

Abstract
Ubiquinone (coenzyme Q10 or CoQ10) is a lipid-soluble component of virtually all cell membranes and has multiple metabolic functions. Deficiency of CoQ10 (MIM 607426) has been associated with five different clinical presentations that suggest genetic heterogeneity, which may be related to the multiple steps in CoQ10 biosynthesis. Patients with all forms of CoQ10 deficiency have shown clinical improvements after initiating oral CoQ10 supplementation. Thus, early diagnosis is of critical importance in the management of these patients. This year, the first molecular defect causing the infantile form of primary human CoQ10 deficiency has been reported. The availability of genetic testing will allow for a better understanding of the pathogenesis of this disease and early initiation of therapy (even presymptomatically in siblings of patients) in this otherwise life-threatening infantile encephalomyopathy.
Keywords: CoQ10, Mitochondria, Encephalopathy, Myopathy, Cerebellar ataxia
Introduction
Coenzyme Q10 (CoQ10) is the predominant human form of endogenous ubiquinone. Synthesized in the mitochondrial inner membrane, CoQ10 is comprised of a ubiquinone head group attached to a tail of 10 five-carbon isoprenoid units, that anchors the molecule to the membranes [1]. In additions to its central role in the mitochondrial respiratory chain as electrons carrier from complex I and II to complex III [2], CoQ10 is now thought to be involved in a number of cellular functions (Table 1) [1]. Intracellular synthesis is the major source of CoQ10, although a small proportion is acquired through diet. The complexity of the biosynthesis (Fig. 1) suggests that defects in different biosynthetic enzymes or regulatory proteins may cause different clinical syndromes.
Table 1.
Functions of coenzyme Q (modified from [1])
| Electron carrier in mitochondrial respiratory chain [3] |
| Extra-mitochondrial electron transport [4] |
| Antioxidant [5] |
| Regulation of mitochondrial permeability transition pore [6] |
| Regulation of physiochemical properties of membranes [7] |
| Modulation of the amount of β-integrins on the surface of blood monocytes [8] |
| Modulator of endothelial function [9] |
| Oxidation of sulfide in yeast [10] |
| Introduction of disulfide bonds in bacteria [11] |
Fig. 1.
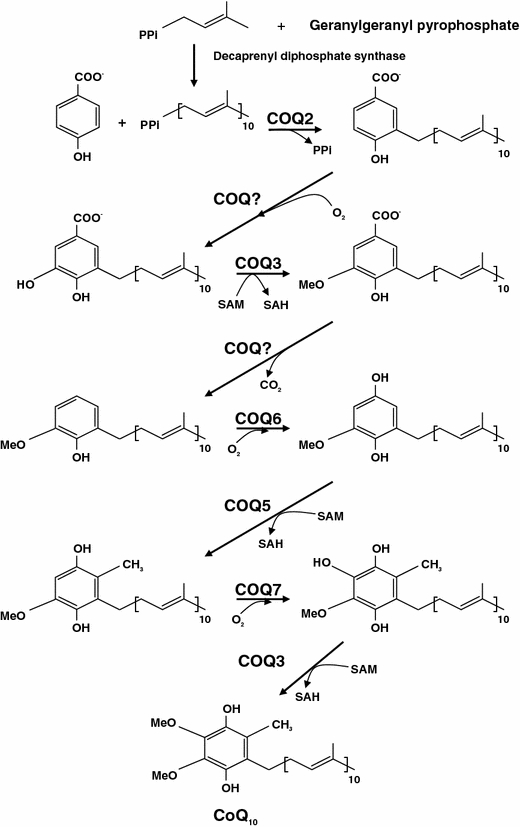
CoQ10 biosynthetic pathway with eight known biosynthetic enzymes denoted as COQ1-8. CoQ10 is composed of a benzoquinone and a decaprenyl side chain. While the quinone ring is derived from amino acids tyrosine or phenylalanine, the isoprenoid side chain is produced by addition of isopentenyl pyrophosphate molecules to geranylgeranyl pyrophosphate (derived from mevalonate pathway) by decaprenyl diphosphate synthase. After para-hydroxybenzoate and decaprenyl pyrophosphate are produced, at least seven enzymes (encoded by COQ2-8) catalyze condensation, methylation, decarboxylation, and hydroxylation reactions to synthesize CoQ10
Primary CoQ10 deficiency is an autosomal recessive condition with a clinical spectrum that encompasses at least five major phenotypes: (1) encephalomyopathy characterized by the triad of recurrent myoglobinuria, brain involvement and ragged red fibers; (2) severe infantile multisystemic disease; (3) cerebellar ataxia; (4) Leigh syndrome with growth retardation, ataxia and deafness; and (5) isolated myopathy [12–26].
