversion of labeled glutamine to glutamate following an excitotoxic insult of the hippocampus. Results indicated significant conversion of glutamine to glutamate in experimental versus control animals, and consequently support the concept of glutaminase induced neuronal damage (180).
In HAD, significant numbers of MP migrate into the CNS where they are productively infected as well as activated. Glutamate is secreted in large quantities by macrophage (20, 30, 181). The MP cell population expresses glutaminase at significant levels (30). Viral infection leads to formation of multinucleated giant cells, as well as necrotic cell death. This type of cellular stress has the potential to disrupt membrane stability leading to release of mitochondrial glutaminase. Our group recently demonstrated a glutamine dependent upregulation of glutamate production by HIV-1 infected macrophage cultures. This glutamate increase relates to cell viability, and was nearly eliminated in the presence of antiviral treatment (30). Thus, HIV-1 may lead to increased enzyme activity or release of enzyme into a glutamine rich substrate with little product feedback, allowing excess glutamate generation from macrophage populations. Thus in HAD, a system of immune cell recruitment, activation and infection causing MP stress and death may then lead to poor regulation of glutaminase, producing an excitotoxic environment. How this process is regulated and the glutaminase subtypes involved remains unclear; however, the elucidation of such pathways may help us better understand MP mediated neuronal injury in neurodegeneration (Fig. 4).
Figure 4.
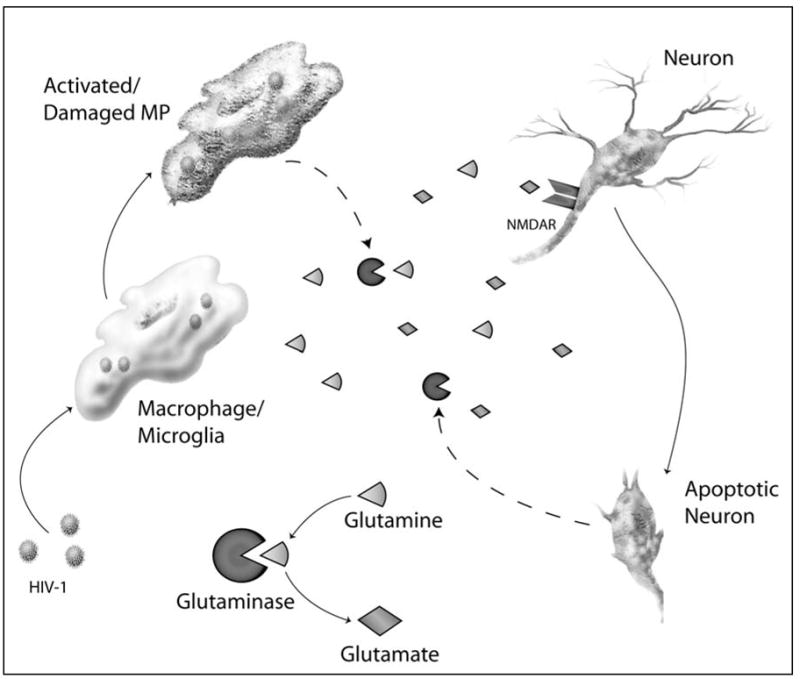
Model for Glutaminase Activity in HAD. Infected and/or activated mononuclear phagocytes (MP) release functional glutaminase to the glutamine rich extracellular space. Glutaminase then converts glutamine to glutamate in an energy free process. This increase in glutamate leads to over-stimulation of glutamate receptors, notably NMDA-R, causing excitotoxic neuron death.
Outside of a disease setting, the regulation of glutaminase and the enzyme conducting the reverse reaction has been investigated as a potential dynamic equilibrium between neurons and astrocytes. Hippocampal measurements of glutaminase activity demonstrated a decrease upon excitotoxic stimulation with AMPA (182). Glutamine synthetase has been shown to be upregulated in response to inflammatory and excitotoxic stimuli in glia; however, the adaptive response is attenuated by inflammatory cytokines (183). This complex interplay adds a new variable to the well-established phenomenon of excitotoxicity in neurodegenerative disorders.
Future Directions, 7
The critical role of excitotoxicity is supported by the effects of glutamate receptor antagonists in various neurodegenerative processes. In EAE, glutamate receptor antagonists reduced clinical symptoms and axonal damage (184, 185). Memantine, an open channel NMDA receptor blocker, has been shown to be effective in gp120 transgenic mice, HIVE SCID mice, and appears to have some clinical benefit in Alzheimer’s (186–189). This type of partial glutamate receptor blockade may have therapeutic benefit in a variety of neurodegenerative disorders. However, blocking the harmful effects of glutamate is not a simple endeavor. Excessive blockade of excitatory amino acid receptors causes side effects including neural degeneration and psychogenic properties (190–192). While limiting excitotoxicity is clearly a desired outcome, interruption of the physiologic roles of glutamate in the brain or elsewhere is an unacceptable side effect.
Glutaminase is not a newly discovered enzyme, it was first described 30 years ago (193), yet its biological significance in disease may not yet be fully appreciated. While there is still much to be determined concerning the mechanisms of glutaminase activity in CNS disorders, glutaminase presents a potential site for therapeutic intervention in a wide range of disorders (194, 195). If glutaminase release from activated or damaged cells proves to be typical of neuroinflammation, targeting extracellular glutaminase would avoid the complications of blocking glutamate receptors and may also avoid interfering with proper signal transduction within the CNS. Through the understanding of how glutamate is generated, cycled between cells, and removed, we may gain a better understanding of how dysfunction at various points in the disease process leads to neuronal damage.
