

Abstract
We describe a concise solid support-based synthetic method for the preparation of cyclic D,L α-peptides bearing 1,4,5,8-naphthalenetetracarboxylic diimide (NDI) side chains. Studies of the structural and photoluminescence properties of these molecules in solution show that the hydrogen bond directed self-assembly of the cyclic D,L α-peptide backbone promotes intermolecular NDI excimer formation. The efficiency of NDI charge transfer in the resulting supramolecular assemblies is shown to depend on the length of the linker between the NDI and the peptide backbone, the distal NDI substituent, and the number of NDIs incorporated in a given structure. The design rationale and synthetic strategies described here should provide a basic blueprint for a series of self-assembling cyclic D,L α-peptide nanotubes with interesting optical and electronic properties.
Keywords: charge transfer, cyclic peptides, self-assembly, supramolecular chemistry, nanotubes
Introduction
Cyclic D,L α-peptides are the archetypical member of a growing class of organic tubular supramolecular structures[1–8] with promising applications in biological[9] and materials[10] settings. Due to increasing interest in the field of molecular electronics[11], theoretical calculations have been used to assess the conductive properties of self-assembling peptide nanotubes.[12] It has been proposed that the cyclic D,L α-peptide structures possess a wide band gap (Eg > 4 eV) that could potentially be tuned by appropriate modifications to the cyclic peptide.[12d] However, it has been determined that natural amino acid side chains cannot be used to decrease the band gap to levels sufficient for potential use in molecular electronics applications. Considering that many organic semiconducting and conducting materials are based on one dimensional stacking of aromatic molecules through π-π interactions and partial charge transfer[13], we envisioned that cyclic peptide self-assembly might be useful in controlling and directing the stacking of aromatic side chains and offer a level of control over the long range order in the resulting materials.[1, 10b, 14]
Among aromatic molecules that have found utility in the design of conductive materials, naphthalenediimide (NDI) derivatives have drawn considerable attention due to their tendency to form n-type materials, as opposed to most other organic molecules used to fabricate p-type semiconductors.[15] High conductivity materials based on NDI derivatized dendrimers and polymers have also been reported.[16] We postulated that the intermolecular interactions of NDIs could be directed and/or promoted by the display of such species as side chains of cyclic D,L α-peptides. It was envisioned that controlling aromatic ring interactions and geometry through peptide backbone directed self-assembly[17] might provide a rational supramolecular approach to conductive materials engineering. Towards these goals, in the studies reported here we present a model system for probing hydrogen bond directed intermolecular NDI-NDI interactions in solution. We report the methods developed for the synthesis of NDI modified cyclic D,L α-peptides, examine their propensity for self-assembly by NMR, and probe the efficiency of intermolecular NDI-NDI charge transfer by fluorescence measurements.
Results and Discussion
Design
Cyclic D,L α-peptides can form cylindrical β-sheet ensembles through hydrogen bond directed ring stacking.[2] Interrupting the hydrogen bonding network on one face of the cyclic peptide subunit through backbone N-alkylation restricts the self-assembly to antiparallel cylindrical dimers (Figure 1a).[18, 19] Such assemblies represent the asymmetric unit of the parent extended peptide nanotube and have proven to be useful models for probing the nanotube structure, self-assembly process, and functional properties.[10a, 17–20] We have used this strategy here to investigate the design and optical properties of NDI functionalized self-assembling cyclic D,L α-peptides.
Figure 1.
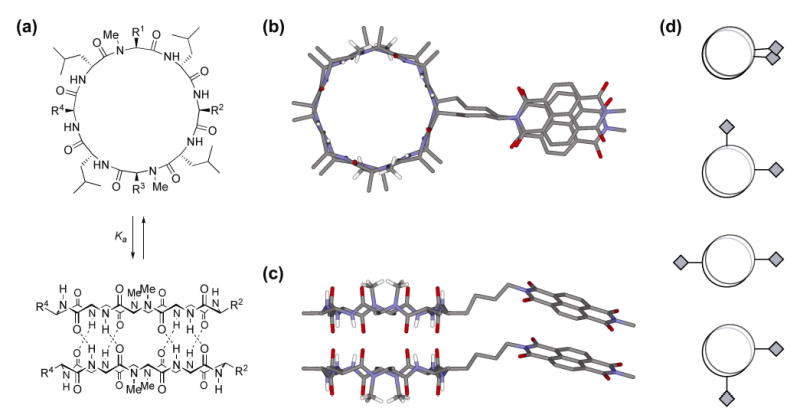
(a) Schematic representation of hydrogen bond directed dimerization of backbone N-methylated cyclic D,L α-peptides (for clarity most side chains are not depicted in dimeric assembly) with generic side chains R1-R4; (b) calculated model of one possible dimer of peptide 3 viewed from the top and (c) from the side (most side chains and protons omitted for clarity); (d) schematic illustration depicting overlap of NDI side chains (diamonds) in the four possible dimeric assemblies available to the C1 symmetric peptides 1, 2, and 3.
Modeling suggests that NDI modified side chains could adopt productive charge transfer geometries in the context of the cyclic D,L α-peptide dimer (Figure 1b, c). Accordingly, peptides 1–5 (Figure 2) were designed in order to probe the influence of linker length, distal NDI substituent, and number of NDI amino acid side chains on the dimer self-assembly and optical properties. Peptides 1–3, which bear a single NDI side chain, were designed to evaluate the effects of steric bulk of the distal NDI substituent (methyl in 3 vs. isobutyl in 1 and 2). The effect of linker length was examined by comparing properties of cylindrical dimers derived from peptides 1 and 2. Peptides 4 and 5, which bear two NDI side chain functionalities, were designed similarly to examine the role of multiple NDI substitutions and linker length. It should be noted that ring symmetry is an important design consideration in these molecules as it determines the number of non-equivalent dimeric assemblies that a given cyclic peptide can form.[17, 19] In the flat ring conformation adopted by an eight residue cyclic D,L α-peptide in the context of a nanotube, the backbone is C4 symmetric with the axis of rotation perpendicular to the plane of the ring.[2b, 19] Accordingly, the dimer shown in Figure 1a depicts only one of four possible diastereomeric assemblies which differ in the cross-strand pairing of homochiral amino acids. Depending on the peptide sequence, one or more of these dimers may be equivalent. Therefore, the C1 symmetric peptides 1–3 can each form four different diastereomeric assemblies (Figure 1d), whereas the C2 symmetric eight residue peptides 4 and 5 can form a maximum of two diastereomers. This is an important consideration as inter-subunit NDI-NDI interactions should only be possible in dimeric species where the aromatic side chains are juxtaposed across each other on opposite rings.
Figure 2.
