

Abstract
Acetylcholine excites many central and autonomic neurons through inhibition of M-channels, slowly activating, noninactivating voltage-gated potassium channels. We here provide information regarding the in vivo distribution and biochemical characteristics of human brain KCNQ2 and KCNQ3, two channel subunits that form M-channels when expressed in vitro, and, when mutated, cause the dominantly inherited epileptic syndrome, benign neonatal familial convulsions. KCNQ2 and KCNQ3 proteins are colocalized in a somatodendritic pattern on pyramidal and polymorphic neurons in the human cortex and hippocampus. Immunoreactivity for KCNQ2, but not KCNQ3, is also prominent in some terminal fields, suggesting a presynaptic role for a distinct subgroup of M-channels in the regulation of action potential propagation and neurotransmitter release. KCNQ2 and KCNQ3 can be coimmunoprecipitated from brain lysates. Further, KCNQ2 and KCNQ3 are coassociated with tubulin and protein kinase A within a Triton X-100-insoluble protein complex. This complex is not associated with low-density membrane rafts or with N-methyl-d-aspartate receptors, PSD-95 scaffolding proteins, or other potassium channels tested. Our studies thus provide a view of a signaling complex that may be important for cognitive function as well as epilepsy. Analysis of this complex may shed light on the unknown transduction pathway linking muscarinic acetylcholine receptor activation to M-channel inhibition.
Mutations in ion channel genes have been shown to cause a large number of forms of epilepsy and other episodic disorders in humans and inbred mice (1–3). Independent efforts led by Leppert (4–6) and Steinlein (7, 8) to identify genes responsible for benign neonatal familial convulsions (BNFC), a dominantly inherited epilepsy associated with frequent seizures in the first weeks of life and a 10–16% risk of seizure recurrence after infancy, resulted in the cloning of two potassium channel subunits, KCNQ2 and KCNQ3. KCNQ2 and KCNQ3 have been characterized electrophysiologically with heterologous expression systems (8–12). Coexpression of KCNQ2 and KCNQ3 in Xenopus oocytes resulted in currents that were much larger than those observed when either subunit was expressed alone, leading to the suggestion that heteromultimeric KCNQ2/KCNQ3 channels were expressed in vivo (9, 10). In oocytes or HEK cells, currents mediated by these heterologously expressed KCNQ2/KCNQ3 channels were enhanced by activation of the cAMP-dependent protein kinase (PKA); this effect was abolished by site-directed mutations that eliminated a single PKA consensus site near the amino terminus of the KCNQ2 polypeptide (10). Mutations causing BNFC were found to cause only slight reductions in current compared with wild-type controls, suggesting that small differences in the activity of these KCNQ channels in vivo might be sufficient to cause epilepsy (10). When KCNQ2 and KCNQ3 cDNAs were coexpressed in oocytes, currents were observed that exhibited kinetic and voltage-dependent properties similar to M-currents seen in cultured superior cervical ganglion neurons (9, 13). Indeed, heterologous KCNQ2 or KCNQ3-mediated currents in mammalian cultured cells were inhibited by activation of cotransfected M1 muscarinic acetylcholine receptors (11, 12).
To complement such heterologous expression studies, we raised antibodies against KCNQ2 and KCNQ3 and characterized the composition and distribution of channels formed by these subunits in human central neurons.
Materials and Methods
Antibody Production and Purification.