Acknowledgments
This work was supported in part by research grants by the National Institutes of Health: R01 NS 41858, P20 RR15635 and P01 NS043985 (JZ). We kindly acknowledge Jianxing Zhao, Shelley Herek and Alicia Lopez provided technical support for this work. Robin Taylor provided outstanding administrative support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Piot P, Bartos M, Ghys PD, Walker N, Schwartlander B. The global impact of HIV/AIDS. Nature. 2001 Apr 19;410(6831):968–73. doi: 10.1038/35073639. [DOI] [PubMed] [Google Scholar]
- 2.McArthur JC. Neurologic manifestations of AIDS. Medicine (Baltimore) 1987 Nov;66(6):407–37. doi: 10.1097/00005792-198711000-00001. [DOI] [PubMed] [Google Scholar]
- 3.McArthur JC, Hoover DR, Bacellar H, Miller EN, Cohen BA, Becker JT, et al. Dementia in AIDS patients: incidence and risk factors. Multicenter AIDS Cohort Study. Neurology. 1993;43(11):2245–52. doi: 10.1212/wnl.43.11.2245. [DOI] [PubMed] [Google Scholar]
- 4.Michaels J, Sharer LR, Epstein LG. Human immunodeficiency virus type 1 (HIV-1) infection of the nervous system: a review. Immunodefic Rev. 1988;1(1):71–104. [PubMed] [Google Scholar]
- 5.Koenig S, Gendelman HE, Orenstein JM, Canto MCD, Pezeshkpour GH, Yungbluth M, et al. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986;233(4768):1089–93. doi: 10.1126/science.3016903. [DOI] [PubMed] [Google Scholar]
- 6.Gabuzda DH, Ho DD, Monte MSDL, Rota TR, Sobel RA. Immunohistochemical identification of HTLV-III antigen in brains of patients with AIDS. Ann Neurol. 1986;20:289–95. doi: 10.1002/ana.410200304. [DOI] [PubMed] [Google Scholar]
- 7.Wiley CA, Schrier RD, Nelson JA, Lampert PW, Oldstone MBA. Cellular localization of human immunodeficiency virus infection within the brains of acquired immune deficiency syndrome patients. Proc Natl Acad Sci USA. 1986;83:7089–93. doi: 10.1073/pnas.83.18.7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navia BA, Jordan BD, Price RW. The AIDS dementia complex: I. Clinical features. Annals of Neurology. 1986;19:517–24. doi: 10.1002/ana.410190602. [DOI] [PubMed] [Google Scholar]
- 9.Dore GJ, Hoy JF, Mallal SA, Li Y, Mijch AM, French MA, et al. Trends in incidence of AIDS illnesses in Australia from 1983 to 1994: the Australian AIDS cohort. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;16(1):39–43. doi: 10.1097/00042560-199709010-00006. [DOI] [PubMed] [Google Scholar]
- 10.Sacktor N, Lyles RH, Skolasky R, Kleeberger C, Selnes OA, Miller EN, et al. HIV-associated neurologic disease incidence changes: Multicenter AIDS Cohort Study, 1990–1998. Neurology. 2001;56(2):257–60. doi: 10.1212/wnl.56.2.257. [DOI] [PubMed] [Google Scholar]
- 11.Janssen RS, Saykin AJ, Cannon L, Campbell J, Pinsky PF, Hessol NA, et al. Neurological and neuropsychological manifestations of HIV-1 infection: association with AIDS-related complex but not asymptomatic HIV-1 infection. Ann Neurol. 1989 Nov;26(5):592–600. doi: 10.1002/ana.410260503. [DOI] [PubMed] [Google Scholar]
- 12.Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, McArthur JC, et al. HIV-associated cognitive impairment before and after the advent of combination therapy. J Neurovirol. 2002 Apr;8(2):136–42. doi: 10.1080/13550280290049615. [DOI] [PubMed] [Google Scholar]
- 13.Ellis RJ, Deutsch R, Heaton RK, Marcotte TD, McCutchan JA, Nelson JA, et al. Neurocognitive impairment is an independent risk factor for death in HIV infection. San Diego HIV Neurobehavioral Research Center Group. Arch Neurol. 1997;54(4):416–24. doi: 10.1001/archneur.1997.00550160054016. [DOI] [PubMed] [Google Scholar]
- 14.Cherner M, Masliah E, Ellis RJ, Marcotte TD, Moore DJ, Grant I, et al. Neurocognitive dysfunction predicts postmortem findings of HIV encephalitis. Neurology. 2002 Nov 26;59(10):1563–7. doi: 10.1212/01.wnl.0000034175.11956.79. [DOI] [PubMed] [Google Scholar]
- 15.Ryan LA, Cotter RL, Zink WE, Gendelman HE, Zheng J. Macrophages, Chemokines and Neuronal Injury in HIV-1 Associated Dementia. Cellualr and Molecular Biology. 2002;48(2):125–38. [PubMed] [Google Scholar]
- 16.Belmadani A, Zou J, Schipma MJ, Neafsey EJ, Collins MA. Ethanol pre-exposure suppresses HIV-1 glycoprotein 120-induced neuronal degeneration by abrogating endogenous glutamate/Ca(2+)-mediated neurotoxicity. Neuroscience. 2001;104(3):769–81. doi: 10.1016/s0306-4522(01)00139-7. [DOI] [PubMed] [Google Scholar]
- 17.Zink WE, Zheng J, Persidsky Y, Poluektova L, Gendelman HE. The neuropathogenesis of HIV-1 infection. FEMS Immunol Med Microbiol. 1999;26(3–4):233–41. doi: 10.1111/j.1574-695X.1999.tb01394.x. [DOI] [PubMed] [Google Scholar]
- 18.Giulian D, Wendt E, Vaca K, Noonan CA. The envelope glycoprotein of human immunodeficiency virus type 1 stimulates release of neurotoxins from monocytes. Proc Natl Acad Sci U S A. 1993;90(7):2769–73. doi: 10.1073/pnas.90.7.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pulliam L, Clarke JA, McGuire D, McGrath MS. Investigation of HIV-infected macrophage neurotoxin production from patients with AIDS dementia. Adv Neuroimmunol. 1994;4(3):195–8. doi: 10.1016/s0960-5428(06)80257-3. [DOI] [PubMed] [Google Scholar]
- 20.Jiang Z, Piggee C, Heyes MP, Murphy C, Quearry B, Bauer M, et al. Glutamate is a mediator of neurotoxicity in secretions of activated HIV-1-infected macrophages. J Neuroimmunol. 2001;117(1–2):97–107. doi: 10.1016/s0165-5728(01)00315-0. [DOI] [PubMed] [Google Scholar]
- 21.Cutler RW, Dudzinski DS. Regional changes in amino acid content in developing rat brain. J Neurochem. 1974 Nov;23(5):1005–9. doi: 10.1111/j.1471-4159.1974.tb10752.x. [DOI] [PubMed] [Google Scholar]
- 22.Fonnum F. Glutamate: a neurotransmitter in mammalian brain. J Neurochem. 1984 Jan;42(1):1–11. doi: 10.1111/j.1471-4159.1984.tb09689.x. [DOI] [PubMed] [Google Scholar]
- 23.Orrego F, Villanueva S. The chemical nature of the main central excitatory transmitter: a critical appraisal based upon release studies and synaptic vesicle localization. Neuroscience. 1993 Oct;56(3):539–55. doi: 10.1016/0306-4522(93)90355-j. [DOI] [PubMed] [Google Scholar]
- 24.Olney JW. Glutamate-induced neuronal necrosis in the infant mouse hypothalamus. An electron microscopic study. J Neuropathol Exp Neurol. 1971 Jan;30(1):75–90. doi: 10.1097/00005072-197101000-00008. [DOI] [PubMed] [Google Scholar]
- 25.McCall A, Glaeser BS, Millington W, Wurtman RJ. Monosodium glutamate neurotoxicity, hyperosmolarity, and blood-brain barrier dysfunction. Neurobehav Toxicol. 1979 Winter;1(4):279–83. [PubMed] [Google Scholar]