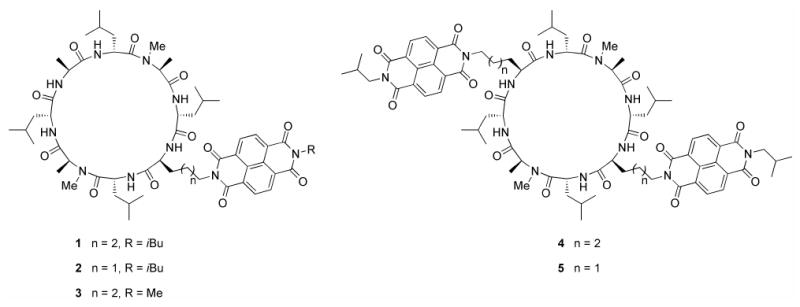
The NDI modified cyclic D,L α-peptide sequences used in this study.
Synthesis
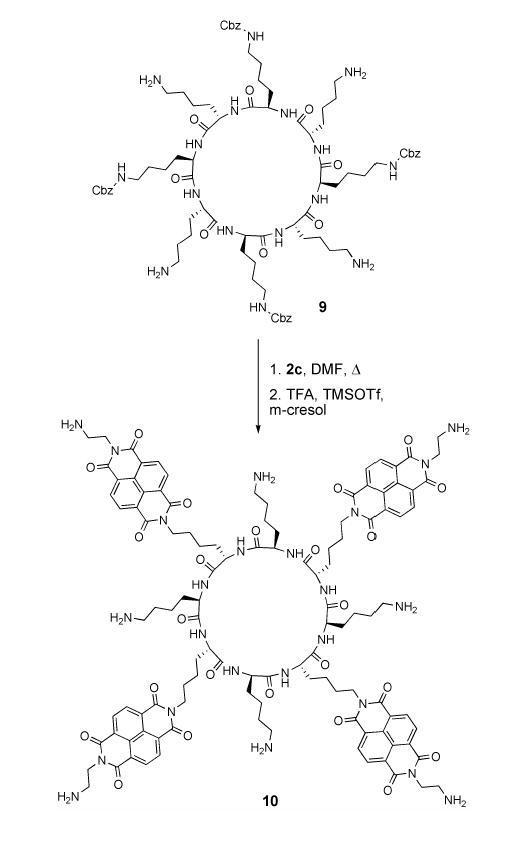
We investigated three synthetic routes toward the desired cyclic peptides bearing NDI side chains. These include preparation from an NDI functionalized amino acid[21], post-synthetic modification[16c] of cyclic D,L α-peptides (Scheme 2), and modification of natural amino acid side chains during the course of solid phase peptide synthesis[22] (Scheme 3). We found for the synthesis of peptides 1–5, the latter approach to be most general and synthetically convenient. Central to each of these approaches is the preparation of unsymmetrically substituted NDIs. Symmetrical NDIs such as N,N’-dibutyl-1,4,5,8-napthalenetetracarboxylic diimide (7) can be prepared by condensation of 1,4,5,8-napthalenetetracarboxylic dianhydride (6) with two equivalents of a primary amine (Scheme 1).[23] Unsymmetrical NDIs present a greater challenge and have been typically prepared from anhydride 6 by condensation with a 1:1 mixture of two different primary amines, leading to a statistical mixture of products. We have found that a modification of a reported synthesis of NDI cyclophanes[24] to be a convenient alternative to this approach (Scheme 1). Following complete hydrolysis of a suspension of 6 (20 mM in water) with KOH, reacidification to pH 6.2 with H3PO4 leads to the formation of the napthalenetetracarboxylic monoanhydride diacid. Addition of one equivalent of a primary amine to the same reaction vessel followed by overnight refluxing yields naphthalene monoimides 8a–c in good isolated yield and purity by a simple filtration workup.
Scheme 2.
Scheme 3.
Scheme 1.
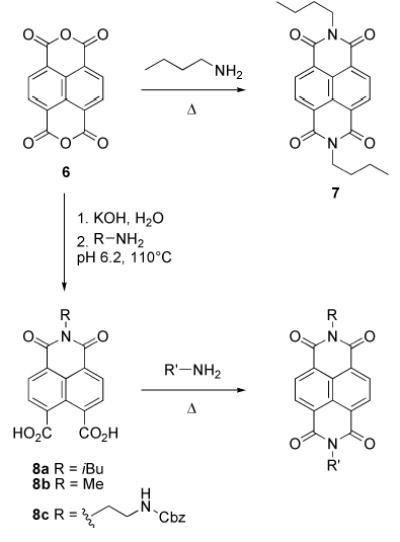
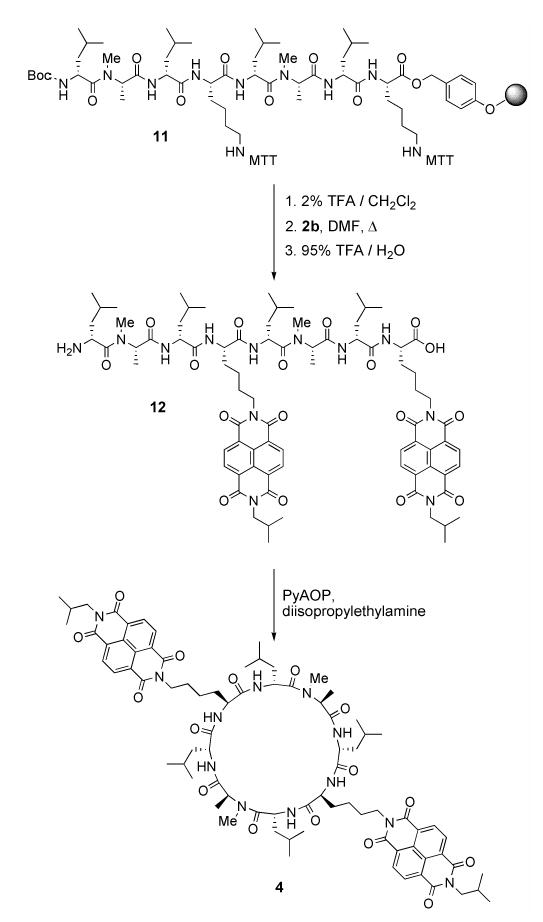
We found that the dicarboxylic acid moieties in 8a–c could be directly converted to imides by condensation with primary amines in DMF at temperatures above 110°C without the need to first isolate the corresponding anhydrides.[16c, 24] In one example of the use of monoimides (8) in the synthesis of cyclic D,L α-peptides (Scheme 2), the four free amine lysine side chains of peptide 9 were converted to diimides by heating at 115°C with Cbz-ethylenediamine modified monoimide 8c in DMF. Deprotection of the Cbz protecting groups followed by purification yielded tetra-NDI substituted peptide 10 in 38% overall yield for the two steps. Naphthalene monoimides were also incorporated onto peptides via on-resin conversion of lysine or ornithine side chain amines to imides (Scheme 3). A representative synthesis began with preparation of linear peptide 11 using standard Fmoc solid phase peptide synthesis protocols, employing ɛ-4-methyltrityl (MTT) protected lysine. The resin bearing 11 was treated with dilute TFA to remove the MTT protecting group, and the resulting free amines converted to imides by stirring the resin with an excess of 8b in DMF at 110°C. The NDI modified linear peptide was cleaved from the resin by treatment with TFA and purified by preparative HPLC to yield 12 in 28% overall yield. Treatment of 12 with (7-azabenzotriazole-1-yloxy) tripyrrolidinophosphonium hexafluorophosphate (PyAOP) and diisopropylethylamine at 0.7 mM in DMF led to formation of the desired cyclic product 4 in 71% isolated yield. This synthetic approach was also used to prepare peptides 1, 2, 3, and 5.