Generalized weakness, exercise intolerance, and recurrent myoglobinuria are the distinctive features in the first four patients reported with the encephalomyopathic form, [12–15]; however, myoglobinuria was absent in another patient [16]. All the patients described so far have proximal muscle weakness [12–16] The brain involvement is variable; seizures are a common finding [12, 13], in association with cognitive impairment [12] and cerebellar symptoms [12, 15, 16].
The second variant, described in three families, manifests as an infantile encephalopathy with renal involvement. Rötig and colleagues initially described two siblings with defects of multiple quinone-dependent enzymes ascribed to a deficiency of CoQ10 in various tissues [17]. The patients presented with retinitis pigmentosa, optic nerve atrophy, bilateral sensorineuronal deafness, nephrotic syndrome, progressive ataxia, and cardiomyopathy. In Rahman’s patient, hypothermia, lactic acidosis, cerebral and cerebellar atrophy, and developmental delay were associated with renal tubulopathy and ventricular hypertrophy [18]. Last year, we described two siblings who presented with nephrotic syndrome due to glomerulosclerosis, and the older sibling developed hypotonia, psychomotor delay, seizures, stroke-like episodes, and cerebellar and cerebral atrophy [19]. Interestingly, in these infantile-onset patients, deficiency of CoQ10 is present not only in skeletal muscle, but also in other tissues and cells including skin fibroblasts. In addition, two adult sisters with encephalopathy, growth retardation, infantilism, ataxia, deafness and lactic acidosis have been described by Van Maldergam [23].
Cerebellar ataxia is the most common phenotype associated with CoQ10 deficiency with 21 reported patients [20–22, 26]. Cerebellar ataxia and cerebellar atrophy are present in all the patients. Epilepsy is the most common associated feature, but pyramidal signs, mental retardation, myopathic weakness, and delayed motor milestones are other variably associated symptoms and signs. Except for two adult brothers with cerebellar ataxia and hypogonadism [22], the condition begins in childhood or adolescence. Muscle morphology does not show ragged-red fibers and lipid storage myopathy and CoQ10 is moderately reduced in skin fibroblasts.
The clinical presentation of the variant isolated myopathy, recently described in four patients, is subacute onset of exercise intolerance and proximal limb weakness at variable ages. All patients have lipid storage and ragged-red fibers in muscle, as well as increased serum lactate and creatine kinase (CK) [24, 25]. In all the patients with different phenotypes, respiratory chain enzymes analyses show reduced activities of complex I + III and II + III with normal activities of isolated complex I and III.
In most of the reported patients, the exact site and nature of the defects in the biosynthesis of CoQ10 have not yet been identified. Because ubiquinone biosynthesis is complex and not fully defined (Fig. 1), identification of the molecular genetic defect is not straightforward.
Undetectable CoQ10 and low levels of decaprenylpyrophosphate, an intermediate compound in the synthesis of the lateral chain of CoQ10 were found in the fibroblasts of the patient reported by Rötig and colleagues suggesting a defect in the synthesis of the decaprenyl side-chain, but they did not find any mutation in the cDNA of COQ1, the gene encoding transprenyltransferase, the enzyme that synthesizes decaprenylpyrophosphate [17].
This year, in two siblings with the infantile form of CoQ10 deficiency, we reported a homozygous missense mutation (Y297C) in the COQ2 gene, which encodes para-hydroxybenzoate (PHB) polyprenyl transferase. PHB-polyprenyl transferase mediates the conjugation of the benzoquinone ring with the decaprenyl side chain. Biochemical assays measuring incorporation of radiolabeled PHB or decaprenyl-PP into CoQ10 in skin fibroblasts from the proband showed 23–25% of control fibroblasts activity [27] confirming the defect of CoQ10 biosynthesis.
The finding of a homozygous stop codon mutation in the APTX gene which is known to cause ataxia-oculomotor-aprataxia 1 (AOA1), in three siblings with cerebellar ataxia and CoQ10 deficiency, supports the hypothesis that the ataxic form is a genetically heterogeneous disease in which CoQ10 deficiency can be secondary [28]. Aprataxin is a member of the histidine triad superfamily and may be involved in nuclear DNA single strand break repair [29–31]. Despite the lack of an obvious link between aprataxin and regulation of CoQ10 synthesis or catabolism, our findings, coupled with the clinical improvement of patients after CoQ10 supplementation, suggest that CoQ10 deficiency plays a pathogenic role in AOA1. Intriguingly, both CoQ10 and cholesterol share a common biosynthetic pathway, therefore, in AOA1, altered levels of these molecules could be due to aberrant biosynthesis.