- 26.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988 Oct;1(8):623–34. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 27.Newcomb R, Sun X, Taylor L, Curthoys N, Giffard RG. Increased production of extracellular glutamate by the mitochondrial glutaminase following neuronal death. J Biol Chem. 1997 Apr 25;272(17):11276–82. doi: 10.1074/jbc.272.17.11276. [DOI] [PubMed] [Google Scholar]
- 28.Ollenschlager G, Jansen S, Schindler J, Rasokat H, Schrappe-Bacher M, Roth E. Plasma amino acid pattern of patients with HIV infection. Clin Chem. 1988 Sep;34(9):1787–9. [PubMed] [Google Scholar]
- 29.Droge W, Eck HP, Betzler M, Naher H. Elevated plasma glutamate levels in colorectal carcinoma patients and in patients with acquired immunodeficiency syndrome (AIDS) Immunobiology. 1987 Aug;174(4–5):473–9. doi: 10.1016/s0171-2985(87)80019-0. [DOI] [PubMed] [Google Scholar]
- 30.Zhao J, Lopez AL, Erichsen D, Herek S, Cotter RL, Curthoys NP, et al. Mitochondrial glutaminase enhances extracellular glutamate production in HIV-1-infected macrophages: linkage to HIV-1 associated dementia. J Neurochem. 2004 Jan;88(1):169–80. doi: 10.1046/j.1471-4159.2003.02146.x. [DOI] [PubMed] [Google Scholar]
- 31.Wurdig S, Kugler P. Histochemistry of glutamate metabolizing enzymes in the rat cerebellar cortex. Neurosci Lett. 1991 Sep 16;130(2):165–8. doi: 10.1016/0304-3940(91)90388-a. [DOI] [PubMed] [Google Scholar]
- 32.Nicklas WJ, Zeevalk G, Hyndman A. Interactions between neurons and glia in glutamate/glutamine compartmentation. Biochem Soc Trans. 1987 Apr;15(2):208–10. doi: 10.1042/bst0150208. [DOI] [PubMed] [Google Scholar]
- 33.Ward HK, Thanki CM, Bradford HF. Glutamine and glucose as precursors of transmitter amino acids: ex vivo studies. J Neurochem. 1983 Mar;40(3):855–60. doi: 10.1111/j.1471-4159.1983.tb08058.x. [DOI] [PubMed] [Google Scholar]
- 34.Curthoys NP, Watford M. Regulation of glutaminase activity and glutamine metabolism. Annu Rev Nutr. 1995;15:133–59. doi: 10.1146/annurev.nu.15.070195.001025. [DOI] [PubMed] [Google Scholar]
- 35.Kvamme E, Svenneby G, Hertz L, Schousboe A. Properties of phosphate activated glutaminase in astrocytes cultured from mouse brain. Neurochem Res. 1982 Jun;7(6):761–70. doi: 10.1007/BF00965528. [DOI] [PubMed] [Google Scholar]
- 36.Holcomb T, Taylor L, Trohkimoinen J, Curthoys NP. Isolation, characterization and expression of a human brain mitochondrial glutaminase cDNA. Brain Res Mol Brain Res. 2000 Mar 10;76(1):56–63. doi: 10.1016/s0169-328x(99)00331-9. [DOI] [PubMed] [Google Scholar]
- 37.Lipton SA, Gendelman HE. Dementia associated with the acquired immunodeficiency syndrome. New Engl J Med. 1995;16:934–40. doi: 10.1056/NEJM199504063321407. [DOI] [PubMed] [Google Scholar]
- 38.Shi B, Girolami UD, He J, Wang S, Lorenzo A, Busciglio J, et al. Apoptosis induced by HIV-1 infection of the central nervous system. J of Clinical Investigation. 1996;98(9):1979–90. doi: 10.1172/JCI119002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nath A, Conant K, Chen P, Scott C, Major EO. Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. J Biol Chem. 1999;274(24):17098–102. doi: 10.1074/jbc.274.24.17098. [DOI] [PubMed] [Google Scholar]
- 40.Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410(6831):988–94. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- 41.Vitkovic L, da Cunha A, Tyor WR. Cytokine expression and pathogenesis in AIDS brain. Res Publ Assoc Res Nerv Ment Dis. 1994;72:203–22. [PubMed] [Google Scholar]
- 42.Wesselingh SL, Power C, Glass JD, et al. Intracerebral cytokine messenger RNA expression in acquired immunedeficiency syndrome dementia. Ann Neurol. 1993;33:576–82. doi: 10.1002/ana.410330604. [DOI] [PubMed] [Google Scholar]
- 43.Nuovo G, Alfieri M. AIDS dementia is associated with massive, activated HIV-1 infection and concomitant expression of several cytokines. Mol Med. 1996;2:358–66. [PMC free article] [PubMed] [Google Scholar]
- 44.Tyor WR, Glass JD, Griffin JW, Becker PS, McArthur JC, Bezman L, et al. Cytokine expression in the brain during acquired immune deficiency syndrome. Ann Neurol. 1992;31(4):349–60. doi: 10.1002/ana.410310402. [DOI] [PubMed] [Google Scholar]
- 45.Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, et al. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci USA. 1998;95:3117–21. doi: 10.1073/pnas.95.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Snyder SH, Bredt DS. Biological roles of nitric oxide. Sci Am. 1992;266(5):68–71. 4–7. doi: 10.1038/scientificamerican0592-68. [DOI] [PubMed] [Google Scholar]
- 47.Heyes MP, Brew BB, Martin A, Price RW, Salazar A, Sidtis JJ, et al. Quinolinic acid in cerebrospinal fluid and serum in HIV-1 infection: relationship to clinical and neurological status. Ann Neurol. 1991;29:202–9. doi: 10.1002/ana.410290215. [DOI] [PubMed] [Google Scholar]
- 48.Heyes MP, Rubinow D, Lane C, Markey SP. Cerebrospinal fluid quinolinic acid concentrations are increased in acquired immune deficiency syndrome. Ann Neurol. 1989;26:275–7. doi: 10.1002/ana.410260215. [DOI] [PubMed] [Google Scholar]
- 49.Nath A, Geiger J. Neurobiological aspects of human immunodeficiency virus infection: neurotoxic mechanisms. Prog Neurobiol. 1998;54(1):19–33. doi: 10.1016/s0301-0082(97)00053-1. [DOI] [PubMed] [Google Scholar]
- 50.Genis P, Jett M, Bernton E, Boyle T, Gelbard H, Dzenko K, et al. Cytokines and arachidonic metabolites produced during human immunodeficiency virus (HIV)-infected macrophage-astroglia interactions: implications for the neuropathogenesis of HIV disease. J Exp Med. 1992;176:1703–18. doi: 10.1084/jem.176.6.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dawson TM, Bredt DS, Fotuhi M, Hwang PM, Snyder SH. Nitric oxide synthase and neuronal NADPH diaphorase are identical in brain and peripheral tissues. Proc Natl Acad Sci USA. 1991;88:7797–801. doi: 10.1073/pnas.88.17.7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Minagar A, Shapshak P, Fujimura R, Ownby R, Heyes M, Eisdorfer C. The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J Neurol Sci. 2002 Oct 15;202(1–2):13–23. doi: 10.1016/s0022-510x(02)00207-1. [DOI] [PubMed] [Google Scholar]
- 53.Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005 Jan;5(1):69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- 54.Giulian D, Noonan C, Vaca K. HIV-1 infected mononuclear phagocytes release neurotoxins. Science. 1990;250:1593–5. doi: 10.1126/science.2148832. [DOI] [PubMed] [Google Scholar]