NMR Structural Characterization
NMR was utilized to probe the structure and assembly of peptides 1–5 in CDCl3. 1HNMR spectra at low concentrations (~200 μM) are sharp and consistent with the preponderance of monomeric species in solution. Spectra of peptides 4 and 5 showed peaks for only four different residues, indicating that these peptides maintain an apparent C2 symmetry in solution. For each peptide, increasing the concentration leads to a new set of peaks consistent with hydrogen bonded dimeric species in slow exchange with the monomer on the NMR time scale.[19a] The concentration dependent monomer to dimer ratio was used to determine the apparent self-association constants (Ka) (Table 1). In the calculation of the association constants, all possible diastereomeric ensembles formed by a given peptide were considered equivalent. Thus, the Ka values in Table 1 reflect the overall propensity for self-association demonstrated by each peptide. 1HNMR data fit well to the self-association model, and the results of the calculations show that peptides form dimeric species with Ka ranging from 51 to 192 M−1 in CDCl3.
Table 1.
Association constants as determined by NMR.
| compound | Sequence[a] | Ka[b] (M−1) |
|---|---|---|
| 1 | cyclo[Ala-Leu-MeAla-Leu-LysNDI-iBu-Leu-MeAla-Leu] | 116 |
| 2 | cyclo[Ala-Leu-MeAla-Leu-OrnNDI-iBu-Leu-MeAla-Leu] | 192 |
| 3 | cyclo[Ala-Leu-MeAla-Leu-LysNDI-Me-Leu-MeAla-Leu] | 119 |
| 4 | cyclo[(LysNDI-iBu-Leu-MeAla-Leu)2] | 52 |
| 5 | cyclo[(OrnNDI-iBu-Leu-MeAla-Leu)2] | 87 |
| 7 | - | 0.4 |
underlined residues indicate D-enantiomers; MeAla = N-methyl-alanine;
details of the calculations are found in the experimental.
Direct comparison of the association constants for the non-symmetric peptides 1–3 with that of the C2 symmetric peptides 4 and 5 is complicated by two factors. The non-equivalent diastereomeric assemblies that can be formed from the dimerization of the peptide give rise to an entropy of mixing term that varies with the number and population of those assemblies.[25] In addition, a free energy term arising from the symmetry difference between the peptide monomer and dimer contributes a symmetry component to the observed Ka that is independent of the inherent chemical propensity to self-associate.[26] Performing calculations for the C1 and C2 symmetric peptides, however, shows that the energetic contribution from these two terms cancel assuming that a statistical mixture of dimeric assemblies is formed.
The differences in the binding constants of the various molecules provide some insight into the effect of the NDI modification on assembly of the peptide backbone. Comparison of the single NDI containing peptides 1 and 2 to the bis-NDI peptides 4 and 5, respectively, show 2-fold decrease in the binding constants accompanying incorporation of an additional NDI side chain. This suggests that multiple NDIs on a cyclic peptide tend to destabilize dimer formation. Comparison of 4 to 5 and 1 to 2 show ~60% decrease in Ka for the lysine based linkers, indicating that the dimer is more stable when the NDI group is tethered closer to the peptide backbone. The similar association constants for 1 and 3 indicate that the distal NDI substituent has little impact on intermolecular peptide self-association.
Fluorescence Characterization
Charge delocalization as a consequence of aromatic ring stacking provides the basis for the electronic properties observed in high order NDI aggregates. Charge transfer within an NDI dimer in solution leads to the formation of excimers that can be conveniently monitored by fluorescence spectroscopy.[27] Thus, the role of cyclic peptide backbone self-assembly in enhancing NDI-NDI interactions was probed through fluorescence measurements. Figure 3a shows the fluorescence spectra for single NDI containing peptides 1, 2, and 3 as well as the control compound 7 at 1.65 mM in CDCl3. The formation of excimers is indicated by the presence of a broad peak with a maximum at 492 nm, red shifted with respect to the monomeric peaks at wavelengths of 434 and 408 nm due to charge delocalization. As expected from the peptide self-assembly observed by NMR, all three peptides (1–3) show enhanced excimer peak intensity relative to the control compound 7. These effects can be seen throughout the concentration range (0.1 – 5 mM) studied (Figure 3b).
Figure 3.
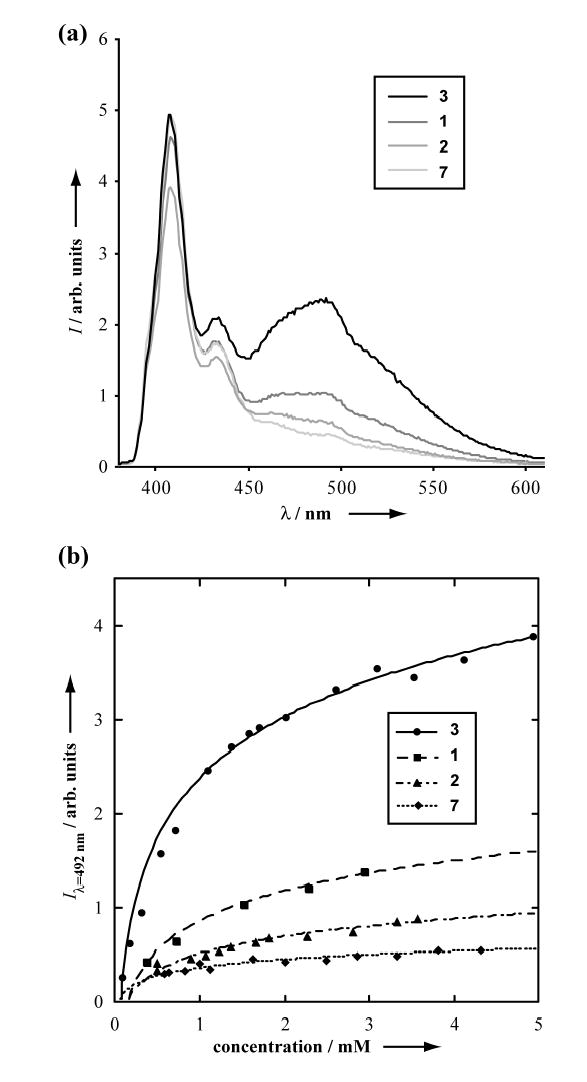
(a) Fluorescence spectra of 1.65 mM solutions of peptides 1, 2, 3, and control compound 7 in CDCl3 (λex = 330 nm); (b) the change in excimer intensity as a function of peptide concentration (lines are shown to guide the eye).