Patients with all forms of CoQ10 deficiency have shown clinical improvement with oral CoQ10 supplementation. The beneficial effects of exogenous CoQ10 require high doses and long-term administration [32, 33]; its oral bioavailability is poor due to its extreme hydrophobicity; therefore, only a small fraction of orally administered CoQ10 reaches the circulatory system, and augmentation of mitochondrial CoQ10 content is even less effective. Beneficial effects of CoQ10 supplementation are supported by in vitro correction of biochemical and histological abnormalities [12–14, 17]. However, while muscle abnormalities improve clinically and biochemically, in general, cerebral symptoms are only partially ameliorated [16]. This difference could be explained by the presence of irreversible structural brain damage before treatment or the poor penetration of CoQ10 across the blood-brain barrier. Increases in plasma concentrations of CoQ10 after oral supplementation in both humans and animals has been well-documented, but early work questioned whether CoQ10 accumulates in tissues [34] and studies of 45-day-old rodents showed little or no change in rodent brain CoQ10 concentrations after oral administration perhaps because levels of CoQ10 are tightly regulated in young animals or may be saturated in membranes [34, 35]. In contrast, administration of very high doses of CoQ10 (200 mg/kg daily) to 12–24 month-old rats produced significant increases in brain CoQ10 levels [36], but a more recent study of the regional distribution of CoQ in 16 week-old rat brain before and after 4 weeks of moderately high exogenous dietary CoQ10 (20 mg/kg daily) supplementation showed that the concentration of CoQ was essentially unchanged in the cortex and in the striatum [37]. That study also demonstrated that the cerebellum, of the 7 brain regions dissected, contains the lowest levels of CoQ. If human brain has the same regional distribution of CoQ10 as rat brain, the cerebellum may have the narrowest safety margin and therefore would be the first tissue to suffer from a pathological shortage of CoQ10. Moreover, to function as an antioxidant, CoQ10 must be in the reduced form but normally only 20% of the lipid is reduced in the brain. The high proportion of oxidized CoQ10 in the brain could be a reflection of the high oxygen consumption in this tissue, causing an increased demand of anti-oxidants [38]. Therefore, CoQ10 deficiency could be another form of inherited ataxia due to oxidative damage, as vitamin E deficiency.
Oxidative damage and mitochondrial dysfunction have also been implicated in neurodegenerative diseases such Parkinson disease (PD), Alzheimer disease (AD), and Friedreich’s ataxia (FRDA) [39–41]. Preclinical studies have indicated that CoQ10 can protect the nigrostriatal dopaminergic system and high doses (1200 mg daily) of CoQ10 for 16 months appeared to slow progression PD [40, 42]. In 10 FRDA patients treated for 47 months high doses of CoQ10 (400 mg daily) with vitamin E (2100 IU daily), improved bioenergetics in heart and skeletal muscle, improved cardiac function by echocardiography, and, possibly less clinical decline compared to historical controls [43]. Larger studies are necessary to better define the short- and long-term effects of CoQ10 as primary or adjunctive therapy in neurodegenerative diseases.
Deficiency of CoQ10 has been associated with a variety of neurological disorders that respond to oral supplementation. We have demonstrated that CoQ10 deficiency can be primary due to a defect of biosynthesis or secondary as in patients with APTX mutations. The response of both groups of patients to oral supplementation suggests that exogenous CoQ10 is taken up by affected tissues and can correct biochemical derangements. Further studies to define CoQ10 deficiency syndrome will reveal new causes, enhance our understanding of its pathogenesis, and may provide novel insights that may be relevant to other neurodegenerative diseases.
Footnotes
Special issue dedicated to John P. Blass.