- 55.Zheng J, Thylin MR, Cotter RL, Lopez AL, Ghorpade A, Persidsky Y, et al. HIV-1 infected and immune competent mononuclear phagocytes induce quantitative alterations in neuronal dendritic arbor: relevance for HIV-1-associated dementia. Neurotoxi Res. 2001;3:443–59. doi: 10.1007/BF03033203. [DOI] [PubMed] [Google Scholar]
- 56.Xiong H, Zeng YC, Zheng J, Thylin M, Gendelman HE. Soluble HIV-1 infected macrophage secretory products mediate blockade of long-term potentiation: a mechanism for cognitive dysfunction in HIV-1-associated dementia. J NeuroVirol. 1999;5:519–28. doi: 10.3109/13550289909045381. [DOI] [PubMed] [Google Scholar]
- 57.Xiong H, Zeng YC, Lewis T, Zheng J, Persidsky Y, Gendelman HE. HIV-1 infected mononuclear phagocyte secretory products affect neuronal physiology leading to cellular demise: relevance for HIV-1-associated dementia. J Neurovirol. 2000;6(Suppl 1):S14–23. [PubMed] [Google Scholar]
- 58.Yeh MW, Kaul M, Zheng J, Nottet HS, Thylin M, Gendelman HE, et al. Cytokine-stimulated, but not HIV-infected, human monocyte-derived macrophages produce neurotoxic levels of l -cysteine. J Immunol. 2000;164(8):4265–70. doi: 10.4049/jimmunol.164.8.4265. [DOI] [PubMed] [Google Scholar]
- 59.Mattson M. Calcium as sculptor and destroyer of neural circuitry. Exp Gerontol. 1992;27:29–49. doi: 10.1016/0531-5565(92)90027-w. [DOI] [PubMed] [Google Scholar]
- 60.Dawson VL, Dawson TM, Uhl GR, Snyder SH. Human immunodeficiency virus type 1 coat protein neurotoxicity mediated by nitric oxide in primary cortical cultures. Proc Natl Acad Sci USA. 1993;90:3256–9. doi: 10.1073/pnas.90.8.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gelbard HA, Dzenko K, Diloreto D, Cerro CD, Cerro MD, Epstein LG. Neurotoxic effects of tumor necrosis factor in primary human neuronal cultures are mediated by activation of the glutamate AMPA recetor subtypes implications for AIDS neuropathogenesis. Dev Neurosci. 1993;15:417–22. doi: 10.1159/000111367. [DOI] [PubMed] [Google Scholar]
- 62.Lawrence CB, Allan SM, Rothwell NJ. Interleukin-1beta and the interleukin-1 receptor antagonist act in the striatum to modify excitotoxic brain damage in the rat. Eur J Neurosci. 1998 Mar;10(3):1188–95. doi: 10.1046/j.1460-9568.1998.00136.x. [DOI] [PubMed] [Google Scholar]
- 63.Wang P, Barks JD, Silverstein FS. Tat, a human immunodeficiency virus-1-derived protein, augments excitotoxic hippocampal injury in neonatal rats. Neuroscience. 1999 Jan;88(2):585–97. doi: 10.1016/s0306-4522(98)00242-5. [DOI] [PubMed] [Google Scholar]
- 64.Hermann GE, Rogers RC, Bresnahan JC, Beattie MS. Tumor necrosis factor-alpha induces cFOS and strongly potentiates glutamate-mediated cell death in the rat spinal cord. Neurobiol Dis. 2001 Aug;8(4):590–9. doi: 10.1006/nbdi.2001.0414. [DOI] [PubMed] [Google Scholar]
- 65.Ma XC, Gottschall PE, Chen LT, Wiranowska M, Phelps CP. Role and mechanisms of interleukin-1 in the modulation of neurotoxicity. Neuroimmunomodulation. 2002;10(4):199–207. doi: 10.1159/000068322. [DOI] [PubMed] [Google Scholar]
- 66.Xu Y, Tao YX. Involvement of the NMDA receptor/nitric oxide signal pathway in platelet-activating factor-induced neurotoxicity. Neuroreport. 2004 Feb 9;15(2):263–6. doi: 10.1097/00001756-200402090-00010. [DOI] [PubMed] [Google Scholar]
- 67.Lipton SA, Rosenberg PA. Excitatory Amino Acids as a Final Common Pathway for Neurologic Disorders. New England Journal of Medicine. 1994;330:613–22. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 68.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65(1):1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 69.Arriza JL, Eliasof S, Kavanaugh MP, Amara SG. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci U S A. 1997;94(8):4155–60. doi: 10.1073/pnas.94.8.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hertz L, Dringen R, Schousboe A, Robinson SR. Astrocytes: glutamate producers for neurons. J Neurosci Res. 1999;57(4):417–28. [PubMed] [Google Scholar]
- 71.Martinez-Hernandez A, Bell KP, Norenberg MD. Glutamine synthetase: glial localization in brain. Science. 1977;195(4284):1356–8. doi: 10.1126/science.14400. [DOI] [PubMed] [Google Scholar]
- 72.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 73.Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995 Jan;34(1):1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 74.Monaghan D, Bridges R, Cotman C. The excitatory amino acid receptors. Annu Rev Pharmacol Toxicol. 1989;29:365–402. doi: 10.1146/annurev.pa.29.040189.002053. [DOI] [PubMed] [Google Scholar]
- 75.Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci. 2004 Mar;61(6):657–68. doi: 10.1007/s00018-003-3319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gil Z, Amitai Y. Adult thalamocortical transmission involves both NMDA and non-NMDA receptors. J Neurophysiol. 1996 Oct;76(4):2547–54. doi: 10.1152/jn.1996.76.4.2547. [DOI] [PubMed] [Google Scholar]
- 77.Li XF, Phillips R, LeDoux JE. NMDA and non-NMDA receptors contribute to synaptic transmission between the medial geniculate body and the lateral nucleus of the amygdala. Exp Brain Res. 1995;105(1):87–100. doi: 10.1007/BF00242185. [DOI] [PubMed] [Google Scholar]
- 78.Choi DW, Koh J-y, Peters S. Pharmacology of Glutamate Neuorotoxicity in Cortical Cell Culture: Attenuation by NMDAv Antagonists. Teh journal of Neuroscience. 1988;8(1):185–96. doi: 10.1523/JNEUROSCI.08-01-00185.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993 May;13(5):2085–104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992 Jan;8(1):189–98. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- 81.Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA--gated glutamate receptor channels depends on subunit composition. Science. 1991 May 10;252(5007):851–3. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 82.Kohr G, Melcher T, Seeburg PH. Candidate editases for GluR channels in single neurons of rat hippocampus and cerebellum. Neuropharmacology. 1998 Oct–Nov;37(10–11):1411–7. doi: 10.1016/s0028-3908(98)00149-x. [DOI] [PubMed] [Google Scholar]
- 83.Berman FW, Murray TF. Domoic acid neurotoxicity in cultured cerebellar granule neurons is mediated predominantly by NMDA receptors that are activated as a consequence of excitatory amino acid release. J Neurochem. 1997 Aug;69(2):693–703. doi: 10.1046/j.1471-4159.1997.69020693.x. [DOI] [PubMed] [Google Scholar]
