

Abstract
Asparaginyl endopeptidase (AEP), also known as legumain, is a cysteine protease that has been ascribed roles in antigen presentation yet its exact role in human biology remains poorly understood. We report here the use of a positional scanning combinatorial library of peptide AOMKs containing a P1 aspartic acid to probe the P2, P3 and P4 subsite specificity of endogenous legumain. Using inhibitor specificity profiles of cathepsin B and legumain, we designed fluorescent ABPs that are highly selective, cell permeable reagents for monitoring legumain activity in complex proteomes.
Asparaginyl Endopeptidase (AEP), or legumain, was originally identified in leguminous seeds as a cysteine protease with specificity for asparagine residues in the P1 position[1]. The mammalian legumain homologue is a lysosomal cysteine protease that is a member of the clan CD protease family which includes the caspases, separase and the gingipains[2]. Mammalian legumain has been ascribed a role in the initiation of invariant chain processing during MHC class II mediated antigen presentation[3, 4]. Although the nature of this activity remains controversial, legumain is undoubtedly a key player in lysosomal proteolysis, contributing to the processing of antigenic peptides as well as the processing of the papain family cathepsins[5].
Like all endocytic proteases, legumain is synthesized as an inactive zymogen, and its activity is regulated by post-translational activation events. Therefore, tools that can be used to monitor legumain's activity are necessary in order to understand its functional role. Activity based probes (ABPs) are reagents that can specifically label active proteases, thus allowing their activity, and more importantly their regulation, to be directly monitored[6, 7]. Our laboratory recently reported a synthesis strategy based on the general solid phase methods developed by Ellman and co-workers[8] for the production of peptidyl acyloxymethyl ketone ABPs for diverse cysteine protease activities[9].
We have previously demonstrated that the biotinylated ABP b-hex-D-AOMK efficiently labels endogenous legumain in 816 B cell lysates[9]. However this reagent lacks cell permeability and its overall selectivity towards legumain had not been extensively examined. We therefore set out to develop fluorescent ABPs based on this general scaffold, with the goal of producing cell permeable ABPs with increased potency and selectivity for legumain. We first assessed the ability of peptide AOMKs containing either a single Asp residue (f-hex-D-AOMK) or VAD peptide (f-hex-VAD-AOMK) linked to a short aliphatic spacer and fluorescein tag to function as ABPs for legumain (Fig. 1a, b). Both ABPs potently labeled a ∼38 kDa protein in acidic proteomes from 816 B cells or RAW264.7 monocytes. This activity was confirmed to be legumain by immunoprecipitation using antisera specific for legumain. In addition to the ∼38 kDa predominant active form of legumain, a faint ∼40 kDa protein was labeled by both probes and was also immunodepleted by legumain specific antisera. This protein likely corresponds to the p46 intermediate form of legumain that has been reported to retain enzymatic activity[10]. A ∼50 kDa polypeptide was labeled in the RAW264.7 extracts, presumably corresponding to the ∼56 kDa proenzyme of legumain[10]. Previous studies using saturating concentration of ABPs have demonstrated labeling of inactive protease zymogens due to flexibility of pro-peptide binding in the active site[11].
Figure 1.