Unique regions of the deduced peptide sequences of KCNQ2 and KCNQ3 corresponding to amino acids 397–423 (Q2C1) and 713–737 (Q2C2) of the full-length KCNQ2 (5) sequence and amino acids 524–548 (Q3C1) of KCNQ3 (6) were conjugated to keyhole limpet hemocyanin. Antibodies were raised by standard protocols after injection into guinea pig (anti-Q2C1 and anti-Q2C2) and rabbit (anti-Q3C1). Antibodies were affinity-purified from sera by using the peptide immunogen immobilized on SulfoLink resin (Pierce). Control Western blots against transfected cells and human brain membranes showed that KCNQ2 and KCNQ3 immunoreactivity was absent from preimmune sera and was eliminated by preincubation of affinity-purified antibodies with excess immunogenic peptide. Rabbit antibodies against N-methyl-d-aspartate (NMDA)-R1, and NMDA-R2 have been characterized (15); antibodies against PSD-95 were the kind gift of David Bredt (University of California, San Francisco). Mouse mAbs were purchased from Transduction Laboratories (Lexington, KY) (anti-calcium and calmodulin-dependent protein kinase II, anti-protein kinase A catalytic and regulatory subunits, anticalcineurin, anti-AKAP79), Sigma (anti-tubulin), and Amersham Pharmacia (anti-actin) and used at dilutions suggested by the suppliers.
Collection and Processing of Human Brain Specimens.
Specimens of human brain used in these studies were collected for research purposes according to procedures approved by the University of California, San Francisco Committee on Human Research. Brain specimens for biochemical experiments were obtained from patients undergoing surgery for epilepsy or tumor resection. Samples were washed briefly in ice-cold saline and then either frozen on dry ice and stored at −80°C or used immediately for membrane preparation. P2 membranes were prepared by standard procedures. All biochemical procedures were performed in the presence of protease inhibitors (PMSF, aprotinin, leupeptin, EGTA, EDTA, or Boehringer-Mannheim Complete) and 1 mM DTT. Immunocytochemical experiments were performed on surgical specimens after immersion fixation in 4% paraformaldehyde/0.1% glutaraldehyde and vibratome or cryostat sectioning (data not shown), or on paraffin-embedded autopsy specimens from adults that died of nonneurological disorders (see Figs. 2 and 3).
Figure 2.

Immunolocalization of KCNQ2 and KCNQ3 in human cortex and hippocampus. (A) Low-power view (bar equals 1 mm) of transverse section through human hippocampal formation after immunoperoxidase staining using Q2C2 antibodies and light counterstaining with hematoxylin. Subregions indicated are the dentate gyrus (DG), sections CA3 and CA1 of the hippocampus, and the subiculum (sub). Regions shown in red boxes represent locations shown at higher power in E–H. (B) Control experiment showing immunostaining was abolished by preincubation of antibodies with antigenic peptide. Adjoining sections were incubated with Q2C2 antibodies in the presence (Left) or absence (Right) of excess Q2C2 peptide. (Left) Only purple–blue counterstain of neurons and glia. (Right) Brown immunoperoxidase staining of neuronal somata, proximal dendrites, and lighter neuropil staining. (C) Confocal images of Q2C2 (Left) and Q3C1 (Right) staining of temporal cortex (parahippocampal gyrus) neurons. Red shows channel antibody staining in a punctate somotodendritic distribution; green shows brain autofluorescence, mostly associated with lipofuscin in perinuclear vesicular structures. (Scale bar equals 10 μm.) (D) Q2C2 staining of mossy fiber bundles in the dentate hilar region. (E) Q2C2 (Left) and Q3C1 (Right) staining of polymorphic neurons in the dentate hilar region. (Arrows indicate stained neuronal somata; asterisks indicate neuropil, which is stained more heavily with Q2C2 than Q3C1.) (F) Q2C2 (Left) and Q3C1 (Right) staining of neurons in the CA3 pyramidal cell layer. (G) Q2C2 (Left) and Q3C1 (Right) staining of pyramidal cells in the subiculum. (H) Staining of the neuropil in the dentate hilar and inner molecular layers by Q2C2 but not Q3C1. Arrows indicate hilar neuronal somata, labeled by both antibodies; brackets indicate inner molecular layer staining by Q2C2 but not Q3C1. (Scale bar equals 100 μm.) Pm, polymorphic layer; gc, granule cell layer; iml, inner (associational-commissural) molecular layer; oml, outer (perforant path) molecular layer. (Scale bar in B equals 20 μm in B, D–G.)
Figure 3.

