

Abstract
Early detection of an O2 deficit in the bloodstream is essential to initiate corrective changes in the breathing pattern of mammals. Carotid bodies serve an essential role in this respect;their type I cells depolarize when O2 levels fall, causing voltage-gated Ca2+ entry. Subsequent neurosecretion elicits increased afferent chemosensory fibre discharge to induce appropriate changes in respiratory function (1). Whilst depolarization of type I cells by hypoxia is known to arise from K+ channel inhibition, the identity of the signalling pathway has been contested and the coupling mechanism is unknown (2). We tested the hypothesis that AMP-activated protein kinase (AMPK)1 is the effector of hypoxic chemotransduction. AMPK is co-localized at the plasma membrane of type I cells with O2-sensitive K+ channels. In isolated type I cells, activation of AMPK using AICAR inhibited O2-sensitive K+ currents (carried by large conductance Ca2+-activated (BKCa) channels and TASK-like channels), leading to plasma membrane depolarization, Ca2+ influx and increased chemosensory fibre discharge. Conversely, the AMPK antagonist compound C reversed the effects of hypoxia and AICAR on type I cell and carotid body activation. These results suggest that AMPK activation is both sufficient and necessary for the effects of hypoxia. Furthermore, AMPK activation inhibited currents carried by recombinant BKCa channels while purified AMPK phosphorylated the α subunit of the channel in immunoprecipitates, an effect that was stimulated by AMP and inhibited by compound C. Our findings demonstrate a central role for AMPK in stimulus-response coupling by hypoxia, and identify for the first time a link between metabolic stress and ion channel regulation in an O2-sensing system.
Chronic and intermittent deficits in O2 supply to the body precipitate a variety of pathologies including dementia (3) and pulmonary hypertension (4). To develop effective therapies, it is necessary to understand the homeostatic mechanisms that monitor O2 supply to the body and elicit corrective changes in respiratory and circulatory function to maintain O2 levels. O2-sensitive ion channels, which were first identified in the carotid body type I cell, play a pivotal role in this respect and have now been reported in a diverse range of highly specialised O2-sensing tissues (5). Within the carotid body, clusters of type I cells lie in presynaptic contact with afferent sensory fibres, whose discharge increases in proportion to the degree of systemic arterial O2 deficit, providing information concerning blood O2 levels to the central respiratory centres (1, 2). This occurs subsequent to hypoxic inhibition of type I cell K+ channels, membrane depolarization, voltage-gated Ca2+ influx (6) and consequent neurotransmitter release. For many years there has existed compelling evidence that mitochondria serve an important role in O2 sensing by type I cells (2, 7). Indeed, mitochondrial inhibitors can both mimic the cellular effects of hypoxia and occlude type I cell O2 sensitivity (8, 9). These effects, and the downstream cellular responses, are manifest at relatively high tissue O2 levels, which would not be limiting in other, non-specialised cells (10). Thus, type I cell metabolism may be the key to the mechanism by which hypoxia is coupled to neurosecretion. We therefore investigated the potential role in this process of AMPK, a key sensor of metabolic stress that is universally expressed in eukaryotes. When cells are exposed to metabolic stresses, a rise in the cellular ADP/ATP ratio is partly compensated by the adenylate kinase reaction, which precipitates a much larger increase in the AMP/ATP ratio even though the fall in ATP may be negligible (11, 12). AMPK is activated via multiple mechanisms that make the system acutely sensitive to relatively small changes in the AMP/ATP ratio (13). Although originally proposed to regulate energy balance within single cells (14), it has recently become clear that AMPK also performs this function at the level of the whole animal, through its regulation of hypothalamic neurones that drive food intake and by its ability to stimulate energy expenditure in peripheral tissues (15). The hypothesis that AMPK also underpins the response of other physiological systems to stimuli that affect metabolism, e.g. excitation of the carotid body by hypoxia (16), has therefore become attractive. This is particularly so, given that our previous study established that physiologically relevant levels of hypoxia increased the AMP/ATP ratio in O2-sensing cells, leading to concomitant activation of AMPK activity that was primarily associated with heterotrimers containing the AMPK-α1 catalytic subunit isoform (16). Furthermore, this study suggested that activation of AMPK by AICAR induces transmembrane calcium influx into carotid body type I cells and thereby increases afferent fibre discharge from the isolated carotid body (16). However, we have yet to determine whether this is achieved by the same mechanisms that are engaged by hypoxia. Here we demonstrate that AMPK elicits carotid body activation by mimicking precisely the effects of hypoxia on carotid body type I cells, and that inhibition of AMPK reverses these effects.
Experimental Procedures
All experiments were performed under the UK Animals (Scientific Procedures) Act 1986.
Carotid body type I cell isolation
Neonatal rats (10-21 days) were killed and carotid bodies rapidly removed and placed in ice cold, oxygenated phosphate buffered saline without Ca2+ or Mg2+ (Sigma). These were then enzymically dissociated and cultured as described previously (9).
HEK 293 cell culture
HEK 293 cells which express human brain αβ-BKCa channels (17) were cultured as described previously (18). The co-expressed α and β subunits were KCNMA1 (Genbank accession number U11717) and KCNMB1 (Genbank accession number U42600) respectively.
Immunocytochemistry
Type I cells were fixed, immunostained and visualised as described previously (16). The primary antibodies were selective for the AMPKα1 subunit (1:500, sheep polyclonal, D.G. Hardie, Dundee, raised against peptide residues 344-358 of rat AMPKα1) and the BKCaα subunit (1:100, mouse monoclonal, BD Transduction Laboratories, raised against peptide residues 995-1113 of human brain BKCaα). Images were deconvolved and analysed off-line via Softworx (Applied Precision) and Volocity (Improvision).