Although the practical range of peptide concentration that can be studied (as determined by the magnitude of Ka) is above where rigorous quantitative analysis of the fluorescence data is possible, some qualitative differences are clear. Differences in excimer peak intensity suggest that, in addition to the enhanced effective concentration brought about by peptide backbone directed self-assembly, other structural parameters seem to play a role in the formation of NDI excimers. It is evident that peptide 3 has a much higher propensity for the formation of NDI excimers than 1. Since the NMR data show similar association constants for the two peptides, the difference in excimer formation can be attributed to the reduced steric hindrance from the distal NDI methyl group in 3 versus the branched isobutyl moiety in 1 allowing more productive interactions between the aromatic rings.
The length of the tether between the peptide backbone and the NDI substituent seems to also influence NDI excimer formation. Peptide 1 shows an increase in excimer formation relative to the control compound 7, while 2, which has an association constant almost twice that of 1 by NMR, shows excimer peak intensity closer to that of the control compound. This suggests that the shorter ornithine based linker between the NDI and the cyclic peptide backbone in 2 does not allow the necessary flexibility for the NDI side chains to adopt an orientation on adjacent peptide strands suitable for an efficient charge transfer. In contrary to the effect of linker length on peptide dimerization, in this case the additional degrees of freedom offered by the lysine based linker allow more favorable intermolecular NDI interactions relative to the shorter ornithine.
As discussed above, in the non-symmetrically substituted peptide 3, the dimer in which the NDI side chains are juxtaposed across from each other represents only one of four possible diastereomeric assemblies (Figure 1d). In calculation of the apparent association constants reported in Table 1, these dimers were all considered equivalent. It is reasonable to assume that only the assembly in which the NDI side chains overlap would be productive with respect to intermolecular excimer formation (Figure 1b, c). Yet, this dimer is likely the least stable and therefore least populated of the four, as the other three have the bulky NDI moiety paired with less sterically demanding alanine side chains. Although peptides 4 and 5 show Ka two orders of magnitude larger than that of 7 by NMR, efforts to probe the efficiency of the intermolecular NDI charge transfer in these C2 symmetric peptides were not informative due to interference by intramolecular NDI interactions. The fluorescence spectrum of each exhibits a large concentration independent excimer peak (data not shown). This peak remains even at concentrations where the 1HNMR indicates that only monomers exist in solution for both peptides, suggesting that there is enough flexibility in the peptide backbone to allow productive intramolecular interaction between NDI side chains. As expected, this intramolecular NDI excimer formation depends on linker length and is more pronounced in 4 than 5.
In conclusion, the above studies indicate that cyclic D,L α-peptide self-assembly can be an effective process for templating directed aromatic side chain-side chain interactions. It is therefore expected that the design rationale described here coupled with the efficient synthetic process should allow access to a variety of cyclic D,L α-peptide sequences bearing NDI side chains for use in the construction of self-assembling peptide nanotube analogs with novel optical and electronic properties.
Experimental Section
General
The Fmoc-amino acids, resins used for solid phase peptide synthesis, and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were all purchased from Novabiochem. NMR solvents were obtained from Cambridge Isotope Labs. N-Cbz-ethylene-diamine-hydrochloride was purchased from TCI America. (7-azabenzotriazole-1-yloxy) tripyrrolidinophosphonium hexafluorophosphate (PyAOP) and all other solvents and reagents were purchased from Aldrich. DMF used in coupling and cyclization steps was dried over 4 Å molecular sieves. All other reagents and solvents were used as received. Reverse phase HPLC was carried out on C18 columns using gradients between 99 : 1 : 0.1 (water/acetonitrile/TFA) and 10 : 90 : 0.1 (water/acetonitrile/TFA). NMR spectra were obtained on Bruker AMX-400, DRX-500, or DRX-600 spectrometers. UV-VIS measurements were made on a Cary 100 Bio UV-visible Spectrophotometer. Fluorescence spectra were acquired on an Aminco Bowman Series 2 Luminescence Spectrometer.
Monoimide 8a
1,4,5,8-napthalenetetracarboxylic dianhydride (6) (2.0 g, 7.46 mmol) was weighed into a 500-ml flask. Water (350 ml) was added, followed by 1 M KOH (35 ml). In some cases, heat, sonication, and/or additional KOH was used to completely dissolve the starting material. After the solid dissolved, the solution was acidified to pH 6.4 with 1 M H3PO4. Isobutylamine (0.741 ml, 7.46 mmol) was added, and the solution was again acidified to pH 6.4 with 1 M H3PO4. The reaction vessel was fitted with a reflux condenser, heated to 110 °C, and stirred overnight. The reaction was then cooled to room temperature and filtered. Acetic acid (5 ml) was added to the filtrate and a white solid precipitated from the solution. The solid was collected by filtration, washed with water, and dried under high vacuum to yield 1.714 g (67%) of the desired product as an off-white solid. 1HNMR (400MHz, DMSO-d6): δ8.51 (d, J = 7 Hz, 2H), 8.07 (d, J = 7 Hz, 2H), 3.90 (d, J = 7 Hz, 2H), 2.12 (m, 1H), 0.90 (d, J = 7 Hz, 6H); 13CNMR (100MHz, DMSO-d6): δ 169.7, 163.3, 130.3, 128.6, 128.1, 128.0, 125.5, 123.4, 46.6, 26.9, 20.2; MALDI-FTMS: [M+H]+ obsd. = 342.0972 (calc. = 342.0964).
Monoimide 8b
Prepared by the reaction of 1,4,5,8-napthalenetetracarboxylic dianhydride (6) (1.0 g, 3.73 mmol) with methylamine-hydrochloride (252 mg, 3.73 mmol) as described for 8a. The reaction yielded 890 mg (80%) of the product as a pale yellow solid. 1HNMR (500 MHz, D2O): δ 8.56 (d, J = 8 Hz, 2H), 8.05 (d, J = 8 Hz, 2H), 3.47 (s, 3H); 13CNMR (125 MHz, NaOD/D2O, DSS as internal standard): δ 178.0, 169.1, 147.6, 134.0, 131.6, 129.5, 127.5, 124.8, 29.6; ESI-TOF MS: [M-H]− obsd. = 298.0360 (calc. = 298.0357).