Heteromeric human brain KCNQ2/KCNQ3 channels are contained within a Triton X-100-resistant subcellular fraction. (A) Insolubility of KCNQ2 and KCNQ3. Human cortical membranes were solubilized in 1% Triton X-100, and soluble and insoluble fractions were separated by centrifugation. Starting membranes (M) and insoluble (P, pellet) and soluble (S) fractions were probed with antibodies against KCNQ subunits, Shaker family potassium channel subunits (Kv1.2, Kvl.4), a glutamate receptor subunit (NMDA-R1), and PSD-95. The Shaker subunits were efficiently solubilized by Triton X-100; the KCNQ subunits, NMDA-R1 and PSD-95 were poorly solubilized. (B) Solubilization by SDS at 4°C. Human brain membranes were solubilized with SDS at the indicated concentrations on ice as described in Materials and Methods, and soluble and insoluble fractions were separated by centrifugation. KCNQ3, NMDA-R1, and PSD-95 controls were efficiently solubilized by cold SDS, but the majority of KCNQ2 remained associated with the insoluble fraction. (C) SDS-solubilized KCNQ2 and KCNQ3 are not coassociated. After solubilization of brain membranes using 1% SDS/4°C, Q2C1 and Q3C2 immunoprecipitates were probed with the indicated antibodies. (D) Solubilization of KCNQ2/KCNQ3 complexes (but not PSD-95) by Triton X-100 after low salt washes, high salt, and pH 11. Membranes were washed as described in Materials and Methods, then solubilized as indicated with octyl glucoside or Triton X-100 at 4°C or 37°C as indicated. KCNQ2 and KCNQ3 (not shown) were partly solubilized by Triton X-100 after low-salt washes, sequential low/high-salt washes, or pH 11, but not by octyl glucoside, and solubilization with Triton X-100 at 37°C after low-salt washes was not more effective than at 4°C. PSD-95 remained in pellet after salt or pH 11 treatment. (E) Coimmunoprecipitation of KCNQ2 and KCNQ3 by either Q2C2 or Q3C1 antibodies. Membranes were low-salt washed three times, extracted at pH 11, solubilized with Triton X-100, and cleared of insoluble material by centrifugation. KCNQ2 and KCNQ3 (but not PSD-95) were both immunoprecipitated by antibodies specific for either subunit.
Heterologous Expression.
KCNQ2 cDNA in pTLN (8) (identical to ref. 4 except for alternative splicing at two sites that result in deletion of 30 aa in the C-terminal portion of the protein) was subcloned into pcDNA3 (Invitrogen). HEK293 cells were transfected with pcDNA3/Q2 and pcDNA3/Q3 (11) using FuGene (Roche Molecular Biochemicals). Cells were harvested for biochemical experiments or fixed with 4% paraformaldehyde for immunostaining 36–60 h after transfection.
Subcellular Fractionation.
Attempts to solubilize KCNQ subunits were performed on rodent or human P2 membranes (1–2 mg/ml protein; Bio-Rad protein assay) resuspended in solubilization buffer [calcium and magnesium free (CMF)-PBS supplemented with protease inhibitors and 1 mM DTT]. Triton X-100 extraction was performed by addition of detergent to a final concentration of 1%, tumbling at 4°C for 30 min, and centrifugation at 20,800 × g for 30 min. Solubilization of KCNQ2 with 2% Triton X-100, 1% deoxycholate, or RIPA buffer (0.15 M NaCl/0.05 M Tris⋅HCl, pH 7.2/1% Triton X-100/1% sodium deoxycholate/0.1% SDS) (data not shown) gave identical results to 1% Triton. For membrane extraction (see Fig. 3D), P2 membranes were washed twice with 10 mM Tris, pH 7.4, then either resuspended in solubilization buffer and solubilized with 1% Triton at 4°C or 37°C or 60 mM octyl glucoside (16) at 4°C for 30 min. Parallel 10 mM Tris-washed samples were resuspended in PBS/CMF supplemented with NaCl to 1 M, or 0.1 M Na2Co3, pH 11.0, tumbled for 30 min at 4°C, and centrifuged (20,800 × g, 20 min). Pellets were resuspended in solubilization buffer and solubilized with 1% Triton X-100. Affinity chromatography using anti-KCNQ2 antibodies was performed by adding antibodies to 1% Triton lysates (without centrifugation to remove insoluble material), incubation for 2 h at 4°C, isolation using protein A Sepharose beads, and washing five times using 100 bead volumes of lysis buffer.