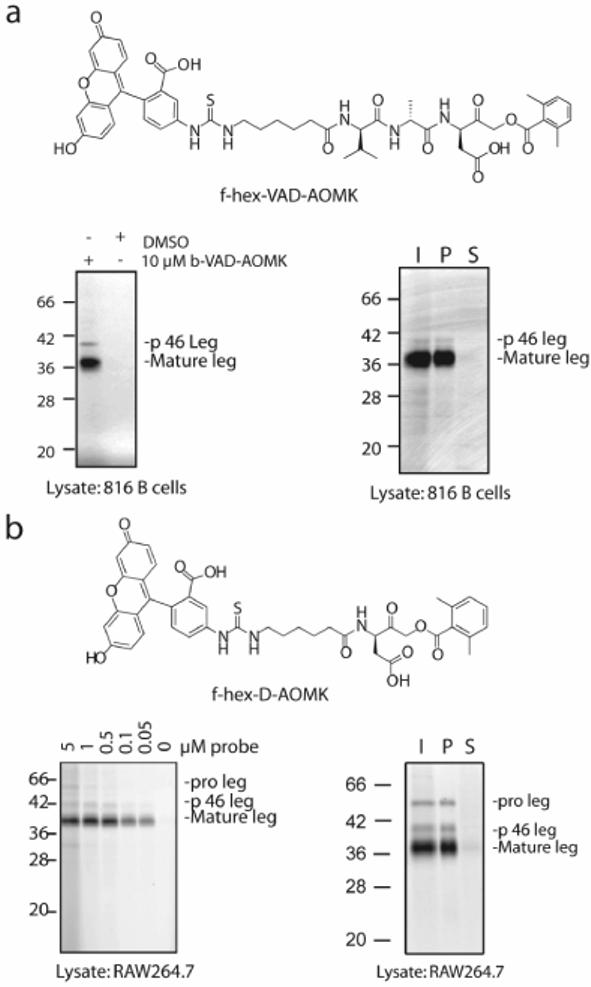
Detection of endogenous legumain activity in complex proteomes (a). Labeling of lysates from 816 B cells with the fluorescent ABP f-hex-VAD-AOMK. Lysates were pre-treated with 10 μM of the broad-spectrum caspase inhibitor b-VAD-AOMK or DMSO followed by labeling for 30 min. with 1 μM f-hex-VAD-AOMK. Labeled proteins were separated by SDS-PAGE and visualized using a flatbed laser scanner. Labeled proteins we identified as leguamin by immunoprecipitation. I=input, P=immunoprecipitated pellet, S=supernatant following immunoprecipitation. (b). Labeling of RAW 264.7 extracts with the P1-only probe f-hex-D-AOMK. Extracts were treated with the probe at the indicated concentrations and labeled proteins were visualized and identified as legumain by immunoprecipitation as in (a).
Since the D-AOMK and VAD-AOMK containing probes were originally designed to target caspases, we reasoned that it should be possible to further optimize the peptide sequence and improve potency towards legumain. To achieve this, we screened a series of positional scanning combinatorial libraries (PSCLs) of peptide AOMKs containing a fixed P1 aspartic acid residue. For each sub-library either the P2, P3 or P4 position was held constant as a single amino acid while the remaining positions were coupled with an equal mixture of 19 amino acids (all 20 natural amino acids minus cysteine and methionine to avoid dimerization and oxidation problems and plus norleucine as a structural analog for methionine) as has been reported previously[12]. Scanning of the natural amino acid sequences through each of the P2-P4 positions provided a specificity fingerprint for legumain that could then be used to select optimal residues for the design of legumain-directed probes. Libraries were screened in 816 B cell lysates by preincubation with 200 nanomolar concentrations of each sub-library followed by labeling of residual legumain activity with the general probe f-hex-VAD-AOMK (Fig. 2).
Figure 2.
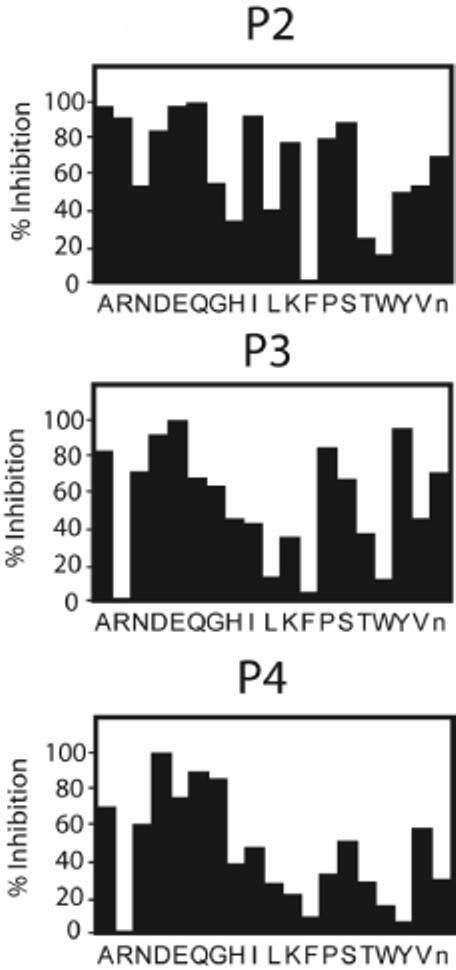
Profiling subsite specificity of endogenous legumain using positional scanning combinatorial libraries of peptide AOMKs. Quantification of results from screening of P2-P4 fixed PSCLs..Values for percent inhibition were calculated by dividing intensity of residual labeled p36 legumain after library treatment by the intensity of labeled p36 legumain in DMSO control samples.