Monoimide 8c
Prepared by the reaction of 1.163 g (4.34 mmol) of 1,4,5,8-napthalenetetracarboxylic dianhydride (6) with 1.0 g (4.33 mmol) N-Cbz-ethylene-diamine-hydrochloride as described for 2a. The reaction yielded 1.129 g (56%) of the product as a pale tan solid. 1HNMR (400 MHz, DMSO-d6) δ 8.50 (d, J = 8 Hz, 2H), 8.07 (d, J = 8 Hz, 2H), 7.4–7.2 (m, 6H), 4.94 (s, 2H), 4.16 (t, J = 6 Hz, 2H), 3.60 (q, J = 6 Hz, 2H); 13CNMR (150 MHz, DMSO-d6 with 4 eq. triethylamine to improve solubility) δ 170.8, 163.6, 156.2, 146.3, 137.2, 129.9, 128.7, 128.1, 127.5, 127.4, 126.3, 125.7, 121.6, 64.9, 38.2; ESI-TOF MS (m/z) [M-H]− obsd. = 461.0989 (calc. =461.0990).
Cyclic Peptide 9
Peptide 9 was prepared from Fmoc-Lys-OAllyl attached via the amine of the lysine side chain to 2-Cl-Trt resin using methodology previously reported.[28] The crude material was purified by preparative RP-HPLC. ESI-TOF MS (m/z) [M+H]+ obsd. = 1561.9134 (calc. = 1561.9141).
NDI derivitization of a peptide in solution – Synthesis of 10
Peptide 9 (11 mg, 5.5 μmol assuming a salt with 4 molecules of trifluoroacetate per peptide) and 8c (12.6 mg, 27.3 μmol) were suspended in DMF (5 ml). The mixture was heated to 115°C under argon and stirred overnight. The resulting solution was cooled to room temperature and concentrated to dryness. The remaining solid was washed (2 × 2 ml) with pH 8.0 phosphate buffer, the supernatant discarded, and the solid dried under high vacuum. The crude solid was dissolved in 10:2:2 TFA/trimethylsilyl trifluoromethanesulfonate/m-cresol (200 μl). After 1 hr, ether (2 ml) was added, and the resulting suspension centrifuged. The supernatant was discarded, and the solid washed twice more with ether (2 ml). The crude peptide was purified by preparative RP-HPLC to yield 6.4 mg (38% assuming a salt with 8 molecules of trifluoroacetate per peptide) of the product as an off white solid. 1HNMR (600 MHz, CDCl3/TFA, all signals were broad, likely due to aggregation; integration of amide NH at 6.93 ppm was lower than 8 due to partial exchange with TFA) δ 8.93 (br s, 16H), 6.93 (br s, 7 H), 4.91 (br s, 8 H), 4.79 (br s, 8 H), 4.31 (br s, 8H), 3.82 (br s, 8H), 3.37 (br s, 8H), 2.00 (br s, 32 H), 1.65 (br s, 16H); ESI-TOF MS (m/z) [M+2H]2+ obsd. = 1097.4841 (calc. = 1097.4840).
Representative on-resin NDI derivitization of peptides – Synthesis of 12
1. Protected linear peptide on resin
Fmoc-Lys(MTT) Wang polystyrene resin (139 mg, 0.100 mmol) was weighed into a sintered glass peptide synthesis vessel and allowed to swell in CH2Cl2 for 45 min, then in DMF for 10 min. The resin was treated with 20% piperidine/DMF (2 x 8 min) to remove the Fmoc group, and washed with DMF (3x). A solution of Fmoc-D-leucine (142 mg, 0.40 mmol), HBTU (148 mg, 0.39 mmol), and diisopropylethylamine (175 μl, 1.0 mmol) in DMF (3 ml) was allowed to prereact for 2 min then added to the resin. The resin suspension was agitated for 30 min, drained, and washed with DMF (3x). This deprotection – coupling cycle was repeated to obtain the full length linear peptide 11. Coupling reactions following N-methyl-alanine residues were performed twice to ensure complete reaction.
2. Conversion of Resin bound Lys(MTT) to Lys(NDI) and cleavage from resin
The resin bearing 11 was washed with CH2Cl2 (3 x 1 min) and treated with a mixture of 93 : 5 : 2 CH2Cl2/triethylsilane/trifluoroacetic acid (3 x 2 min followed by 3 x 10 min) to remove the lysine MTT protecting groups. The resin was then washed sequentially with CH2Cl2, DMF, 95:5 DMF/disopropylethylamine, and again with DMF. The resin was transferred to a 20 ml vial to which 8a (1.5 eq relative to resin bound amines, 102 mg, 0.30 mmol) was added followed by DMF (8 ml). The suspension was heated to 110 °C and stirred for 6 hr. The reaction was cooled to room temperature, and filtered through a fritted syringe. The resin was washed thoroughly (5 x DMF, 3 x CH2Cl2, 3 x MeOH) and dried under high vacuum. The peptide was cleaved from the resin by treatment with 95:5 TFA/H2O for 3 hr. The cleavage mixture was collected by filtration and the resin was washed twice with TFA. The filtrates were combined and concentrated under vacuum to an oily residue. Water was added and the mixture was sonicated to produce a suspension of cream colored solid. This mixture was frozen and lyophilized to yield crude product. Purification by preparative RP-HPLC on a C18 column yielded 46 mg (28%) of the trifluoroacetate salt of the product as an off-white solid. MALDI-FTMS: [M+H]+ obsd. = 1507.7926 (calc. = 1507.7871).
Cyclic 4
Linear peptide 12 (46 mg, 28 μmol) was dissolved in DMF (40 ml). PyAOP (44 mg, 85 μmol) was added followed by diisopropylethylamine (30 μl, 170 μmol). The reaction was stirred for 2.5 hr at room temperature and concentrated under vacuum. Water/acetonitrile/TFA (8 ml of 1 : 1 : 0.001) was added and the residue was sonicated to create a suspension. After centrifugation, the supernatant was removed. The solid was washed twice more with the same procedure. The resulting pellet was suspended in water, sonicated, and lyophilized to yield 30 mg (71%) of the product as an off white solid. 1HNMR (600 MHz, CDCl3): δ 8.55 (s, 8H), 7.59 (d, J = 8 Hz, 2H), 6.82 (d, J = 10 Hz, 2H), 6.55 (d, J = 7 Hz, 2H), 5.25 (q, J = 7 Hz, 2H), 4.87 (dd, J = 7, 8 Hz, 2H), 4.65 (dd, J = 6, 6 Hz, 2H), 4.46 (dt, J = 4, 10 Hz, 2H), 4.10 (m, 4H), 4.00 (d, J = 7 Hz, 4H), 2.83 (s, 6H), 2.19 (m, 2H), 1.96 (m, 2H), 1.9–1.7 (m, 8H), 1.68–1.36 (m, obscured by H2O), 1.32 (d, J = 7 Hz, 6H), 0.97–0.93 (overlapping doublets, 24H), 0.90 (d, J = 7 Hz, 6H), 0.85 (d, J = 7 Hz, 6H); MALDI-FTMS: [M+H]+ obsd. = 1489.7760 (calc. = 1489.7766).