Carotid Body Electrophysiology
Recordings were made using the amphotericin perforated-patch configuration of the whole-cell patch-clamp technique. Data were aquired using an Axopatch 200B amplifier (Axon Instruments, CA) controlled by Clampex 6.0 or Fetchex 6.0 software (Axon Instruments, CA). Currents were digitised at 6.6 kHz and filtered at 1-2 kHz, membrane potential recordings were digitised at 0.1 kHz and filtered at 1 kHz. Experiments were performed at 35-36°C. Cells were bathed in a solution A of composition (mM): KCl, 4.5; NaCl, 140; CaCl2, 2.5; MgCl2, 1; glucose, 11; HEPES, 20, adjusted to pH 7.4. Pipette solution was (mM): K2SO4, 55; KCl, 30; MgCl2, 5; EGTA, 1; glucose, 10; HEPES, 20, adjusted to pH 7.2. Pipette solution contained Amphotericin B (240μg ml-1, Sigma). Liquid junction potential was approximately 6 mV. Results were not leak subtracted, seal resistance was typically > 5 Gohms, access resistance was typically 15-20 Mohms and was not compensated for, holding currents were typically less than 5 pA. All data were analysed using Clampfit and Fetchan programs (pClamp6, Axon Instruments, CA).
HEK 293 cell electrophysiology
Recordings were made using the conventional whole-cell configuration of the patch-clamp technique. Data were aquired using a Multiclamp 700A amplifier (Axon Instruments, CA) controlled by Clampex 9.0 software (Axon Instruments, CA). Data were digitised at 5 kHz and filtered at 1 kHz. All experiments were performed at 22°C. Cells were bathed in a solution of composition (mM): KCl, 5; NaCl, 150; CaCl2, 2; MgCl2, 2; glucose, 10; HEPES, 10, adjusted to pH 7.4. Pipette solution was (mM) KSCN, 140; EGTA, 5; MgCl2, 1; CaCl2, 0.5; HEPES, 10; MgATP, 3; NaGTP, 0.3, adjusted to pH 7.25. Liquid junction potential was approximately 6.5 mV. Results were leak subtracted, seal resistance was typically > 7 Gohms, access resistance was typically 8-12 Mohms and series resistance was compensated by 60-80 %, holding currents were 20-50 pA. All data were analysed using Clampfit 9 (Axon Instruments, CA).
Ca2+ imaging
Cells were incubated for 30 min (22-24°C, dark) in a Ca2+-free solution (mM): KCl, 4.5; NaCl, 140; MgCl2, 1; glucose, 11; EGTA, 1; HEPES, 20, adjusted to pH 7.4, containing 5μM FURA-2AM (Molecular Probes) then allowed to equilibrate for 20 min in solution A (35-36°C). Hypoxic solutions were generated by gassing solution A with 100% N2, pO2 was monitored using an ISO2 oxygen meter (WPI). Changes in [Ca2+]i were monitored using alternate excitation wavelengths of 340 nm and 380 nm (F340 & F380, 20 ms exposure). Emitted fluorescence was recorded as described previously (16) at 0.05 Hz with background subtraction carried out on line.
Isolated carotid body
Rats were anaesthetised with 1-4 % halothane in O2 and carotid bodies with attached carotid sinus nerve excised as described previously (19). Extracellular recordings of single or few-fibre afferent fibre action potentials were recorded on videotape and sampled digitally via Spike2 software (CED) for analysis of discharge frequency.
Co-immunoprecipitation
Cells were harvested in PBS and centrifuged at 1000 × g, after which the pellet was homogenised in 1ml ice-cold buffer A (50mM Tris-HCl (pH 7.4), 140mM KCl, 1mM EGTA, 1mM MgCl2, 1mM phenylmethanesulfonylfluoride (PMSF) and 50μg/ml aprotinin). This was followed by centrifugation (1000 × g, 5min, 4°C). The pellet was then solubilized in 0.5ml ice cold buffer B (5mM Tris-HCl (pH 7.4), 500mM NaCl, 1.5% v/v Triton X-100, with 1mM PMSF and 50μg/ml aprotinin). Antibodies (either anti-BKCa α (5μg) or anti-AMPKα1 subunits (6μg), as used for immunocytochemistry) were added to solubilized suspensions and incubated overnight at 4°C before addition of 50μl protein G beads. Following further incubation (>4h, 4°C, with rotation), the beads were pelleted at 7000 × g (5min) and washed 3 times in 1ml ice-cold buffer C (5mM Tris-HCl (pH 7.4), 20mM NaCl, 0.5% v.v Triton X-100, with 1mM PMSF and 50μg/ml aprotinin).
Pelleted immunoprecipitates were taken up into 50μl sample buffer (62.5mM Tris-HCl (pH 6.8), 10% v/v glycerol, 0.2% w/v SDS 5% v/v β-mercaptoethanol, 0.02% bromophenol blue), heated for 3min at 100°C and loaded onto 10% (w/v) SDS polyacrylamide gels and separated at 100V for 1h. Proteins were transferred to PVDF membrane at 250mA for 1 hour and membranes blocked overnight with 10 % (w/v) non-fat powdered milk in PBS at 4°C. After 3 × washing membranes in PBS/0.05% Tween 20, they were incubated with anti-AMPKα1 antibody (1:500) or anti-BKCa α antibody (1:250) in PBS / 0.5% non-fat powdered milk for 1h at 21°C. Membranes were then washed extensively with PBS before incubation with anti-mouse or anti-sheep peroxidase-linked secondary antibody (1:10,000 in PBS, Pierce) for 1h. Proteins were visualized using enhanced chemiluminescence (Amersham Biosciences, U.K.). Molecular masses were estimated using calibrated pre-stained standards (Bio-Rad Labs, Hemel Hempstead, U.K.).