- 84.Doble A. The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol Ther. 1999 Mar;81(3):163–221. doi: 10.1016/s0163-7258(98)00042-4. [DOI] [PubMed] [Google Scholar]
- 85.Prehn JH, Lippert K, Krieglstein J. Are NMDA or AMPA/kainate receptor antagonists more efficacious in the delayed treatment of excitotoxic neuronal injury? Eur J Pharmacol. 1995 Jan 13;292(2):179–89. doi: 10.1016/0926-6917(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 86.Lu YM, Yin HZ, Weiss JH. Ca2+ permeable AMPA/kainate channels permit rapid injurious Ca2+ entry. Neuroreport. 1995 May 30;6(8):1089–92. doi: 10.1097/00001756-199505300-00004. [DOI] [PubMed] [Google Scholar]
- 87.Brorson JR, Manzolillo PA, Miller RJ. Ca2+ entry via AMPA/KA receptors and excitotoxicity in cultured cerebellar Purkinje cells. J Neurosci. 1994 Jan;14(1):187–97. doi: 10.1523/JNEUROSCI.14-01-00187.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Carriedo SG, Yin HZ, Weiss JH. Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J Neurosci. 1996 Jul 1;16(13):4069–79. doi: 10.1523/JNEUROSCI.16-13-04069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999 Mar;51(1):7–61. [PubMed] [Google Scholar]
- 90.Portera-Cailliau C, Price DL, Martin LJ. Non-NMDA and NMDA receptor-mediated excitotoxic neuronal deaths in adult brain are morphologically distinct: further evidence for an apoptosis-necrosis continuum. J Comp Neurol. 1997 Feb 3;378(1):88–104. [PubMed] [Google Scholar]
- 91.Ferkany JW, Zaczek R, Coyle JT. Kainic acid stimulates excitatory amino acid neurotransmitter release at presynaptic receptors. Nature. 1982 Aug 19;298(5876):757–9. doi: 10.1038/298757a0. [DOI] [PubMed] [Google Scholar]
- 92.Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999 Jan;29(1):83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- 93.Pin JP, Acher F. The metabotropic glutamate receptors: structure, activation mechanism and pharmacology. Curr Drug Targets CNS Neurol Disord. 2002 Jun;1(3):297–317. doi: 10.2174/1568007023339328. [DOI] [PubMed] [Google Scholar]
- 94.Lan JY, Skeberdis VA, Jover T, Zheng X, Bennett MV, Zukin RS. Activation of metabotropic glutamate receptor 1 accelerates NMDA receptor trafficking. J Neurosci. 2001 Aug 15;21(16):6058–68. doi: 10.1523/JNEUROSCI.21-16-06058.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gereau RWt, Conn PJ. Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. J Neurosci. 1995 Oct;15(10):6879–89. doi: 10.1523/JNEUROSCI.15-10-06879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nicoletti F, Bruno V, Copani A, Casabona G, Knopfel T. Metabotropic glutamate receptors: a new target for the therapy of neurodegenerative disorders? Trends Neurosci. 1996 Jul;19(7):267–71. doi: 10.1016/S0166-2236(96)20019-0. [DOI] [PubMed] [Google Scholar]
- 97.McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med. 1998;4(3):291–7. doi: 10.1038/nm0398-291. [DOI] [PubMed] [Google Scholar]
- 98.Olney JW. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science. 1969;164(880):719–21. doi: 10.1126/science.164.3880.719. [DOI] [PubMed] [Google Scholar]
- 99.Coyle J, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–95. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 100.Budd SL, Tenneti L, Lishnak T, Lipton SA. Mitochondrial and extramitochondrial apoptotic signaling pathways in cerebrocortical neurons. Proc Natl Acad Sci U S A. 2000;97(11):6161–6. doi: 10.1073/pnas.100121097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tenneti L, D'Emilia DM, Troy CM, Lipton SA. Role of caspases in N-methyl-D-aspartate-induced apoptosis in cerebrocortical neurons. J Neurochem. 1998;71(3):946–59. doi: 10.1046/j.1471-4159.1998.71030946.x. [DOI] [PubMed] [Google Scholar]
- 102.Ghatan S, Larner S, Kinoshita Y, Hetman M, Patel L, Xia Z, et al. p38 MAP kinase mediates bax translocation in nitric oxide-induced apoptosis in neurons. J Cell Biol. 2000;150(2):335–47. doi: 10.1083/jcb.150.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Atlante A, Calissano P, Bobba A, Giannattasio S, Marra E, Passarella S. Glutamate neurotoxicity, oxidative stress and mitochondria. FEBS Lett. 2001 May 18;497(1):1–5. doi: 10.1016/s0014-5793(01)02437-1. [DOI] [PubMed] [Google Scholar]
- 104.Craven SE, Bredt DS. PDZ proteins organize synaptic signaling pathways. Cell. 1998;93(4):495–8. doi: 10.1016/s0092-8674(00)81179-4. [DOI] [PubMed] [Google Scholar]
- 105.Bossy-Wetzel E, Schwarzenbacher R, Lipton SA. Molecular pathways to neurodegeneration. Nat Med. 2004 Jul;10(Suppl):S2–9. doi: 10.1038/nm1067. [DOI] [PubMed] [Google Scholar]
- 106.Watanabe H, Bannai S. Induction of cysteine transport activity in mouse peritoneal macrophages. J Exp Med. 1987;165:628–40. doi: 10.1084/jem.165.3.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci. 1996 Oct 1;16(19):6125–33. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fiskum G. Mitochondrial participation in ischemic and traumatic neural cell death. J Neurotrauma. 2000 Oct;17(10):843–55. doi: 10.1089/neu.2000.17.843. [DOI] [PubMed] [Google Scholar]
- 109.Fiskum G, Starkov A, Polster BM, Chinopoulos C. Mitochondrial mechanisms of neural cell death and neuroprotective interventions in Parkinson's disease. Ann N Y Acad Sci. 2003;991:111–9. doi: 10.1111/j.1749-6632.2003.tb07469.x. [DOI] [PubMed] [Google Scholar]
- 110.Bellizzi MJ, Lu S, Gelbard HA. Protecting the synapse: evidence for a rational strategy to treat HIV-1 associated neurologic disease. Journal of Neuroimmune Pharmacology. 2006 doi: 10.1007/s11481-005-9006-y. in press. [DOI] [PubMed] [Google Scholar]
- 111.Masliah E, DeTeresa RM, Mallory ME, Hansen LA. Changes in pathological findings at autopsy in AIDS cases for the last 15 years. Aids. 2000;14(1):69–74. doi: 10.1097/00002030-200001070-00008. [DOI] [PubMed] [Google Scholar]
- 112.Garden GA, Budd SL, Tsai E, Hanson L, Kaul M, D'Emilia DM, et al. Caspase cascades in human immunodeficiency virus-associated neurodegeneration. J Neurosci. 2002;22(10):4015–24. doi: 10.1523/JNEUROSCI.22-10-04015.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zheng J, Zhuang W, Yan N, Kou G, Peng H, McNally C, et al. Classification of HIV-1 Mediated Neuronal Dendritic and Synaptic Damage Using Multiple Criteria Linear Programming. Neuroinformatics. 2004;2(3):303–26. doi: 10.1385/ni:2:3:303. [DOI] [PubMed] [Google Scholar]
- 114.Park JS, Bateman MC, Goldberg MP. Rapid alterations in dendrite morphology during sublethal hypoxia or glutamate receptor activation. Neurobiol Dis. 1996;3(3):215–27. doi: 10.1006/nbdi.1996.0022. [DOI] [PubMed] [Google Scholar]