Linear precursor to 1
Linear peptide was synthesized from Fmoc-Ala Wang polystyrene resin (141 mg, 0.10 mmol) and purified as described above for 12 to yield 27 mg (21%) of the trifluoroacetate salt of the product as a white solid. MALDI-FTMS: [M+H]+ obsd. = 1145.6621 (calc. = 1145.6605).
Cyclic 1
Linear precursor (27 mg, 21.4 μmol) was cyclized as described for 4. The product was purified by RP-HPLC on a C18 column to yield 12 mg (50%) of the desired product as a white solid. 1HNMR (600 MHz, CDCl3): δ 8.75(d, J = 2 Hz, 4H), 7.58 (d, J = 8 Hz, 1H), 7.51 (d, J = 9 Hz), 6.86 (d, J = 9 Hz, 1H), 6.80 (d, J = 10 Hz, 1H), 6.66 (d, J = 6 Hz, 1H), 6.57 (d, J = 7 Hz, 1H), 5.24 (m, 2H), 4.85 (m, 2H), 4.59 (m, 2H), 4.46 (m, 2H), 4.15 (t, J = 8 Hz, 2H), 4.06 (d, J = 7 Hz, 2H), 2.82 (overlapping s, 6H), 2.23 (m, 1H), 1.37 (d, J = 7 Hz, 3H), 1.32 (d, J = 7 Hz, 6H), 1.95–1.40 (m, obscured by H2O), 1.0–0.8 (overlapping d, 30H); MALDI-FTMS: [M+Na]+ obsd. = 1149.6313 (calc. = 1149.6319).
Linear precursor to 2
Linear peptide was synthesized from Fmoc-Ala Wang polystyrene resin (141 mg, 0.10 mmol) and purified as described above for 12 to yield 8 mg (6%) of the trifluoroacetate salt of the product as a white solid. MALDI-FTMS: [M+H]+ obsd. = 1131.6441 (calc. = 1131.6448).
Cyclic 2
Linear precursor (8 mg, 6.4 μmol) was cyclized as described for 4. The product was purified by RP-HPLC on a C18 column to yield 4.2 mg (59%) of the desired product as an off-white solid. 1HNMR (600 MHz, CDCl3): δ 8.75 (d, J = 8 Hz, 2H), 8.72 (d, J = 8 Hz, 2H), 7.61 (d, J = 7 Hz, 1H), 7.46 (d, J = 8 Hz, 1H), 6.83 (d, J = 9 Hz, 1H), 6.77 (d, J = 8 Hz, 1H), 6.66 (br s, 1H), 6.57 (br s, 1H), 5.21 (br s, 2H), 4.83 (m, 2H), 4.66 (m, 1H), 4.56 (m, 1H), 4.48 (m, 1H), 4.44 (m, 1H), 4.15 (m, 2H), 4.06 (d, J = 8 Hz, 2H), 2.28 (s, 6H), 2.0–1.2 (overlapping multiplets obscured by H2O peak), 1.35 (d, J = 7 Hz, 3H), 1.31 (d, J = 7 Hz, 6H), 1.0–0.87 (overlapping doublets, 36H); MALDI-FTMS: [M+H]+ obsd. = 1113.6343 (calc. = 1113.6343).
Linear precursor to 3
Linear peptide was synthesized (using monoimide 8b) from Fmoc-Ala Wang polystyrene resin (141 mg, 0.10 mmol) and purified as described above for 12 to yield 16 mg (13%) of the trifluoroacetate salt of the product as a white solid. MALDI-FTMS: [M+H]+ obsd. = 1103.6113 (calc. = 1103.6135).
Cyclic 3
Linear precursor (16 mg, 13.1 μmol) was cyclized as described for 4. The product was purified by RP-HPLC on a C18 column to yield 8 mg (56%) of the desired product as an off-white solid. 1HNMR (600 MHz, CDCl3) δ 8.76 (s, 4H), 7.57 (d, J = 8 Hz, 1H), 7.49 (d, J = 8 Hz, 1H), 6.85 (d, J = 10 Hz, 1H), 6.79 (d, J = 10 Hz, 1H), 6.63 (d, J = 6 Hz, 1H), 6.55 (d, J = 7 Hz, 1H), 5.24 (m, 2H), 4.86 (m, 2H), 4.59 (m, 2H), 4.46 (m, 2H), 4.15 (t, J = 8 Hz, 2H), 3.59 (s, 3H), 2.82 (s, 3H), 2.81 (s, 3H), 1.9–1.4 (overlapping multiplets), 1.36 (d, J = 7 Hz, 3H), 1.31 (d, J = 7 Hz, 6H), 1.23 (s, 3H), 0.98–0.84 (overlapping doublets, 24H); ESI-TOF MS: [M+H]+ obsd. = 1085.6030 (calc. = 1085.6030).
Linear precursor to 5
Linear peptide was synthesized from Fmoc-Orn(MTT) Wang polystyrene resin (204 mg, 0.10 mmol) and purified as described above for 12 to yield 39 mg (24%) of the trifluoroacetate salt of the product as a white solid. MALDI-FTMS: [M+H]+ obsd. = 1479.7511 (calc. = 1479.7558).
Cyclic 5
Linear precursor (39 mg, 24.4 μmol) was cyclized as described for 4 to yield 33 mg (93%) of the product as an off-white solid. 1HNMR (500 MHz, CDCl3): δ 8.70 (s, 8H), 7.55 (d, J = 8 Hz, 2H), 6.78 (d, J = 10 Hz, 2H), 6.55 (d, J = 7 Hz, 2H), 5.21 (q, J = 7 Hz, 2H), 4.84 (dd, J = 7, 8 Hz, 2H), 4.72 (dd, J = 6, 6 Hz, 2H), 4.47 (td, J = 10, 4 Hz), 4.19 (m, 4H), 4.04 (d, J = 7 Hz, 4H), 2.81 (s, 6H), 2.2 (m, 2H), 1.98 (m, 2H), 1.85–1.40 (m, 18H), 1.30 (d, J = 7 Hz, 6H), 0.98 (d, J = 7 Hz, 12H), 0.92 (m, 24H); MALDI-FTMS: [M+Na]+ obsd. = 1483.7228 (calc. = 1483.7272).