Purification of BKCa from HEK293 cells
BKCa α was immunoprecipitated from HEK-293 cells stably expressing the channel (20). Culture dishes (4 × 15 cm) of cells were grown to 90% confluency. Cells were washed once in 10 ml ice-cold phosphate-buffered saline (PBS) then lysed in situ by the addition of 2 ml lysis buffer (50 mM Tris-HCl, pH 7.4 at 4°C, 150 mM NaCl, 50 mM NaF, 5 mM Na pyrophosphate, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol (DTT), 0.1 mM benzamidine, 0.1 mM phenylmethane sulphonylfluoride, 5 μg/ml soybean trypsin inhibitor, 2% Triton X-100, 0.25 M mannitol). Lysate was then pre-cleared by incubating on a roller mixer with 2 ml protein G-Sepharose (Amersham, 50% slurry in lysis buffer) for two hr at 4°C followed by centrifugation (230xg, 5 min). BKCa α was subsequently immunoprecipitated from the supernatant using 20 μg of anti-BKCa-α antibody (BD Transduction Laboratories) pre-bound to 40 μl protein G-Sepharose. After 2 hr at 4°C, the precipitate was collected by centrifugation (230xg, 5 min). The immunoprecipitate was washed 5x in lysis buffer containing 1 M NaCl, followed by 5x in lysis buffer without NaCl.
Phosphorylation of BKCa α subunit using purified rat liver AMPK
Immunopurified BKCa was washed 6x in assay buffer (50 mM Na-Hepes, pH 7.2, 1 mM dithiothreitol, 0.02% (w/v) Brij-35) containing 1 M NaCl, followed by 8 washes in assay buffer without NaCl. The immune complexes were incubated for 30 min at 30°C with 4.5 μg/ml recombinant PP1γ in assay buffer. They were washed 5x in assay buffer containing 1 M NaCl, followed by 5 further washes in assay buffer. They were then incubated for 30 min at 30°C with 5 mM MgCl2 and 200 μM [γ-32P]ATP (300 cpm/pmol) in 50 mM Na-Hepes, pH 7.2, 1 mM dithiothreitol, 0.02% (w/v) Brij-35, with or without purified rat liver AMPK (Hawley et al (1996) JBC 271: 27879--27887), with or without AMP (200 μM) or compound C (100 μM). The reactions were stopped by the addition of LDS loading dye and reducing agent (Invitrogen). The samples were analysed by SDS-PAGE using the 3-8% gradient Tris-acetate system (Invitrogen) and gels stained with Colloidal Blue staining kits (Invitrogen). Destained gels were dried and subject to autoradiography. The BKCa α band was excised and the incorporation of 32P-phosphate was determined by Cerenkov counting. The amount of BKCa α subunit was estimated by running known amounts of bovine serum albumin on the same gel, comparing intensities of Commassie Blue stained bands by densitometry, and assuming that the same amount of dye bound per μg of protein.
Identification of the BKCa α subunit by mass spectrometry
A gel slice was excised and washed successively for 15 min in water, CH3CN, 100 mM NH4HCO3, and finally in 20 mM NH4HCO3/CH3CN (50:50 v/v). The gel slice was dehydrated in CH3CN for 5 min. The supernatant was dried in a SpeedVac concentrator and resuspended in 20 μl of 12.5 μg/ml trypsin (Roche, modified sequencing grade) in 20 mM NH4HCO3/0.1% n-octylglucoside. Digestion occurred overnight at 30°C on a shaking platform. An equal volume of CH3CN was then added to the digest and incubated for 30 min at 30°C on a shaking platform. An aliquot (10%) of the peptide digest was then analysed by MALDI-TOF-TOF MS-MS (Applied Biosystems 4700 Proteomics Analyser). The MS and MS-MS data generated were used to search the Celera Discovery System human database (Applied Biosystems) using the Mascot search algorithm (http://www.matrixscience.com). This confirmed the identity of the 125 kDa band as the BKCa α subunit (SwissProt Accession; KCNMA1).
Statistics
All data are presented as mean ± standard error of the mean. Statistical significance was assessed using 2 tailed Bonferroni-Dunn post hoc test (in vitro carotid body) or Student’s paired or unpaired t test (all other data) as appropriate. P< 0.05 was considered significant.