- 115.Hasbani MJ, Hyrc KL, Faddis BT, Romano C, Goldberg MP. Distinct roles for sodium, chloride, and calcium in excitotoxic dendritic injury and recovery. Exp Neurol. 1998 Nov;154(1):241–58. doi: 10.1006/exnr.1998.6929. [DOI] [PubMed] [Google Scholar]
- 116.Swann JW, Al-Noori S, Jiang M, Lee CL. Spine loss and other dendritic abnormalities in epilepsy. Hippocampus. 2000;10(5):617–25. doi: 10.1002/1098-1063(2000)10:5<617::AID-HIPO13>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 117.Hasbani MJ, Schlief ML, Fisher DA, Goldberg MP. Dendritic spines lost during glutamate receptor activation reemerge at original sites of synaptic contact. J Neurosci. 2001 Apr 1;21(7):2393–403. doi: 10.1523/JNEUROSCI.21-07-02393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mattson MP, Keller JN, Begley JG. Evidence for synaptic apoptosis. Exp Neurol. 1998;153(1):35–48. doi: 10.1006/exnr.1998.6863. [DOI] [PubMed] [Google Scholar]
- 119.Ying W. Deleterious network: a testable pathogenetic concept of Alzheimer's disease. Gerontology. 1997;43(4):242–53. doi: 10.1159/000213856. [DOI] [PubMed] [Google Scholar]
- 120.Cotter R, Williams C, Ryan L, Erichsen D, Lopez A, Peng H, et al. Fractalkine (CX3CL1) and brain inflammation: Implications for HIV-1-associated dementia. J Neurovirol. 2002;8(6):585–98. doi: 10.1080/13550280290100950. [DOI] [PubMed] [Google Scholar]
- 121.Gartner S. HIV infection and dementia. Science. 2000;287(5453):602–4. doi: 10.1126/science.287.5453.602. [DOI] [PubMed] [Google Scholar]
- 122.Zheng J, Thylin MR, Persidsky Y, Williams CE, Cotter RL, Zink W, et al. HIV-1 infected immune competent mononuclear phagocytes influence the pathways to neuronal demise. Neurotoxi Res. 2001;3:461–84. doi: 10.1007/BF03033204. [DOI] [PubMed] [Google Scholar]
- 123.Floden AM, Li S, Combs CK. Beta-amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor alpha and NMDA receptors. J Neurosci. 2005;25(10):2566–75. doi: 10.1523/JNEUROSCI.4998-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bellizzi MJ, Lu SM, Masliah E, Gelbard HA. Synaptic activity becomes excitotoxic in neurons exposed to elevated levels of platelet-activating factor. J Clin Invest. 2005;115(11):3185–92. doi: 10.1172/JCI25444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003 Sep 24;23(25):8692–700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fontana G, Valenti L, Raiteri M. Gp120 can revert antagonism at the glycine site of NMDA receptors mediating GABA release from cultured hippocampal neurons. J Neurosci Res. 1997;49(6):732–8. doi: 10.1002/(SICI)1097-4547(19970915)49:6<732::AID-JNR7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 127.Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann Neurol. 2000;47(2):186–94. [PubMed] [Google Scholar]
- 128.Lee SC, Hatch WC, Liu W, Brosnan CF, Dickson DW. Productive infection of human fetal microglia in vitro by HIV-1. Ann N Y Acad Sci. 1993;693:314–6. doi: 10.1111/j.1749-6632.1993.tb26295.x. [DOI] [PubMed] [Google Scholar]
- 129.An SF, Groves M, Giometto B, Beckett AA, Scaravilli F. Detection and localisation of HIV-1 DNA and RNA in fixed adult AIDS brain by polymerase chain reaction/in situ hybridisation technique. Acta Neuropathol (Berl) 1999;98(5):481–7. doi: 10.1007/s004010051113. [DOI] [PubMed] [Google Scholar]
- 130.Bagasra O, Lavi E, Bobroski L, Khalili K, Pestaner JP, Tawadros R, et al. Cellular reservoirs of HIV-1 in the central nervous system of infected individuals: identification by the combination of in situ polymerase chain reaction and immunohistochemistry. AIDS. 1996;10:573–85. doi: 10.1097/00002030-199606000-00002. [DOI] [PubMed] [Google Scholar]
- 131.Nuovo GJ, Gallery F, MacConnell P, Braun A. In Situ Detection of Polymerase Chain Reaction-Amplified HIV-1 Nucleic Acids and Tumor Necrosis Factor-α RNA in teh Central Nervous System. Am J Pathol. 1994;144:659–66. [PMC free article] [PubMed] [Google Scholar]
- 132.Ranki A, Nyberg M, Ovod V, Haltia M, Elovaara I, Raininko R, et al. Abundant expression of HIV Nef and Rev proteins in brain astrocytes in vivo is associated with dementia. AIDS. 1995;9:1001–8. doi: 10.1097/00002030-199509000-00004. [DOI] [PubMed] [Google Scholar]
- 133.Saito Y, Sharer LR, Epstein LG, Michaels J, Mintz M, Louder M, et al. Overexpression of nef as a marker for restricted HIV-1 infection of astrocytes in postmortem pediatric central nervous tissues. Neurology. 1994;44(3 Pt 1):474–81. doi: 10.1212/wnl.44.3_part_1.474. [DOI] [PubMed] [Google Scholar]
- 134.Stoler M, Eskin T, Benn S, Angerer R, Angerer L. Human T-cell lympotropic virus type III infection of the central nervous system. JAMA. 1986;256:2360–4. [PubMed] [Google Scholar]
- 135.Takahashi K, Wesselingh SL, Griffin DE, McArthur JC, Johnson RT, Glass JD. Localization of HIV-1 in human brain using polymerase chain reaction/in situ hybridization and immunocytochemistry. Ann Neurol. 1996;39:705–11. doi: 10.1002/ana.410390606. [DOI] [PubMed] [Google Scholar]
- 136.Tornatore C, Chandra R, Berger JR, Major EO. HIV-1 infection of subcortical astrocytes in the pediatric central nervous system. Neurology. 1994;44(3 Pt 1):481–7. doi: 10.1212/wnl.44.3_part_1.481. [DOI] [PubMed] [Google Scholar]
- 137.Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001 Nov;36(2):180–90. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- 138.Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, et al. Control of synaptic strength by glial TNFalpha. Science. 2002;295(5563):2282–5. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 139.Iino M, Goto K, Kakegawa W, Okado H, Sudo M, Ishiuchi S, et al. Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science. 2001;292(5518):926–9. doi: 10.1126/science.1058827. [DOI] [PubMed] [Google Scholar]
- 140.Oliet SH, Piet R, Poulain DA. Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science. 2001;292(5518):923–6. doi: 10.1126/science.1059162. [DOI] [PubMed] [Google Scholar]
- 141.Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. 2001;291(5504):657–61. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- 142.Danbolt NC. The high affinity uptake system for excitatory amino acids in the brain. Prog Neurobiol. 1994 Nov;44(4):377–96. doi: 10.1016/0301-0082(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 143.Nicholls D, Attwell D. The release and uptake of excitatory amino acids [see comments] Trends Pharmacol Sci. 1990;11(11):462–8. doi: 10.1016/0165-6147(90)90129-v. [DOI] [PubMed] [Google Scholar]