Immunohistochemistry.
Immunohistochemical staining was performed on 40-μm floating sections or 5-μm paraffin sections with similar results. Methods for immunoperoxidase and immunofluorescent staining of floating sections have been described (17); paraffin-embedded sections were immunoperoxidase-stained by standard methods after microwave irradiation in citrate buffer. Immunofluorescent staining of paraffin sections was performed by using Cy3-tyramide signal amplification (NEN) to reduce lipofuscin autofluorescence (18, 19). Images were collected on a Bio-Rad 1024 laser scanning confocal microscope. For detection of autofluorescence (see Figs. 2 and 3, green), the 488-nm excitation line and 532-nm band pass filter was used. Cy3 was detected by using the 568 excitation line and HQ598/40 band pass filters.
Results
KCNQ2 and KCNQ3 Expression and Coassembly in HEK Cells.
We raised and affinity-purified antibodies directed against unique segments of the C-terminal portions of KCNQ2 and KCNQ3 (Fig. 1A). Experiments using transfected HEK293 cells established the specificity and efficacy of these reagents and confirmed that KCNQ2 and KCNQ3 coassembled when coexpressed heterologously (9–11). Transfection with KCNQ2 cDNA or KCNQ3 cDNA alone resulted in expression of immunoreactive proteins that on SDS gels exhibited mobility consistent with that predicted from the cDNA sequences (Fig. lB). Cells that were cotransfected with both cDNAs expressed markedly higher amounts of KCNQ2 and KCNQ3 protein compared with cells transfected with cDNA for each subunit alone (Fig. 1B). Whole-cell patch clamp recordings from cells cotransfected with both subunits revealed large currents (5–10 nA) with voltage-dependence and kinetics similar to native M-channels (data not shown). Currents were small or undetectable in cells transfected with KCNQ2 or KCNQ3 alone. Lysis of KCNQ2/Q3 cotransfected cells with nonionic detergent released both KCNQ2 and KCNQ3 into solution, and each of the three antibodies tested immunoprecipitated both subunits (Fig. 1C, and data not shown). Confocal images of HEK cells stained for KCNQ2 and KCNQ3 showed similar or identical staining patterns, further suggesting that KCNQ2 and KCNQ3 were efficiently assembled into heteromeric channel complexes in these cells (Fig. 1D).
Figure 1.