Interestingly, our inhibitor specificity data contains several differences from substrate specificity profiles previously reported for recombinant human legumain[13]. Most notably, legumain showed the strongest preference for a P2 threonine as a substrate, yet threonine ranks among the weakest P2 residues for inhibition of legumain. Likewise, legumain showed little preference for charged residues such as glutamate, aspartate and arginine in the P2 position of substrates, but was highly reactive with these residues in the context of an AOMK inhibitor scaffold. In the S3 subsite our results demonstrate a preference for glutamate followed by tyrosine, whereas very little preference for either of these residues in the P3 position was reported for the substrate profiling data.
Based on the library screening data we chose to generate an optimized fluorescent ABP for legumain containing P2 glutamic acid, P3 proline and P4 valine. These residues were chosen as a compromise between potency and anticipated cell permeable properties. Our choice was further supported by substrate binding studies that suggested that a P2 glutamate would be unlikely to cross-react with lysosomal cathepsins[14]. P3 proline and P4 valine were chosen because they are hydrophobic compounds that show relatively potent inhibition of legumain.
We initially compared an ABP containing an extended binding sequence, f-hex-VPED-AOMK, to the original P1-only ABP, f-hex-D-AOMK. Indeed, the extended subsite ABP was approximately six times more reactive against endogenous legumain in B cell lysates than the P1-only probe (Fig. 3a). Although both probes are highly specific for legumain in B cell lysates, recent studies have suggested that B cells contain lower levels of lysosomal protease activity than other antigen presenting cells[15]. We therefore decided to assess the potency and specificity of our ABPs in the murine monocytic cell line RAW264.7 (Fig 3b). The increased potency for legumain afforded by extension of the peptide binding sequence was accompanied by enhanced reactivity with an additional 30 kDa protease. Labeling of this protein could be blocked by pretreatment with the pan-cathepsin inhibitor, JPM-565, as well as the cathepsin B specific inhibitor CA074 (Supplemental Fig 1) suggesting it was in fact cathepsin B. This was a surprising result since profiling studies performed on the cysteine cathepsins had previously demonstrated that these proteases do not tolerate negatively charged amino acid residues in the P2 position in either substrates or epoxide-based inhibitors[12, 14, 16].
Figure 3.

Probes with extended binding sequence have enhanced reactivity for legumain but lose selectivity. (a). Labeling of 816 lysates with f-hex-VPED-AOMK and f-hex-D-AOMK over a range of probe concentrations. Indicated samples were pretreated with 10 μM JPM-565 to block labeling of papain family csyteine proteases. (b). Labeling of RAW264.7 lysates with f-hex-VPED-AOMK and f-hex-D-AOMK. Samples were prepared and treated as in (a). Note that in this proteome, f-hex-VPED-AOMK labels a ∼30 kDa protein that is blocked by pretreatment with the papain family protease inhibitor JPM-565.
In addition to selecting optimal peptide recognition sequences, we examined effects of extension of the linker region used to attach the fluorescent tag. The use of a hexanoic acid spacer in the probe scaffold resulted in an approximately 1.8 fold increase in reactivity of the P1-only ABP with endogenous legumain in RAW264.7 lysates, without any detectable influence on cross-reactivity with cathepsin B (data not shown). We therefore decided to incorporate this scaffold modification into all of our ABPs going forward.
We next decided to evaluate the specificity of endogenous cathepsin B in order to identify residues that could be used to decrease reactivity against this off-target protease activity. We chose to focus on the P2 position as this is the primary structural determinant of specificity for cathepsin B[17]. RAW264.7 lysates were pre-treated with the P2 fixed PSCL and residual cathepsin B active sites were detected by labeling with f-hex-VPED-AOMK (Supplemental Fig. 2). Interestingly, the screening result identified P2 proline as the least effective residue for cathepsin B but one of the most effective residues for legumain. To determine if the P2 residue alone could be used to modulate specificity between legumain and cathepsin B we synthesized fluorescent ABPs containing the extended linker scaffold and either a P2 glutamic acid (f-(hex)2-ED-AOMK) or a P2 proline (f-(hex)2-PD-AOMK). Incubation with RAW264.7 lysate demonstrated that both of these ABPs were more efficient labels of endogenous legumain than f-(hex)2-D-AOMK. Importantly, only f-(hex)2-ED-AOMK showed reactivity with cathepsin B (Fig. 4).