- 144.Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276(5319):1699–702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 145.Hardin-Pouzet H, Krakowski M, Bourbonniere L, Didier-Bazes M, Tran E, Owens T. Glutamate metabolism is down-regulated in astrocytes during experimental allergic encephalomyelitis. Glia. 1997;20(1):79–85. doi: 10.1002/(sici)1098-1136(199705)20:1<79::aid-glia8>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 146.Pitt D, Nagelmeier IE, Wilson HC, Raine CS. Glutamate uptake by oligodendrocytes: Implications for excitotoxicity in multiple sclerosis. Neurology. 2003;61(8):1113–20. doi: 10.1212/01.wnl.0000090564.88719.37. [DOI] [PubMed] [Google Scholar]
- 147.Ohgoh M, Hanada T, Smith T, Hashimoto T, Ueno M, Yamanishi Y, et al. Altered expression of glutamate transporters in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2002 Apr;125(1–2):170–8. doi: 10.1016/s0165-5728(02)00029-2. [DOI] [PubMed] [Google Scholar]
- 148.Fine SM, Angel RA, Perry SW, Epstein LG, Rothstein JD, Dewhurst S, et al. Tumor necrosis factor α inhibits glutamate uptake by primary human astrocytes. J Biol Chem. 1996;271:15303–6. doi: 10.1074/jbc.271.26.15303. [DOI] [PubMed] [Google Scholar]
- 149.Wang Z, Pekarskaya O, Bencheikh M, Chao W, Gelbard HA, Ghorpade A, et al. Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology. 2003;312(1):60–73. doi: 10.1016/s0042-6822(03)00181-8. [DOI] [PubMed] [Google Scholar]
- 150.Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, et al. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391(6664):281–5. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- 151.Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, et al. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4(7):702–10. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- 152.Madl JE, Burgesser K. Adenosine triphosphate depletion reverses sodium-dependent, neuronal uptake of glutamate in rat hippocampal slices. J Neurosci. 1993 Oct;13(10):4429–44. doi: 10.1523/JNEUROSCI.13-10-04429.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nicholls D, Attwell D. The release and uptake of excitatory amino acids. Trends Pharmacol Sci. 1990 Nov;11(11):462–8. doi: 10.1016/0165-6147(90)90129-v. [DOI] [PubMed] [Google Scholar]
- 154.Stover JF, Pleines UE, Morganti-Kossmann MC, Kossmann T, Lowitzsch K, Kempski OS. Neurotransmitters in cerebrospinal fluid reflect pathological activity. Eur J Clin Invest. 1997;27(12):1038–43. doi: 10.1046/j.1365-2362.1997.2250774.x. [DOI] [PubMed] [Google Scholar]
- 155.Sarchielli P, Greco L, Floridi A, Floridi A, Gallai V. Excitatory amino acids and multiple sclerosis: evidence from cerebrospinal fluid. Arch Neurol. 2003;60(8):1082–8. doi: 10.1001/archneur.60.8.1082. [DOI] [PubMed] [Google Scholar]
- 156.Ferrarese C, Aliprandi A, Tremolizzo L, Stanzani L, De Micheli A, Dolara A, et al. Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology. 2001 Aug 28;57(4):671–5. doi: 10.1212/wnl.57.4.671. [DOI] [PubMed] [Google Scholar]
- 157.Dienel GA, Hertz L. Astrocytic contributions to bioenergetics of cerebral ischemia. Glia. 2005;50(4):362–88. doi: 10.1002/glia.20157. [DOI] [PubMed] [Google Scholar]
- 158.Tossman U, Jonsson G, Ungerstedt U. Regional distribution and extracellular levels of amino acids in rat central nervous system. Acta Physiol Scand. 1986 Aug;127(4):533–45. doi: 10.1111/j.1748-1716.1986.tb07938.x. [DOI] [PubMed] [Google Scholar]
- 159.Olalla L, Gutierrez A, Campos JA, Khan ZU, Alonso FJ, Segura JA, et al. Nuclear localization of L-type glutaminase in mammalian brain. J Biol Chem. 2002 Oct 11;277(41):38939–44. doi: 10.1074/jbc.C200373200. [DOI] [PubMed] [Google Scholar]
- 160.Baglietto-Vargas D, Lopez-Tellez JF, Moreno-Gonzalez I, Gutierrez A, Aledo JC. Segregation of two glutaminase isoforms in islets of Langerhans. Biochem J. 2004;381(Pt 2):483–7. doi: 10.1042/BJ20040523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Olalla L, Aledo JC, Bannenberg G, Marquez J. The C-terminus of human glutaminase L mediates association with PDZ domain-containing proteins. FEBS Lett. 2001 Jan 19;488(3):116–22. doi: 10.1016/s0014-5793(00)02373-5. [DOI] [PubMed] [Google Scholar]
- 162.Kovacevic Z, McGivan JD. Mitochondrial metabolism of glutamine and glutamate and its physiological significance. Physiol Rev. 1983;63(2):547–605. doi: 10.1152/physrev.1983.63.2.547. [DOI] [PubMed] [Google Scholar]
- 163.Colquhoun A, Newsholme EA. Aspects of glutamine metabolism in human tumour cells. Biochem Mol Biol Int. 1997;41(3):583–96. doi: 10.1080/15216549700201621. [DOI] [PubMed] [Google Scholar]
- 164.Medina MA, Sanchez-Jimenez F, Marquez J, Rodriguez Quesada A, Nunez de Castro I. Relevance of glutamine metabolism to tumor cell growth. Mol Cell Biochem. 1992;113(1):1–15. doi: 10.1007/BF00230880. [DOI] [PubMed] [Google Scholar]
- 165.Aledo JC, Segura JA, Medina MA, Alonso FJ, Nunez de Castro I, Marquez J. Phosphate-activated glutaminase expression during tumor development. FEBS Lett. 1994;341(1):39–42. doi: 10.1016/0014-5793(94)80236-x. [DOI] [PubMed] [Google Scholar]
- 166.Elgadi KM, Meguid RA, Qian M, Souba WW, Abcouwer SF. Cloning and analysis of unique human glutaminase isoforms generated by tissue-specific alternative splicing. Physiol Genomics. 1999 Aug 31;1(2):51–62. doi: 10.1152/physiolgenomics.1999.1.2.51. [DOI] [PubMed] [Google Scholar]
- 167.Szeliga M, Sidoryk M, Matyja E, Kowalczyk P, Albrecht J. Lack of expression of the liver-type glutaminase (LGA) mRNA in human malignant gliomas. Neurosci Lett. 2005;374(3):171–3. doi: 10.1016/j.neulet.2004.10.051. [DOI] [PubMed] [Google Scholar]
- 168.Mock B, Kozak C, Seldin MF, Ruff N, D'Hoostelaere L, Szpirer C, et al. A glutaminase (gis) gene maps to mouse chromosome 1, rat chromosome 9, and human chromosome 2. Genomics. 1989 Aug;5(2):291–7. doi: 10.1016/0888-7543(89)90060-8. [DOI] [PubMed] [Google Scholar]
- 169.Levy N, Boettger-Tong H, Dohmae K, Agoulnik AI, Ty TI, Nishimune Y, et al. Physical and genetic linkage of glutaminase (Gls), signal transducer and activator of transcription 1 (Stat1), and xeroderma pigmentosum complementation group G (Xpg) on mouse proximal chromosome 1. Genomics. 1998;54(2):355–6. doi: 10.1006/geno.1998.5532. [DOI] [PubMed] [Google Scholar]