RESULTS
Consistent with the hypothesis that AMPK serves a key role in O2 sensing by the carotid body, we found that in isolated carotid body type I cells the AMPK-α1 catalytic subunit isoform and O2-sensitive BKCa channels co-localize at the plasma membrane (Fig. 1Ai-iv). Thus, 75.1 ± 4.4 % of the total AMPK-α1 staining was located within 1 μm of the type I cell plasma membrane (n = 14), where it co-localised with O2-sensitive BKCa channels (21), with 71.0 ± 2.6% of staining for the pore forming BKCa-α subunit coinciding with that for the AMPK-α1 catalytic subunit isoform (Pearson’s correlation 0.65 ± 0.05, n = 5; Fig. 1Aiii-iv). Furthermore, BKCa channel currents were selectively inhibited in acutely isolated type I cells by 5-aminoimidazole-4-carboxamide riboside (AICAR), a nucleoside that is taken up by cells and metabolised to the AMP mimetic, ZMP, thus activating AMPK in the absence of ATP depletion (22). Analysis of current-voltage relationships for macroscopic K+ currents showed that AICAR (1 mM, 10 min) reversibly inhibited voltage-sensitive K+ currents throughout their activation range, e.g. by 40.0 ± 12.2 % during a voltage step from -80 to +30 mV (Fig. 1B; n = 5, P = 0.041). Selective blockade of BKCa channels by iberiotoxin (100 nM) inhibited type I cell macroscopic K+ currents by 60.3 ± 6.8 % at +30 mV (n = 7, P = 0.0069) and, in the presence of the toxin, AICAR had no further inhibitory effect on residual voltage-gated K+ currents (Fig. 1C; n = 3). Rat carotid body type I cells also express O2-sensitive, voltage-independent leak K+ channels (23) in addition to BKCa channels, although the molecular identity of these TASK-like channels is not known. AICAR also inhibited this Ba2+-sensitive, voltage-independent leak conductance (measured during voltage ramps from -100 mV to -40 mV) by 33.0 ± 2.3 %, from 258 ± 24 pS to 171 ± 11 pS (Fig. 1D; n = 4, P = 0.008). Furthermore, in the continued presence of Ba2+, AICAR did not significantly inhibit the residual currents (Fig. 1E; n = 5). These data indicate that AMPK activation, like hypoxia (21, 23), selectively inhibits both the O2-sensing BKCa currents and TASK-like leak K+ currents in rat carotid body type I cells, without affecting O2-insensitive delayed rectifier K+ currents.
Fig. 1.
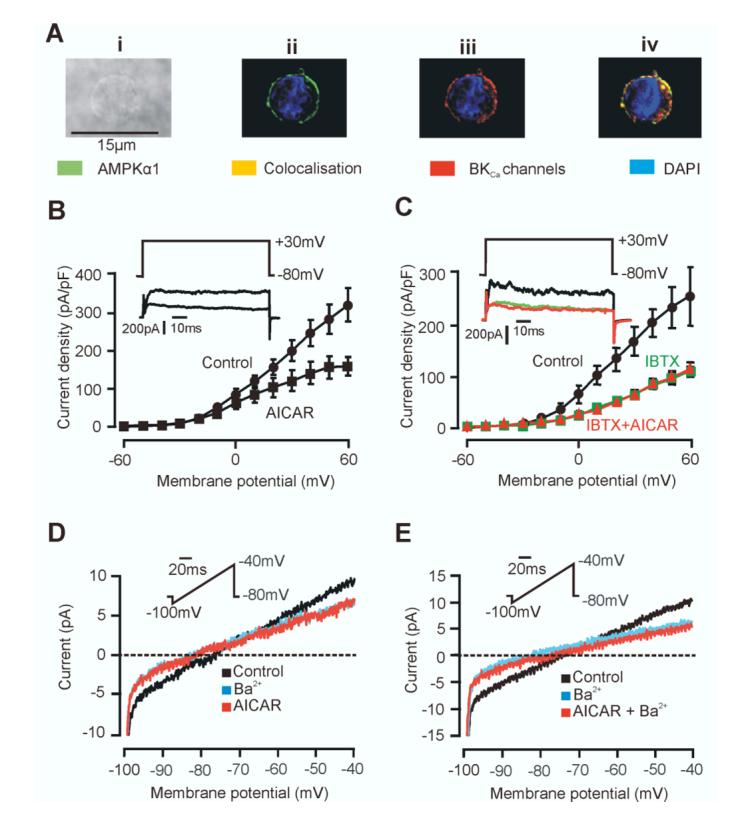
AMPKα1 and BKCa α subunits colocalise and activation of AMPK inhibits O2-sensitive K+ currents in type I cells. Ai, Bright field image of a type I cell. ii, Z-section with immunostaining for AMPKα1 and nuclear staining by DAPI. iii, Z-section with immunostaining for BKCaα subunit and DAPI. iv, Three-dimensional reconstruction showing AMPKα1 and BKCaα colocalisation. B, AICAR inhibits macroscopic current. Data are mean ± SEM. Inset are example currents ± AICAR. C, AICAR inhibits the IBTX-sensitive current. Data are mean ± SEM. Inset are example currents, control, +IBTX and +IBTX and AICAR. D, Ba2+ and AICAR inhibit leak K+ currents. E, AICAR does not inhibit residual currents in the continued presence of Ba2+.
Inhibition of the O2-sensing K+ currents led to reversible depolarisation of type I cells by 12.6 ± 0.9 mV (Fig 2A; n = 6, P = 0.0055) within 12.6 ± 1.9 min of application of AICAR (n = 6). The magnitude of depolarisation was equivalent to that seen in response to hypoxia (24). As expected, AMPK activation triggered voltage-gated Ca2+ influx into carotid body type I cells as monitored by the Ca2+ indicator dye Fura-2 (Fig 2B). Thus, AICAR reversibly increased the Fura-2 fluorescence ratio (F340/F380) from 0.62 ± 0.01 to a peak of 1.71 ± 0.23 (n = 7, P = 0.0028) and this was reversibly inhibited by 84.0 ± 9.3% by Ni2+ (5 mM, n = 7, P = 0.0037), an inhibitor of voltage-gated Ca2+ channels. Likewise, hypoxia (14-21 mm Hg) increased the Fura-2 fluorescence ratio from 0.67 ± 0.02 to 2.5 ± 0.17 (n = 5). Most significantly, the AMPK antagonist compound C (25) (40 μM) reversibly inhibited the increase in intracellular Ca2+ concentration in response to hypoxia by 65.1 ± 9.8 % (Fig. 2C; n = 5, P = 0.00097) and to AICAR by 70.7 ± 4.2 % (Fig. 2D; n = 7, P = 0.0004). In marked contrast, compound C (40 μM) had little effect on voltage-gated Ca2+ influx in response to type I cell depolarisation by K+ (50 mM), the Fura-2 fluorescence ratio increasing from 0.59 ± 0.008 to 2.87 ± 0.23 in its absence and from 0.58 ± 0.01 to 2.22 ± 0.20 in its presence (Fig. 2E, n = 5). Thus, compound C blocked carotid body type I cell excitation in response to hypoxia without affecting basal levels of intracellular Ca2+ concentration, but it did not block membrane depolarisation in response to interventions that reduce the outward K+ conductance nor did it block voltage-gated Ca2+ entry per se.