References
- 1.Turunen M, Olsson J, Dallner G (2004) Metabolism and function of coenzyme Q. Biochim Biophys Acta 1660:171–199 [DOI] [PubMed]
- 2.Crane FL, Hatefi Y, Lester RL, Widmer C (1957) Isolation of a quinone from beef heart mitochondria. Biochim Biophys Acta 25:220–221 [DOI] [PubMed]
- 3.Santos-Ocana C, Do TQ, Padilla S, Navas P, Clarke CF (2002) Uptake of exogenous coenzyme Q and transport to mitochondria is required for bc1 complex stability in yeast coq mutants. J Biol Chem 277:10973–10981 [DOI] [PubMed]
- 4.Sun IL, Sun EE, Crane FL, Morre DJ, Lindgren A, Low H (1992) Requirement for coenzyme Q in plasma membrane electron transport. Proc Natl Acad Sci USA 89:11126–11130 [DOI] [PMC free article] [PubMed]
- 5.Villalba JM, Navas P (2000) Plasma membrane redox system in the control of stress-induced apoptosis. Antioxid Redox Signal 2:213–230 [DOI] [PubMed]
- 6.Walter L, Miyoshi H, Leverve X, Bernardi P, Fontaine E (2002) Regulation of the mitochondrial permeability transition pore by ubiquinone analogs. A progress report. Free Radic Res 36:405–412 [DOI] [PubMed]
- 7.Cornell BA, Keniry MA, Post A, Robertson RN, Weir LE, Westerman PW (1987) Location and activity of ubiquinone 10 and ubiquinone analogues in model and biological membranes. Biochemistry 26:7702–7707 [DOI] [PubMed]
- 8.Turunen M, Wehlin L, Sjoberg M, Lundahl J, Dallner G, Brismar K, Sindelar PJ (2002) Beta2-Integrin and lipid modifications indicate a non-antioxidant mechanism for the anti-atherogenic effect of dietary coenzyme Q10. Biochem Biophys Res Commun 296:255–260 [DOI] [PubMed]
- 9.Watts GF, Playford DA, Croft KD, Ward NC, Mori TA, Burke V (2002) Coenzyme Q(10) improves endothelial dysfunction of the brachial artery in Type II diabetes mellitus. Diabetologia 45:420–426 [DOI] [PubMed]
- 10.Saiki R, Ogiyama Y, Kainou T, Nishi T, Matsuda H, Kawamukai M (2003) Pleiotropic phenotypes of fission yeast defective in ubiquinone-10 production. A study from the abc1Sp (coq8Sp) mutant. Biofactors 18:229–235 [DOI] [PubMed]
- 11.Bader MW, Xie T, Yu CA, Bardwell JC (2000) Disulfide bonds are generated by quinone reduction. J Biol Chem 275:26082–26088 [DOI] [PubMed]
- 12.Ogasahara S, Engel AG, Frens D, Mack D (1989) Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc Natl Acad Sci USA 86:2379–2382 [DOI] [PMC free article] [PubMed]
- 13.Sobreira C, Hirano M, Shanske S, Keller RK, Haller RG, Davidson E, et al. (1997) Mitochondrial encephalomyopathy with coenzyme Q10 deficiency. Neurology 48:1238–1243 [DOI] [PubMed]
- 14.Di Giovanni S, Mirabella M, Spinazzola A, Crociani P, Silvestri G, Broccolini A, et al. (2001) Coenzyme Q10 reverses pathological phenotype and reduces apoptosis in familial CoQ10 deficiency. Neurology 57:515–518 [DOI] [PubMed]
- 15.Boitier E, Degoul F, Desguerre I, Charpentier C, Francois D, Ponsot G, et al. (1998) A case of mitochondrial encephalomyopathy associated with a muscle coenzyme Q10 deficiency. J Neurol Sci 156:41–46 [DOI] [PubMed]
- 16.Aure K, Benoist JF, de Ogier Baulny H, Romero NB, Rigal O, Lombes A (2004) Progression despite replacement of a myopathic form of coenzyme Q10 defect. Neurology 63:727–729 [DOI] [PubMed]
- 17.Rotig A, Appelkvist EL, Geromel V, Chretien D, Kadhom N, Edery P et al. (2000) Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet 356:391–395 [DOI] [PubMed]
- 18.Rahman S, Hargreaves I, Clayton P, Heales S (2001) Neonatal presentation of coenzyme Q10 deficiency. J Pediatr 139:456–458 [DOI] [PubMed]
- 19.Salviati L, Sacconi S, Murer L, Zacchello G, Franceschini L, Laverda AM et al. (2005) Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: a CoQ10-responsive condition. Neurology 65:606–608 [DOI] [PubMed]
- 20.Musumeci O, Naini A, Slonim AE, Skavin N, Hadjigeorgiou GL, Krawiecki N et al. (2001) Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology 56:849–855 [DOI] [PubMed]