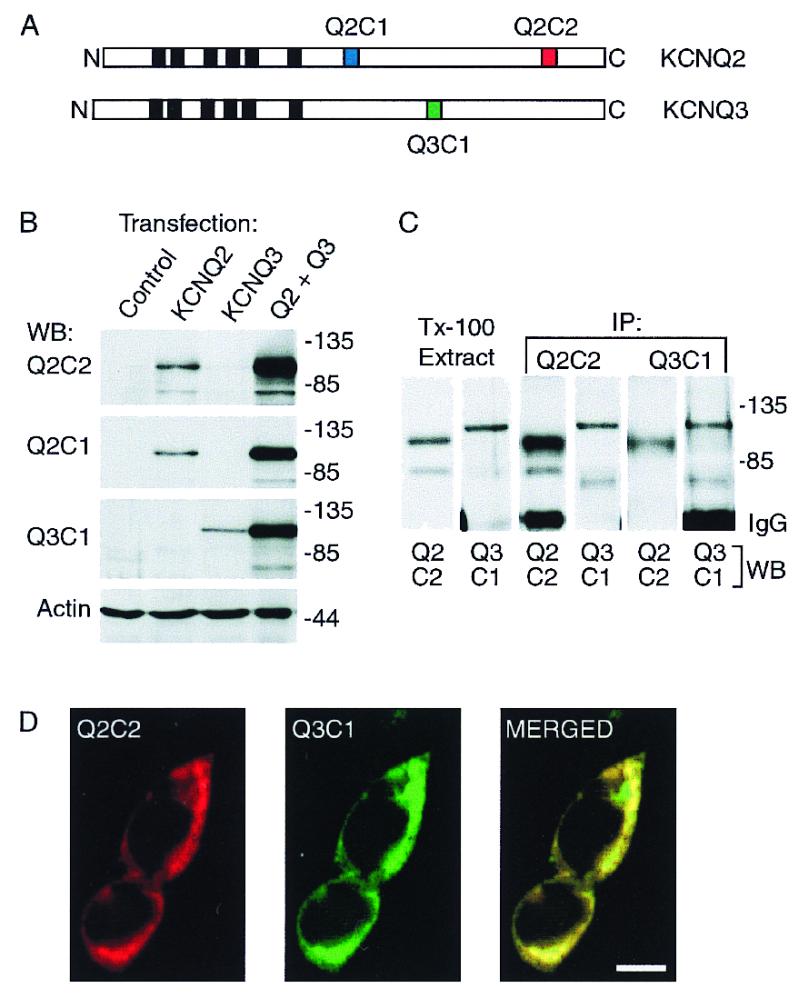
Characterization of heterologously expressed KCNQ2 and KCNQ3 and subunit-specific antibodies. (A) Diagram of the KCNQ2 and KCNQ3 polypeptides predicted by cDNA sequences, showing the position of the six putative membrane-spanning segments (black boxes), and the three segments within the C-terminal domains corresponding to synthetic peptides used for raising subunit-specific antibodies Q2C1, Q2C2, and Q3C1. (B) Immunoblots of whole-cell lysates prepared from HEK293 cells transfected with KCNQ2 alone, KCNQ3 alone, or cotransfected with both subunits. (C) Solubilization and coimmunoprecipitation of KCNQ2 and KCNQ3 expressed in HEK293 cells. Cells cotransfected with KCNQ2 and KCNQ3 were lysed under nondenaturing conditions with 1% Triton X-100. The lysates were cleared of nuclei and insoluble material by centrifugation, then either reserved (Tx-100 extract) or incubated with Q2C2 or Q3C1 antibodies. Whole-cell extracts and immunoprecipitates were probed with Q2C2, then stripped and reprobed with Q3C1. (D) Immunofluorescence staining of cotransfected HEK cells shows predominantly colocalized pattern of expression of KCNQ2 and KCNQ3. (Scale bar equals 10 μm.)
Immunolocalization of KCNQ2 and KCNQ3 in Human Cortex and Hippocampal Formation.
We next examined the cellular and subcellular localization of KCNQ2 and KCNQ3 in human brain using immunohistochemistry (Fig. 2). In the hippocampal formation and cerebral cortex, KCNQ2 and KCNQ3 staining was detected on the somata and dendrites of many polymorphic and pyramidal neurons. Confocal immunofluorescence microscopy revealed that this somatodendritic staining was punctate in character and appeared to label both the cell surface and intracellular components (Fig. 2C). In the hippocampal formation, hilar polymorphic cells (Fig. 2 E and H), CA3 pyramidal cells (Fig. 2F), and subicular pyramidal cells (Fig. 2G) exhibited somatodendritic staining for both subunits. Staining for KCNQ2 (but not for KCNQ3) was detected on mossy fiber bundles traversing the dentate hilus (Fig. 2D) and CA3 and more diffusely in the neuropil of the dentate hilus (Fig. 2 D and E), CA3 (Fig. 2F), CA1 (Fig. 2A), and the subiculum (Fig. 2G). Strong KCNQ2 (but not KCNQ3) neuropil staining also was detected in the inner, but not the outer, dentate molecular layer (Fig. 2H). It is in the dentate inner molecular layer that associational fibers derived from hilar mossy cells form en passante excitatory synapses on granule cell proximal dendrites (20). Because the granule cell dendrites extend radially through both inner and outer molecular layers, but the mossy cell axons and terminals are restricted to the inner layer, the KCNQ2 staining in the inner molecular layer seems most likely presynaptic. Thus, at least two types of hippocampal excitatory neurons (the mossy cells and granule cells) appear to express channels containing KCNQ2 but not KCNQ3 on their axons and/or termini, where they may regulate action potential propagation and neurotransmitter release.