Fig. 2.

Activation of AMPK mimics hypoxia in type I cells and its inhibition reverses the effect of hypoxia. A, Depolarisation induced by AICAR (1 mM). B, Ni2+ (5 mM) attenuates the AICAR (1 mM) induced rise in FURA-2 F340/F380 ratio. C, AMPK antagonist Compound C (40 μM) attenuates the hypoxia (14-21 mmHg) induced rise in FURA-2 F340/F380 ratio. D, Compound C (40 μM) attenuates the AICAR induced rise in FURA-2 F340/F380 ratio. E, K+ (50mM) causes a robust rise in FURA-2 F340/F380 ratio in the presence of Compound C (40 μM).
Recordings from type I cells are not necessarily a reliable indicator of a functional response at the level of the intact carotid body (26, 27). We therefore investigated the role of AMPK in mediating afferent chemosensory fibre discharge from the carotid body in vitro. Consistent with our findings on isolated carotid body type I cells, 1 mM AICAR caused a 7-fold increase in spontaneous, single afferent fibre discharge frequency (Fig 3A). AICAR at 100μM or 300μM was without effect upon basal discharge. The excitatory effect of AICAR at 1mM was significantly inhibited by around 50% after pre-incubation with compound C (40 μM) for 10-20 minutes (Fig. 3A). After 30-40 minutes of preincubation with compound C, basal discharge was reduced to essentially zero and the response to AICAR was abolished. Basal carotid body single or few-fibre chemoafferent discharge varied slightly between preparations, reflecting individual fibre excitability, but, in all cases, discharge increased in response to hypoxia (Fig 3B). Hypoxia-augmented afferent chemosensory fibre discharge frequency was also inhibited by preincubation with compound C. At 10-20 minutes, the degree of inhibition was around 50% (see Fig. 3C). Despite a reduction in basal discharge following sustained incubation with compound C, a hypoxic response was retained, albeit reduced by about 90% at 70 minutes. That this was not simply due to ‘run down’ of the preparation was confirmed by control studies which showed that the hypoxia response was largely retained for up to 120 minutes following repeated stimulation trials. The mean data are shown in Fig 3D. Thus, AMPK activation mediates a large component of carotid body type I cell activation by hypoxia, and this is sufficient to trigger an increase in afferent chemosensory fibre discharge that can be substantially blocked by compound C.
Fig. 3.

AMPK inhibition attenuates hypoxia and AICAR induced increases in carotid sinus nerve discharge. A, AICAR (1mM) stimulates discharge and Compound C (40 μM; after 10 mins pre-incubation) reduces this effect on few fibre-afferent discharge (discharge frequency histogram in 1s intervals (top) and extracellular action potentials (below). B, Hypoxia increases chemoafferent discharge. Traces show (from top): discharge frequency histogram in 1s intervals, superfusate pO2 at the carotid body surface and extracellular action potentials. Note that pO2 levels recorded in the superfusate at the surface of the carotid body are related to but do not represent the precise tissue level of pO2 to which the afferent nerve is responding. This would vary with depth from the surface and can be calculated as ca. -1mmHg per μm. Carotid bodies are maintained at a superfusate pO2 of 400mmHg to ensure adequate oxygenation to the core between hypoxic episodes and maximal responses are normally observed as superfusate pO2 falls below ca. 150mmHg. C, Compound C (40 μM; after 20 mins preincubation) reduces the effect of hypoxia on few fibre-afferent discharge (traces as for B). Di, Mean data ± SEM for experiment A. ii, Mean data ± SEM for experiments in B and C. * P<0.0001 vs control: ΨP<0.002 vs AICAR alone or hypoxia alone
We next sought to determine whether inhibition by AMPK of O2-sensing K+ currents was mediated by direct phosphorylation of channel proteins, by examining the regulation of human BKCa channels stably expressed in HEK293 cells (17). Analysis of voltage ramps between -100 mV and +100 mV showed that, as in carotid body type I cells, AMPK activation by AICAR (1 mM, 10 min) inhibited BKCa channel currents throughout their current-voltage range; e.g. by 74.4 % at +30 mV, from 46.1 ± 8.7 pA pF-1 to 11.8 ± 1.9 pA pF-1 (n = 20, P = 0.00057). Inhibition of BKCa channel currents by AICAR was also reversed to 85.5 % of control, i.e. to 39.4 ± 4.4 pA pF-1, by compound C (Fig. 4A; n = 20). Unlike in type I cells, there was no specific co-localization of the α1 subunit of AMPK with the BKCa α subunit at the plasma membrane (not shown) and the two proteins also failed to co-immunoprecipitate (Fig. 4B). Thus, AMPK-dependent regulation of BKCa channels can occur without coupling of these proteins at the molecular level and without precise targeting of AMPK to the plasma membrane protein. It seems likely, therefore, that channel regulation may result from phosphorylation of the BKCa channel protein by AMPK in a manner that may be further facilitated by targeting of AMPK to the plasma membrane in carotid body type I cells. Consistent with this view, when immunoprecipitates made using anti-BKCa α antibodies from HEK293 cells were incubated with [γ-32P]ATP and AMPK purified from rat liver (a mixture of the α1β1γ1 and α2β1γ1 heterotrimers), we obtained phosphorylation of the pore forming BKCa α subunit in the presence of AMPK. This phosphorylation was stimulated by AMP and inhibited by compound C (Fig. 4C). The identity of this phosphorylated band as BKCa α was confirmed by mass spectrometry of tryptic peptides. Estimates of the stoichiometry of phosphorylation showed that this was substantial, reaching 0.7-0.9 mole per mole of α subunit within 30 minutes using 5 U/ml AMPK.