Concentration Dependent NMR for N-Me cyclic peptides
[19a] All spectra were obtained in CDCl3 (99.96% from Cambridge Isotope Labs). A stock solution of freshly dried peptide was made in CDCl3, and this solution was sonicated and centrifuged to remove any insoluble material. The concentration of this stock was determined by absorption spectroscopy (NDI ɛ380 = 29,850 cm−1 · M−1). The stock solution was then diluted to make samples at the desired concentrations (at least three different concentrations for each peptide). 1HNMR spectra were obtained on a Bruker DRX-500 or DRX-600 spectrometer. As the monomer and dimer species are in slow exchange on the NMR time scale, peaks were visible for both species. The data were fit to the following model:
| [1] |
and
| [2] |
where [A] and [A2] are the concentrations of monomer and dimer respectively and Ctot is the total concentration. The integrated intensities of the backbone N-methyl groups were used to measure the ratio of the two species. A variable x was defined according to eq 3:
| [3] |
where Idimer is the integrated intensity of dimer N-methyl peaks and Imonomer is the integrated intensity of monomer N-methyl peaks. Combining equations 1–3 gives eq. 4.
| [4] |
The equilibrium constant for dimerization (K) was found from the inverse of the slope of the linear fit of Ctot against (2x2+x) for each experimental concentration.
Self-association constant of 7 by NMR
[29] NDI control compound 7 demonstrated concentration dependent chemical shifts in the range of 1 to 50 mM. This concentration dependent behavior was modeled as a monomer and dimer in equilibrium and in fast exchange on the NMR time scale. The data was modeled using to the self-association equilibrium shown in equations 1 and 2 described above. For a given Ctot and an arbitrary value of K, [A] can be found from the root of eq. 5
| [5] |
where [A] is greater than 0 and less than Ctot. Assuming equilibrium between monomer and dimer in fast exchange, the chemical shift for a given proton in 7 follows eq [6]
| [6] |
where δ is the observed chemical shift, δ1 is the chemical shift of the monomer, and δ2 is the chemical shift of the dimer. Thus, observed δ for each experimental Ctot are used with an arbitrary K in eq. [7]
| [7] |
to determine δ1 and δ2 by linear regression analysis. These values are used, in turn, to calculate a theoretical δ for each experimental concentration. Numerical minimization of the sum of the squares of the residuals between observed and calculated δ gave the best fit when K = 0.44 M−1.
Fluorescence Measurements
Stock solutions were prepared in CDCl3 as described above for the concentration dependent NMR experiments. Samples were measured in a 0.2 × 1 cm quartz cuvette (0.2 cm along excitation path). The excitation wavelength was 330 nm and the detector voltage was kept the same for all samples.
Acknowledgments
We would like to thank the National Institute of Health (GM52190) and the Air Force Office of Scientific Research (F49620-00-1-0283) for financial support. W.S.H. thanks the National Science Foundation for a Predoctoral Fellowship and N.A. the Fulbright Foundation for a Postdoctoral Fellowship.
References
- 1.For a review, see: Bong DT, Clark TD, Granja JR, Ghadiri MR. Angew Chem. 2001;113:1016–1041. Angew Chem Int Ed. 2001;40:988–1011.
- 2.Cyclic D,L α-peptides: Ghadiri MR, Granja JR, Milligan RA, McRee DE, Khazanovich N. Nature. 1993;366:324–327. doi: 10.1038/366324a0.Hartgerink JD, Granja JR, Milligan RA, Ghadiri MR. J Am Chem Soc. 1996;118:43–50.Rosenthal-Aizman K, Svensson G, Unden A. J Am Chem Soc. 2004;126:3372–3373. doi: 10.1021/ja0372659.
- 3.Cyclic β-peptides: Clark TD, Buehler LK, Ghadiri MR. J Am Chem Soc. 1998;120:651–656. Seebach D, Matthews JL, Meden A, Wessels T, Baerlocher C, McCusker LB. Helv Chim Acta. 1997;80:173–182.
- 4.Cyclic α,β-peptides: Karle IL, Handa BK, Hassall CH. Acta Crystallogr. 1975;B31:555–560.
- 5.Cyclic α,γ -peptides: Amorin M, Castedo L, Granja JR. J Am Chem Soc. 2003;125:2844–2845. doi: 10.1021/ja0296273.
- 6.Cyclic δ-peptides: Gauthier D, Baillargeon P, Drouin M, Dory YL. Angew Chem. 2001;113:4771–4774. doi: 10.1002/1521-3773(20011217)40:24<4635::aid-anie4635>3.0.co;2-d. Angew Chem Int Ed. 2001;40:4635–4638. doi: 10.1002/1521-3773(20011217)40:24<4635::aid-anie4635>3.0.co;2-d.
- 7.Cyclic oligoureas: Ranganathan D, Lakshmi C, Karle IL. J Am Chem Soc. 1999;121:6103–6107. Semetey V, Didierjean C, Briand J-P, Aubry A, Guichard G. Angew Chem. 2002;114:1975–1978. Angew Chem Int Ed. 2002;41:1895–1898. doi: 10.1002/1521-3773(20020603)41:11<1895::aid-anie1895>3.0.co;2-3.
- 8.Cylclic α,ɛ-peptides Horne WS, Stout CD, Ghadiri MR. J Am Chem Soc. 2003;125:9372–9376. doi: 10.1021/ja034358h.