Figure 4.
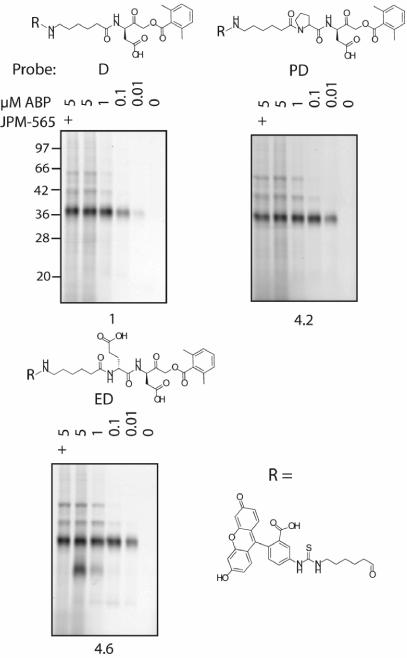
Design of optimized ABPs for legumain. (a). RAW264.7 lysate was pretreated with either 10 μM JPM-565 or DMSO control followed by labeling with f-(hex)2-D-AOMK, f-(hex)2-ED-AOMK or f-(hex)2-PD-AOMK. Intensity of labeling of the p36 legumain species at the highest concentration of each probe relative to the D-only probe is shown at the bottom of gel images.
For the final evaluation of our optimized legumain ABPs we tested if these reagents could be used in live cells. We incubated RAW264.7 cells that had been pretreated for 1 hour with 10 μM of JPM-OEt or DMSO control with 10 μM of either f-(hex)2-D-AOMK, f-(hex)2-ED-AOMK, or f-(hex)2-PD-AOMK. Cells were lysed and labeled proteins visualized by SDS-PAGE followed by fluorescent scanning (Fig. 5). Overall, the in situ labeling profiles observed in live cells were remarkably similar to those observed in cell lysates. Most importantly, ABPs containing either P2 Glu or Pro were more potent towards legumain than the P1-only probe and only f-(hex)2-ED-AOMK showed cross-reactivity towards cathepsin B.
Figure 5.
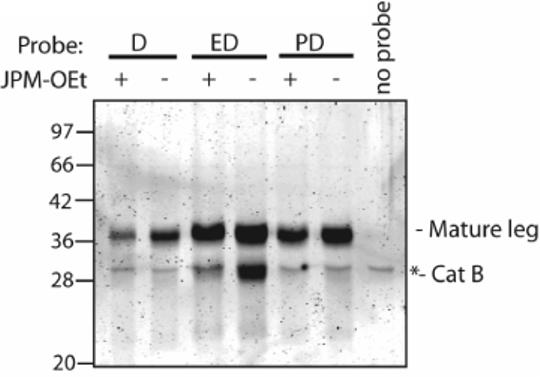
Selectivity of optimized legumain probes in intact cells. Confluent cultures of RAW264.7 cells were treated for 1 hour with 10 μM of papain family protease inhibitor JPM-OEt or DMSO control. Probes from figure 4 were added (1μM final concentration) and incubation continued for 1 hour. Cells were washed three times with PBS and lysed directly in sample buffer. A background band that migrates at the approximate size of cathepsin B was present in cells not treated with inhibitor and is indicated with a *.