Fig. 4.
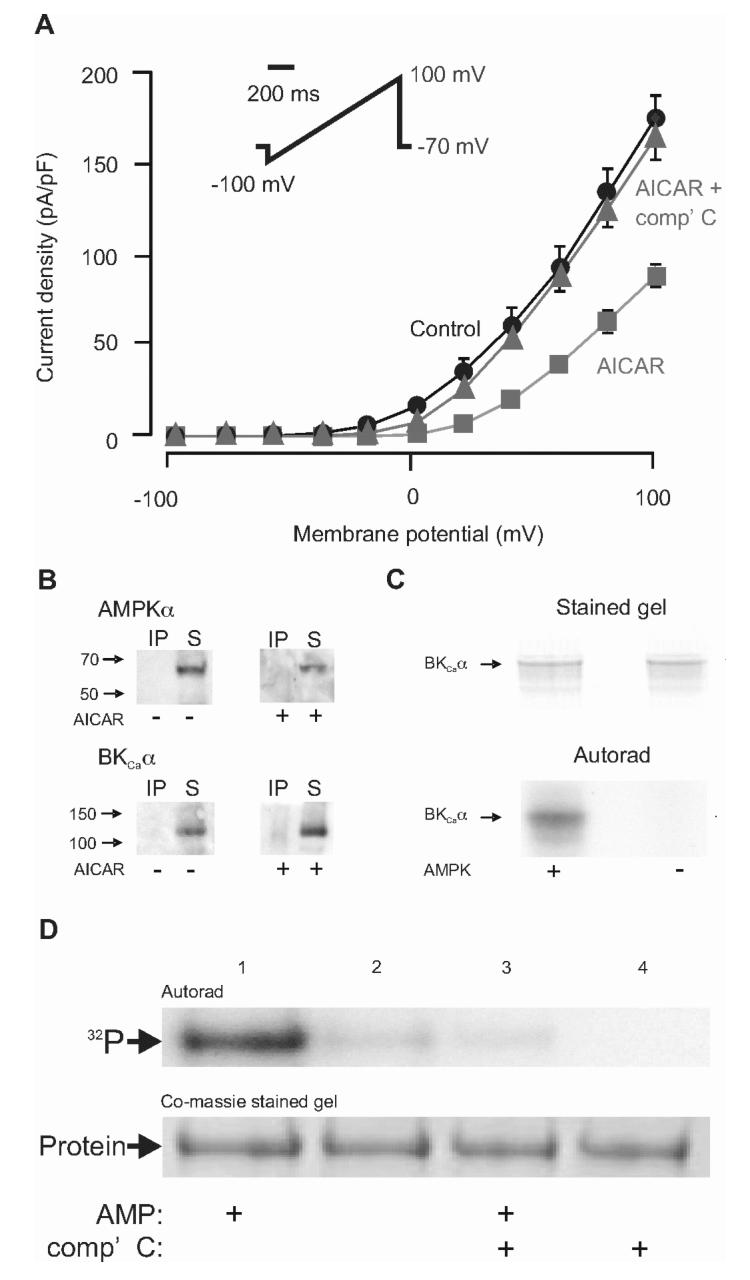
AMPK activation inhibits BKCa channels expressed in HEK 293 cells by direct phosphorylation. A, AICAR (1mM) inhibits BKCa currents, this is attenuated by compound C. Data are mean ± SEM. B, Co-immunoprecipitation with western blot detection, no association of AMPKα with BKCa α. Upper blots show results using anti-BKCa α antibodies, revealing AMPKα ni the supernatant (S) rather than the immunoprecipitate (IP), in extracts from cells treated with or without AICAR (1 mM). Lower blots show results using anti-AMPKα antibodies, revealing BKCa α only in the supernatant. C, Coomassie Blue-stained gel and autoradiogram demonstrating phosphorylation of BKCa α subunits in the presence of AMP (200 μM) with or without AMPK (5 U/ml). D, as C, except the AMPK was reduced to 1 U/ml and the reaction was performed in the presence and absence of AMP (200 μM) or compound C (100 μM).
DISCUSSION
In the present investigation, we have addressed the molecular mechanism by which AMPK effects carotid body excitation by hypoxia. In doing so we have shown that AMPK activation by AICAR precisely mimics the effects of hypoxia on the two key O2-sensitive K+ channels in rat type I cells, i.e. BKCa channels and TASK-like channels. Moreover, we show that activation by hypoxia and AICAR of carotid body type I cells and of the carotid body itself is inhibited, if not abolished, by the AMPK inhibitor, compound C. AICAR is not known to activate any protein kinases other than AMPK, so the finding that compound C reverses carotid body type I cell activation induced by AICAR, but not that induced by depolarisation, provides strong support for the view that its effects are mediated by inhibition of the AMPK pathway.