- 9.a) Ghadiri MR, Granja JR, Buehler LK. Nature. 1994;369:301–304. doi: 10.1038/369301a0. [DOI] [PubMed] [Google Scholar]; b) Granja JR, Ghadiri MR. J Am Chem Soc. 1994;116:10785–10786. [Google Scholar]; c) Sanchez-Quesada J, Isler MP, Ghadiri MR. J Am Chem Soc. 2002;124:10004–10005. doi: 10.1021/ja025983+. [DOI] [PubMed] [Google Scholar]; d) Sanchez-Quesada J, Kim HS, Ghadiri MR. [Google Scholar]; Angew Chem. 2001;113:2571–2574. [Google Scholar]; Angew Chem Int Ed. 2001;40:2503–2506. [Google Scholar]; e) Fernandez-Lopez S, Kim HS, Choi EC, Delgado M, Granja JR, Khasanov A, Kraehenbuehl K, Long G, Weinberger DA, Wilcoxen KM, Ghadiri MR. Nature. 2001;412:452–456. doi: 10.1038/35086601. [DOI] [PubMed] [Google Scholar]
- 10.a) Vollmer MS, Clark TD, Steinem C, Ghadiri MR. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1598::AID-ANIE1598>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]; Angew Chem. 1999;111:1703–1706. [Google Scholar]; Angew Chem Int Ed. 1999;38:1598–1601. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1598::AID-ANIE1598>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]; b) Steinem C, Janshoff A, Vollmer MS, Ghadiri MR. Langmuir. 1999;15:3956–3964. [Google Scholar]; c) Motesharei K, Ghadiri MR. J Am Chem Soc. 1997;119:11306–11312. [Google Scholar]
- 11.a) Bachtold A, Hadley P, Nakanishi T, Dekker C. Science. 2001;294:1317–1320. doi: 10.1126/science.1065824. [DOI] [PubMed] [Google Scholar]; b) Collier CP, Matterstei G, Wong EW, Luo Y, Beverly K, Sampaio J, Raymo FM, Stoddart JF, Heath JR. Science. 2000;289:1172–1175. doi: 10.1126/science.289.5482.1172. [DOI] [PubMed] [Google Scholar]; c) Huang Y, Duan X, Wei Q, Lieber CM. Science. 2001;291:630–633. doi: 10.1126/science.291.5504.630. [DOI] [PubMed] [Google Scholar]; d) Joachim C, Gimzewski JK, Aviram A. Nature. 2000;408:541–548. doi: 10.1038/35046000. [DOI] [PubMed] [Google Scholar]; e) Tour JM. Acc Chem Res. 2000;33:791–804. doi: 10.1021/ar0000612. [DOI] [PubMed] [Google Scholar]
- 12.a) Carloni P, Andreoni W, Parrinello M. Phys Rev Lett. 1997;79:761–764. [Google Scholar]; b) Jishi RA, Braier NC, White CT, Mintmire JW. Phys Rev B. 1998;58:R16009–R16011. [Google Scholar]; c) Lewis JP, Pawley NH, Sankey OF. J Phys Chem B. 1997;101:10576–10583. [Google Scholar]; d) Okamoto H, Nakanishi T, Nagai Y, Kasahara M, Takeda K. J Am Chem Soc. 2003;125:2756–2769. doi: 10.1021/ja0212720. [DOI] [PubMed] [Google Scholar]; e) Okamoto H, Takeda K, Shiraishi K. Phys Rev B. 2001;64:115425/1–115425/17. [Google Scholar]
- 13.Pope M, Swenberg CE. Electronic Processes in Organic Crystals and Polymers. Oxford Science Publications; Oxford: 1999. or recent examples see: Percec V, Glodde M, Bera TK, Miura Y, Shiyanovskaya I, Singer KD, Balagurusamy VSK, Heiney PA, Schnell I, Rapp A, Spiess HW, Hudson SD, Duan H. Nature. 2002;419:384–387. doi: 10.1038/nature01072.Schmidt-Mende L, Fechtenkotter A, Mullen K, Moons E, Friend RH, MacKenzie JD. Science. 2001;293:1119–1122. doi: 10.1126/science.293.5532.1119.
- 14.Rapaport H, Kim HS, Kjaer K, Howes PB, Cohen S, Als-Nielsen J, Ghadiri MR, Leiserowitz L, Lahav M. J Am Chem Soc. 1999;121:1186–1191. [Google Scholar]
- 15.a) Katz HE, Johnson J, Lovinger AJ, Li W. J Am Chem Soc. 2000;122:7787–7792. [Google Scholar]; b) Katz HE, Lovinger AJ, Johnson J, Kloc C, Slegrist T, Li W, Lin YY, Dodabalapur A. Nature. 2000;404:478–481. doi: 10.1038/35006603. [DOI] [PubMed] [Google Scholar]
- 16.a) Miller LL, Mann KR. Acc Chem Res. 1996;29:417–423. [Google Scholar]; b) Miller LL, Duan RG, Tully DC, Tomalia DA. J Am Chem Soc. 1997;119:1005–1010. [Google Scholar]; c) Tabakovic I, Miller LL, Duan RG, Tully DC, Tomalia DA. Chem Mater. 1997;9:736–745. [Google Scholar]
- 17.Bong DT, Ghadiri MR. Angew Chem. 2001;113:2221–2224. [Google Scholar]; Angew Chem Int Ed. 2001;40:2163–2166. [Google Scholar]
- 18.Hexameric, D,L α-peptides Saviano M, Zaccaro L, Lombardi A, Pedone C, Di Blasio B, Sun X, Lorenzi GP. J Inclusion Phenom. 1994;18:27–36.
- 19.Octameric D,L α-peptides: Clark TD, Buriak JM, Kobayashi K, Isler MP, McRee DE, Ghadiri MR. J Am Chem Soc. 1998;120:8949–8962.Ghadiri MR, Kobayashi K, Granja JR, Chadha RK, McRee DE. Angew Chem. 1995;107:76–78. Angew Chem Int Ed Engl. 1995;34:93–95.
- 20.a) Clark TD, Ghadiri MR. J Am Chem Soc. 1995;117:12364–12365. [Google Scholar]; b) Clark TD, Kobayashi K, Ghadiri MR. Chem Eur J. 1999;5:782–792. [Google Scholar]; c) Kobayashi K, Granja JR, Ghadiri MR. Angew Chem Int Ed Engl. 1995;34:95–98. Angew. Chem. 1995, 107, 79–82. [Google Scholar]
- 21.Wagner T, Davis WB, Lorenz KB, Michel-Beyerle ME, Diederichsen U. Eur J Org Chem. 2003:3673–3679. [Google Scholar]
- 22.Mokhir AA, Kraemer R. Bioconjugate Chem. 2003;14:877–883. doi: 10.1021/bc0256345. [DOI] [PubMed] [Google Scholar]
- 23.Sep WJ, Verhoeven JW, De Boer TJ. Tetrahedron. 1975;31:1065–1070. [Google Scholar]
- 24.Buncel E, Mailloux NL, Brown RS, Kazmaier PM, Dust J. Tetrahedron Lett. 2001;42:3559–3562. [Google Scholar]
- 25.Eliel EL, Wilen SH, Mander LN. Stereochemistry of Organic Compounds. John Wiley & Sons; New York: 1994. [Google Scholar]
- 26.Benson SW. J Am Chem Soc. 1958;80:5151–5154. [Google Scholar]
- 27.Barros TC, Brochsztain S, Toscano VG, Berci Filho P, Politi MJ. J Photochem Photobiol, A. 1997;111:97–104. [Google Scholar]
- 28.a) Kates SA, Sole NA, Johnson CR, Hudson D, Barany G, Albericio F. Tetrahedron Lett. 1993;34:1549–1552. [Google Scholar]; b) Redman JE, Wilcoxen KM, Ghadiri MR. J Comb Chem. 2003;5:33–40. doi: 10.1021/cc0200639. [DOI] [PubMed] [Google Scholar]
- 29.Connors KA. Binding Constants: The Measurement of Molecular Complex Stability. John Wiley & Sons; New York: 1987. [Google Scholar]