- 21.Lamperti C, Naini A, Hirano M, De Vivo DC, Bertini E, Servidei S et al (2003) Cerebellar ataxia and coenzyme Q10 deficiency. Neurology 60:1206–1208 [DOI] [PubMed]
- 22.Gironi M, Lamperti C, Nemni R, Moggio M, Comi G, Guerini FR et al. (2004) Late-onset cerebellar ataxia with hypogonadism and muscle coenzyme Q10 deficiency. Neurology 62:818–820 [DOI] [PubMed]
- 23.Van Maldergem L, Trijbels F, DiMauro S, Sindelar PJ, Musumeci O, Janssen A et al. (2002) Coenzyme Q-responsive Leigh’s encephalopathy in two sisters. Ann Neurol 52:750–754 [DOI] [PubMed]
- 24.Lalani SR, Vladutiu GD, Plunkett K, Lotze TE, Adesina AM, Scaglia F (2005) Isolated mitochondrial myopathy associated with muscle coenzyme Q10 deficiency. Arch Neurol 62:317–320 [DOI] [PubMed]
- 25.Horvath R, Schneiderat P, Schoser BG, Gempel K, Neuen-Jacob E, Ploger H et al. (2006) Coenzyme Q10 deficiency and isolated myopathy. Neurology 66:253–255 [DOI] [PubMed]
- 26.Artuch R, Brea-Calvo G, Briones P, Aracil A, Galván M, Espinós C et al. (2006) Cerebellar ataxia with coenzyme Q10 deficiency: Diagnosis and followup after coenzyme Q10 supplementation. J Neurol Sci 246:153–158 [DOI] [PubMed]
- 27.Quinzii C, Naini A, Salviati L, Trevisson E, Navas P, Dimauro S et al. (2006) A Mutation in Para-Hydroxybenzoate-Polyprenyl Transferase (COQ2) Causes Primary Coenzyme Q10 Deficiency. Am J Hum Genet 78:345–349 [DOI] [PMC free article] [PubMed]
- 28.Quinzii CM, Kattah AG, Naini A, Akman HO, Mootha VK, DiMauro S et al. (2005) Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology 64:539–541 [DOI] [PubMed]
- 29.Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S et al. (2001) Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet 29:184–188 [DOI] [PubMed]
- 30.Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T et al. (2001) The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet 29:189–193 [DOI] [PubMed]
- 31.Ahel Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, Caldecott KW, West SC (2006) The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 443:713–716 [DOI] [PubMed]
- 32.James AM, Cocheme HM, Smith RA, Murphy MP (2005) Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J Biol Chem 280:21295–21312 [DOI] [PubMed]
- 33.Bhagavan HN, Chopra RK (2006) Coenzyme Q10: Absorption, tissue uptake, metabolism and pharmacokinetics. Free Radic Res 40:445–453 [DOI] [PubMed]
- 34.Zhang Y, Aberg F, Appelkvist EL, Dallner G, Ernster L (1995) Uptake of dietary coenzyme Q supplement is limited in rats. J Nutr 125:446–453 [DOI] [PubMed]
- 35.Bentinger M, Dallner G, Chojnacki T, Swiezewska E (2003) Distribution and breakdown of labeled coenzyme Q10 in rat. Free Radic Biol Med 34:563–575 [DOI] [PubMed]
- 36.Matthews RT, Yang L, Browne S, Baik M, Beal MF (1998) Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc Natl Acad Sci USA 95:8892–8897 [DOI] [PMC free article] [PubMed]
- 37.Naini A, Lewis VJ, Hirano M, DiMauro S (2003) Primary coenzyme Q10 deficiency and the brain. Biofactors 18:145–152 [DOI] [PubMed]
- 38.Turunen M, Swiezewska E, Chojnacki T, Sindelar P, Dallner G (2002) Regulatory aspects of coenzyme Q metabolism. Free Radic Res 36:437–443 [DOI] [PubMed]
- 39.Beal MF (2004) Mitochondrial dysfunction and oxidative damage in Alzheimer’s and Parkinson’s diseases and coenzyme Q10 as a potential treatment. J Bioenerg Biomembr 36:381–386 [DOI] [PubMed]
- 40.Shults CW, Haas R (2005) Clinical trials of coenzyme Q10 in neurological disorders. Biofactors 25:117–126 [DOI] [PubMed]
- 41.Schapira AH (2006) Mitochondrial diseases. Lancet 1 368:70–82 [DOI] [PubMed]
- 42.Shults C, Oakes D, Kieburtz K, Beal MF, Haas R, Plumb S et al. (2002) Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol 59:1541–1550 [DOI] [PubMed]
- 43.Hart P, Lodi R, Rajagopalan B, Bradley JL, Crilley JG, Turner C et al. (2005) Antioxidant treatment of patients with Friedreich ataxia: four-year follow-up. Arch Neurol 62:621–626 [DOI] [PubMed]