Whilst AICAR does not directly bind to adenosine receptors, it is known to compete with adenosine for the adenosine nucleoside transporter (28) and could therefore increase the extracellular adenosine concentration by competing with released adenosine for uptake into cells. As carotid body afferent discharge is increased by adenosine in a concentration-dependent manner (29, 30), there was a possibility that the effect of AICAR could have been mediated through this mechanism. However, this would only be apparent in the whole carotid body preparation where adenosine might build up in the synaptic cleft and would not occur in isolated, superfused carotid body type I cells. In a separate series of experiments, we showed that the non-selective adenosine receptor antagonist, 8-SPT (300μM), when applied prior to AICAR, reduced the afferent chemosensory response to AICAR in carotid body preparations by 53 ± 12 % (SP and PK unpublished data). Given that the AMPK antagonist, compound C, significantly inhibited AICAR-induced increases in afferent fibre discharge, and that hypoxia elicits carotid body activation by releasing a variety of neurotransmitters including adenosine (31), our findings are consistent with the view that a substantial proportion of the response to AICAR in the whole organ was mediated via AMPK activation and only a small proportion can be attributed to inhibition of adenosine uptake.
Thus, our results suggest that activation of AMPK is necessary as well as sufficient for carotid body activation by hypoxia. We also provide a plausible mechanism for the inhibitory effects on the BKCa channels, by showing that AMPK directly phosphorylates the pore-forming α subunit in immunoprecipitates. As expected for an effect of AMPK, phosphorylation was stimulated by AMP and inhibited by compound C, and the stoichiometry approached 1 mole of phosphate per mole of α subunit, showing that the extent of phosphorylation was significant. We propose, therefore, that the AMPK system acts as the primary metabolic sensor as well as the effector of Ca2+ signalling in response to hypoxia in carotid body type I cells. This mechanism can unite the previous mitochondrial and membrane hypotheses for chemotransduction by hypoxia (2).
Recently, O2 sensing by BKCa channels has been proposed to arise via O2-dependent CO production from the closely associated enzyme, heme oxygenase-2 (HO-2; (32)). Clearly, when this enzyme is active (i.e. in the presence of its substrates, including O2) channel activity is markedly enhanced due to CO production by HO-2. However, the requirement of this mechanism of channel regulation for O2 sensing in the intact carotid body has not been evaluated. Furthermore, the importance of HO-2 in O2 sensing has more recently been questioned, since BKCa channels have been shown to be suppressed by hypoxia in excised membrane patches (33). Collectively, these data suggest that there exist multiple means by which hypoxia might inhibit BKCa channels. However, unlike our proposed mechanism, other mechanisms cannot account for findings that inhibitors of mitochondrial oxidative phosphorylation prevent carotid body activation by hypoxia (8, 9). Furthermore, no other mechanism has been so comprehensively tested with respect to its involvement in O2 sensing at the level of ion channel activity, membrane potential, [Ca2+]i regulation and afferent nerve discharge. On the basis of our present data, therefore, we propose that AMPK is the physiologically relevant mediator of O2 sensing by the carotid body. Furthermore, we propose that the targeting of AMPKα1 complexes to the plasma membrane of carotid body type 1 cells, the acute sensitivity of AMPK to changes in the AMP/ATP ratio, and a reliance of these cells on mitochondrial oxidative phosphorylation for ATP production, underpins the activation of type I cells by relatively small, physiological changes in arterial pO2. Thus, in addition to its ubiquitous role in regulating metabolism in response to metabolic stress at the cellular level, the AMPK signalling pathway has become adapted to mediate the responses of physiological systems that are key to meeting the metabolic needs of the whole body, including hypoxia-response coupling in O2-sensing cells.
Footnotes
The studies of AME and CNW were supported by a Wellcome Trust Project Grant; DGH and KJW were supported by a Wellcome Trust Programme Grant; SAP was supported by a University of Birmingham, Division of Medical Sciences PhD studentship; CP, MLD and LA were supported by the British Heart Foundation, the Alzheimer’s Research Trust and the Alzheimer’s Society.