- 170.Porter D, Hansen WR, Taylor L, Curthoys NP. Differential expression of multiple glutaminase mRNAs in LLC-PK1-F+ cells. Am J Physiol. 1995 Sep;269(3 Pt 2):F363–73. doi: 10.1152/ajprenal.1995.269.3.F363. [DOI] [PubMed] [Google Scholar]
- 171.Porter LD, Ibrahim H, Taylor L, Curthoys NP. Complexity and species variation of the kidney-type glutaminase gene. Physiol Genomics. 2002;9(3):157–66. doi: 10.1152/physiolgenomics.00017.2002. [DOI] [PubMed] [Google Scholar]
- 172.Shapiro RA, Haser WG, Curthoys NP. The orientation of phosphate-dependent glutaminase on the inner membrane of rat renal mitochondria. Arch Biochem Biophys. 1985 Nov 15;243(1):1–7. doi: 10.1016/0003-9861(85)90767-2. [DOI] [PubMed] [Google Scholar]
- 173.Shapiro RA, Farrell L, Srinivasan M, Curthoys NP. Isolation, characterization, and in vitro expression of a cDNA that encodes the kidney isoenzyme of the mitochondrial glutaminase. J Biol Chem. 1991 Oct 5;266(28):18792–6. [PubMed] [Google Scholar]
- 174.Laake JH, Takumi Y, Eidet J, Torgner IA, Roberg B, Kvamme E, et al. Postembedding immunogold labelling reveals subcellular localization and pathway-specific enrichment of phosphate activated glutaminase in rat cerebellum. Neuroscience. 1999;88(4):1137–51. doi: 10.1016/s0306-4522(98)00298-x. [DOI] [PubMed] [Google Scholar]
- 175.Rosenberg PA. Accumulation of extracellular glutamate and neuronal death in astrocyte-poor cortical cultures exposed to glutamine. Glia. 1991;4(1):91–100. doi: 10.1002/glia.440040111. [DOI] [PubMed] [Google Scholar]
- 176.Driscoll BF, Deibler GE, Law MJ, Crane AM. Damage to neurons in culture following medium change: role of glutamine and extracellular generation of glutamate. J Neurochem. 1993;61(5):1795–800. doi: 10.1111/j.1471-4159.1993.tb09818.x. [DOI] [PubMed] [Google Scholar]
- 177.Newcomb R, Pierce AR, Kano T, Meng W, Bosque-Hamilton P, Taylor L, et al. Characterization of mitochondrial glutaminase and amino acids at prolonged times after experimental focal cerebral ischemia. Brain Res. 1998 Nov 30;813(1):103–11. doi: 10.1016/s0006-8993(98)01006-3. [DOI] [PubMed] [Google Scholar]
- 178.Zielke HR, Tildon JT, Zielke CL, Baab PJ, Landry ME. Functional intracellular glutaminase activity in intact astrocytes. Neurochem Res. 1989;14(4):327–32. doi: 10.1007/BF01000035. [DOI] [PubMed] [Google Scholar]
- 179.Werner P, Pitt D, Raine CS. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol. 2001;50(2):169–80. doi: 10.1002/ana.1077. [DOI] [PubMed] [Google Scholar]
- 180.Mena FV, Baab PJ, Zielke CL, Zielke HR. In vivo glutamine hydrolysis in the formation of extracellular glutamate in the injured rat brain. J Neurosci Res. 2000;60(5):632–41. doi: 10.1002/(SICI)1097-4547(20000601)60:5<632::AID-JNR8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 181.Piani D, Frei K, Do K, Cuenod M, Fontana A. Murine brain macrophages induce NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci Lett. 1991;133:159–62. doi: 10.1016/0304-3940(91)90559-c. [DOI] [PubMed] [Google Scholar]
- 182.Ramonet D, Rodriguez MJ, Fredriksson K, Bernal F, Mahy N. In vivo neuroprotective adaptation of the glutamate/glutamine cycle to neuronal death. Hippocampus. 2004;14(5):586–94. doi: 10.1002/hipo.10188. [DOI] [PubMed] [Google Scholar]
- 183.Muscoli C, Visalli V, Colica C, Nistico R, Palma E, Costa N, et al. The effect of inflammatory stimuli on NMDA-related activation of glutamine synthase in human cultured astroglial cells. Neurosci Lett. 2005;373(3):184–8. doi: 10.1016/j.neulet.2004.09.079. [DOI] [PubMed] [Google Scholar]
- 184.Werner P, Pitt D, Raine CS. Glutamate excitotoxicity--a mechanism for axonal damage and oligodendrocyte death in Multiple Sclerosis? J Neural Transm Suppl. 2000;(60):375–85. doi: 10.1007/978-3-7091-6301-6_27. [DOI] [PubMed] [Google Scholar]
- 185.Smith T, Groom A, Zhu B, Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med. 2000;6(1):62–6. doi: 10.1038/71548. [DOI] [PubMed] [Google Scholar]
- 186.Raber J, Toggas SM, Lee S, Bloom FE, Epstein CJ, Mucke L. Central nervous system expression of HIV-1 Gp120 activates the hypothalamic-pituitary-adrenal axis: evidence for involvement of NMDA receptors and nitric oxide synthase. Virology. 1996;226(2):362–73. doi: 10.1006/viro.1996.0664. [DOI] [PubMed] [Google Scholar]
- 187.Anderson ER, Gendelman HE, Xiong H. Memantine protects hippocampal neuronal function in murine human immunodeficiency virus type 1 encephalitis. J Neurosci. 2004 Aug 11;24(32):7194–8. doi: 10.1523/JNEUROSCI.1933-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. Jama. 2004 Jan 21;291(3):317–24. doi: 10.1001/jama.291.3.317. [DOI] [PubMed] [Google Scholar]
- 189.Lipton SA, Chen HS. Paradigm shift in neuroprotective drug development: clinically tolerated NMDA receptor inhibition by memantine. Cell Death Differ. 2004 Jan;11(1):18–20. doi: 10.1038/sj.cdd.4401344. [DOI] [PubMed] [Google Scholar]
- 190.Ellison G. Competitive and non-competitive NMDA antagonists induce similar limbic degeneration. Neuroreport. 1994;5(18):2688–92. doi: 10.1097/00001756-199412000-00070. [DOI] [PubMed] [Google Scholar]
- 191.Muir KW, Grosset DG, Lees KR. Clinical pharmacology of CNS 1102 in man. Ann N Y Acad Sci. 1995;765:336–7. doi: 10.1111/j.1749-6632.1995.tb16609.x. [DOI] [PubMed] [Google Scholar]
- 192.Olney JW, Farber NB. NMDA antagonists as neurotherapeutic drugs, psychotogens, neurotoxins, and research tools for studying schizophrenia. Neuropsychopharmacology. 1995;13(4):335–45. doi: 10.1016/0893-133X(95)00079-S. [DOI] [PubMed] [Google Scholar]
- 193.Curthoys NP, Kuhlenschmidt T. Phosphate-independent glutaminase from rat kidney. Partial purification and identity with gamma-glutamyltranspeptidase. J Biol Chem. 1975 Mar 25;250(6):2099–105. [PubMed] [Google Scholar]
- 194.Zhao JLA, Herek S, Curthoys N, Tsukamoto T, Ferraris D, Zheng J. Society for Neuroscience. Washington, D.C.: 2005. 2005. Glutaminase mediated glutamate neurotoxicity by HIV-1 infected macrophage. [Google Scholar]
- 195.Kenny J, Bao Y, Hamm B, Taylor L, Toth A, Wagers B, et al. Bacterial expression, purification, and characterization of rat kidney-type mitochondrial glutaminase. Protein Expr Purif. 2003 Sep;31(1):140–8. doi: 10.1016/s1046-5928(03)00161-x. [DOI] [PubMed] [Google Scholar]
- 196.Aledo JC, Rosado A, Olalla L, Campos JA, Marquez J. Overexpression, purification, and characterization of glutaminase-interacting protein, a PDZ-domain protein from human brain. Protein Expr Purif. 2001 Dec;23(3):411–8. doi: 10.1006/prep.2001.1522. [DOI] [PubMed] [Google Scholar]