Thus, we have identified multiple cell permeable, highly selective ABPs for the CD clan cysteine protease legumain. These tools will prove useful for monitoring legumain activity in living cells and have the potential to be applied to in vivo imaging studies of legumain function as demonstrated for the papain family cathepsins[18]. We believe these reagents will prove to be valuable tools for future studies of legumain function
Acknowledgements
This work was supported by an NIH National Technology Center for Networks and Pathways grant U54 RR020843 (to M.B.), NIH grant R01-EB005011 (to MB), a Department of Defense Breast Cancer Center of Excellence grant DAMD-17-02-0693 (to M.B.) and a Susan G. Koman post-doctoral fellowship (to K.B.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kembhavi AA, Buttle DJ, Knight CG, Barrett AJ. Arch. Biochem. Biophys. 1993;303:208–213. doi: 10.1006/abbi.1993.1274. [DOI] [PubMed] [Google Scholar]
- 2.Chen JM, Rawlings ND, Stevens RA, Barrett AJ. FEBS Lett. 1998;441:361–365. doi: 10.1016/s0014-5793(98)01574-9. [DOI] [PubMed] [Google Scholar]
- 3.Manoury B, Mazzeo D, Li DN, Billson J, Loak K, Benaroch P, Watts C. Immunity. 2003;18:489–498. doi: 10.1016/s1074-7613(03)00085-2. [DOI] [PubMed] [Google Scholar]
- 4.Maehr R, Hang HC, Mintern JD, Kim YM, Cuvillier A, Nishimura M, Yamada K, Shirahama-Noda K, Hara-Nishimura I, Ploegh HL. J. Immunol. 2005;174:7066–7074. doi: 10.4049/jimmunol.174.11.7066. [DOI] [PubMed] [Google Scholar]
- 5.Shirahama-Noda K, Yamamoto A, Sugihara K, Hashimoto N, Asano M, Nishimura M, Hara-Nishimura I. J. Biol. Chem. 2003;278:33194–33199. doi: 10.1074/jbc.M302742200. [DOI] [PubMed] [Google Scholar]
- 6.Speers AE, Cravatt BF. Chembiochem. 2004;5:41–47. doi: 10.1002/cbic.200300721. [DOI] [PubMed] [Google Scholar]
- 7.Berger AB, Vitorino PM, Bogyo M. Am. J. Pharmacogenomics. 2004;4:371–381. doi: 10.2165/00129785-200404060-00004. [DOI] [PubMed] [Google Scholar]
- 8.Lee A, Huang L, Ellman JA. J. Am. Chem. Soc. 1999;121:9907–9914. [Google Scholar]
- 9.Kato D, Boatright KM, Berger AB, Nazif T, Blum G, Ryan C, Chehade KAH, Salvesen GS, Bogyo M. Nat. Chem. Biol. 2005;1:33–38. doi: 10.1038/nchembio707. [DOI] [PubMed] [Google Scholar]
- 10.Li DN, Matthews SP, Antoniou AN, Mazzeo D, Watts C. J. Biol. Chem. 2003;278:38980–38990. doi: 10.1074/jbc.M305930200. [DOI] [PubMed] [Google Scholar]
- 11.Mikolajczyk J, Boatright KM, Stennicke HR, Nazif T, Potempa J, Bogyo M, Salvesen GS. J. Biol. Chem. 2003;278:10458–10464. doi: 10.1074/jbc.M210564200. [DOI] [PubMed] [Google Scholar]
- 12.Greenbaum DC, Arnold WD, Lu F, Hayrapetian L, Baruch A, Krumrine J, Toba S, Chehade K, Bromme D, Kuntz ID, Bogyo M. Chem. Biol. 2002;9:1085–1094. doi: 10.1016/s1074-5521(02)00238-7. [DOI] [PubMed] [Google Scholar]
- 13.Mathieu MA, Bogyo M, Caffrey CR, Choe Y, Lee J, Chapman H, Sajid M, Craik CS, McKerrow JH. Mol. Biochem. Parasitol. 2002;121:99–105. doi: 10.1016/s0166-6851(02)00026-9. [DOI] [PubMed] [Google Scholar]
- 14.Hasnain S, Hirama T, Huber CP, Mason P, Mort JS. J. Biol. Chem. 1993;268:235–240. [PubMed] [Google Scholar]
- 15.Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES. Science. 2005;307:1630–1634. doi: 10.1126/science.1108003. [DOI] [PubMed] [Google Scholar]
- 16.Maciewicz RA, Etherington DJ. Biochem. J. 1988;256:433–440. doi: 10.1042/bj2560433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGrath ME. Annu. Rev. Biophys. Biomol. Struc. 1999;28:181–204. doi: 10.1146/annurev.biophys.28.1.181. [DOI] [PubMed] [Google Scholar]
- 18.Joyce JA, Baruch A, Chehade K, Meyer-Morse N, Giraudo E, Tsai FY, Greenbaum DC, Hager JH, Bogyo M, Hanahan D. Cancer Cell. 2004;5:443–453. doi: 10.1016/s1535-6108(04)00111-4. [DOI] [PubMed] [Google Scholar]