- AMPK
- adenosine monophosphate-activated protein kinase
- TASK
- tandem pore, acid-sensing potassium channel
- BKCa
- large conductance voltage and Ca2+ -activated potassium channel
- AICAR
- 5-aminoimidazole-4-carboxamide riboside
- pO2
- partial pressure of oxygen
REFERENCES
- 1.Gonzalez C, Almaraz L, Obeso A, Rigual R. Physiol Rev. 1994;74(4):829–898. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- 2.Prabhakar NR. Exp Physiol. 2006;91(1):17–23. doi: 10.1113/expphysiol.2005.031922. [DOI] [PubMed] [Google Scholar]
- 3.Desmond DW, Moroney JT, Sano M, Stern Y. Stroke. 2002;33(9):2254–2260. doi: 10.1161/01.str.0000028235.91778.95. [DOI] [PubMed] [Google Scholar]
- 4.Voelkel NF. Am Rev Respir Dis. 1986;133(6):1186–1195. doi: 10.1164/arrd.1986.133.6.1186. [DOI] [PubMed] [Google Scholar]
- 5.Lopez-Barneo J, del Toro R, Levitsky KL, Chiara MD, Ortega-Saenz P. J Appl Physiol. 2004;96(3):1187–1195. doi: 10.1152/japplphysiol.00929.2003. discussion 1170-1182. [DOI] [PubMed] [Google Scholar]
- 6.Weir EK, Lopez-Barneo J, Buckler KJ, Archer SL. N Engl J Med. 2005;353(19):2042–2055. doi: 10.1056/NEJMra050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mills E, Jobsis FF. J Neurophysiol. 1972;35(4):405–428. doi: 10.1152/jn.1972.35.4.405. [DOI] [PubMed] [Google Scholar]
- 8.Mulligan E, Lahiri S, Storey BT. J Appl Physiol. 1981;51(2):438–446. doi: 10.1152/jappl.1981.51.2.438. [DOI] [PubMed] [Google Scholar]
- 9.Wyatt CN, Buckler KJ. J Physiol. 2004;556(Pt 1):175–191. doi: 10.1113/jphysiol.2003.058131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duchen MR, Biscoe TJ. J Physiol. 1992;450:13–31. doi: 10.1113/jphysiol.1992.sp019114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wyatt CN, Kumar P, Aley P, Peers C, Hardie DG, Evans AM. Adv Exp Med Biol. 2006;580:191–196. doi: 10.1007/0-387-31311-7_29. discussion 351-199. [DOI] [PubMed] [Google Scholar]
- 12.Hardie DG. J Cell Sci. 2004;117(Pt 23):5479–5487. doi: 10.1242/jcs.01540. [DOI] [PubMed] [Google Scholar]
- 13.Hardie DG, Salt IP, Hawley SA, Davies SP. Biochem J. 1999;338(Pt 3):717–722. [PMC free article] [PubMed] [Google Scholar]
- 14.Hardie DG, Carling D. Eur J Biochem. 1997;246(2):259–273. doi: 10.1111/j.1432-1033.1997.00259.x. [DOI] [PubMed] [Google Scholar]
- 15.Kahn BB, Alquier T, Carling D, Hardie DG. Cell Metab. 2005;1(1):15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 16.Evans AM, Mustard KJ, Wyatt CN, Peers C, Dipp M, Kumar P, Kinnear NP, Hardie DG. J Biol Chem. 2005;280(50):41504–41511. doi: 10.1074/jbc.M510040200. [DOI] [PubMed] [Google Scholar]
- 17.Ahring PK, Strobaek D, Christophersen P, Olesen SP, Johansen TE. FEBS Lett. 1997;415(1):67–70. doi: 10.1016/s0014-5793(97)01096-x. [DOI] [PubMed] [Google Scholar]
- 18.Lewis A, Peers C, Ashford ML, Kemp PJ. J Physiol. 2002;540(Pt 3):771–780. doi: 10.1113/jphysiol.2001.013888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pepper DR, Landauer RC, Kumar P. J Physiol. 1995;485(Pt 2):531–541. doi: 10.1113/jphysiol.1995.sp020749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartness ME, Brazier SP, Peers C, Bateson AN, Ashford ML, Kemp PJ. J Biol Chem. 2003;278(51):51422–51432. doi: 10.1074/jbc.M309463200. [DOI] [PubMed] [Google Scholar]
- 21.Peers C. Neurosci Lett. 1990;119(2):253–256. doi: 10.1016/0304-3940(90)90846-2. [DOI] [PubMed] [Google Scholar]
- 22.Corton JM, Gillespie JG, Hawley SA, Hardie DG. Eur J Biochem. 1995;229(2):558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- 23.Buckler KJ. 498(Pt 3):649–662. doi: 10.1113/jphysiol.1997.sp021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wyatt CN, Peers C. J Physiol. 1995;483(Pt 3):559–565. doi: 10.1113/jphysiol.1995.sp020606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. J Clin Invest. 2001;108(8):1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donnelly DF. Respir Physiol. 1999;115(2):151–160. doi: 10.1016/s0034-5687(99)00021-3. [DOI] [PubMed] [Google Scholar]
- 27.Donnelly DF. J Appl Physiol. 1996;81(2):657–664. doi: 10.1152/jappl.1996.81.2.657. [DOI] [PubMed] [Google Scholar]
- 28.Gadalla AE, Pearson T, Currie AJ, Dale N, Hawley SA, Sheehan M, Hirst W, Michel AD, Randall A, Hardie DG, Frenguelli BG. J Neurochem. 2004;88(5):1272–1282. doi: 10.1046/j.1471-4159.2003.02253.x. [DOI] [PubMed] [Google Scholar]
- 29.McQueen DS, Ribeiro JA. Br J Pharmacol. 1981;74(1):129–136. doi: 10.1111/j.1476-5381.1981.tb09964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vandier C, Conway AF, Landauer RC, Kumar P. J Physiol. 1999;515(Pt 2):419–429. doi: 10.1111/j.1469-7793.1999.419ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conde SV, Monteiro EC. Adv Exp Med Biol. 2006;580:179–184. doi: 10.1007/0-387-31311-7_27. discussion 351-179. [DOI] [PubMed] [Google Scholar]
- 32.Williams SE, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, Peers C, Kemp PJ. Science. 2004;306(5704):2093–2097. doi: 10.1126/science.1105010. [DOI] [PubMed] [Google Scholar]
- 33.McCartney CE, McClafferty H, Huibant JM, Rowan EG, Shipston MJ, Rowe IC. Proc Natl Acad Sci U S A. 2005;102(49):17870–17876. doi: 10.1073/pnas.0505270102. [DOI] [PMC free article] [PubMed] [Google Scholar]