Biochemical Characterization of KCNQ2 and KCNQ3 Proteins.
We next characterized brain KCNQ subunit proteins expressed in vivo by Western analysis and subcellular fractionation. Western analysis of human brain membranes using the anti-Q2 and anti-Q3 antibodies revealed bands of similar mobility to those seen in the transfected HEK cells (Fig. 3). However, when we solubilized the brain membranes under conditions that efficiently solubilized both KCNQ2 and KCNQ3 from HEK cells (e.g., 1% Triton X-100), both polypeptides were completely retained within the insoluble pellet (Fig. 3A). By contrast, KCNA4 and KCNA2, human homologues of the Shaker family voltage-gated potassium channels Kvl.4 and Kv1.2, were efficiently solubilized by this treatment. Human PSD-95 and NMDA receptor subunit NR1 were, like KCNQ2 and KCNQ3, triton-insoluble. PSD-95 and NRl are contained within a triton-insoluble postsynaptic complex that can be solubilized by incubation of membranes with the ionic detergent SDS at 4°C, followed by addition of excess Triton X-100 (21). NMDA subunits can subsequently be coimmunoprecipitated by antibodies against PDZ domain-containing proteins such as PSD-95, providing evidence for tight association between these glutamate receptors, which are heteromeric integral membrane proteins, and membrane-associated scaffolding proteins, such as PSD-95, that serve as bridges to the cytoskeleton. We therefore incubated human brain membranes with SDS at 4°C, diluted the mixture with excess cold Triton X-100, and separated soluble and insoluble fractions by centrifugation. PSD-95, NMDA-R1, and KCNQ3 were completely solubilized by this procedure, but most of KCNQ2 remained insoluble (Fig. 3B). Anti-KCNQ2 antibodies immunoprecipitated KCNQ2, but not KCNQ3, from this SDS-treated fraction (Fig. 3C).
Because polypeptides within a quaternary complex may be dissociated by SDS, even at 4°C, we tried other methods of solubilization for the purpose of testing whether KCNQ2 and KCNQ3 form heteromeric channels. Conditions known to strip membranes of associated proteins and the cytoskeleton (washing with hypotonic solution, 1 M NaCl, or 0. 1 M Na2CO3 at pH 11) allowed a significant fraction of KCNQ2 and KCNQ3 to be solubilized during subsequent exposure to Triton X-100 at 4°C (Fig. 3D). Many signaling molecules are colocalized to low-density, membrane microdomains (“lipid rafts”) that cannot be solubilized by Triton X-100 at 4°C, but are efficiently solubilized by Triton X-100 at 37°C or by octyl glucoside at 4°C (16). We therefore also attempted to solubilize KCNQ2 and KCNQ3 under these conditions known to disrupt lipid rafts and release associated proteins into solution. Treatment of low-salt washed human cortical membranes with octyl glucoside was completely ineffective at solubilizing KCNQ2 and KCNQ3; Triton X-100 was no more effective at 37°C than at 4°C (Fig. 3D). Using membranes stripped with high salt or pH 11 treatments, KCNQ2 and KCNQ3 could be solubilized and coimmunoprecipitated by antibodies directed against either subunit, suggesting the presence of heteromeric KCNQ2/KCNQ3 channels in human brain (Fig. 3E). PSD-95 was inefficiently solubilized under these conditions (Fig. 3D) and was absent from the anti-KCNQ immunoprecipitates (Fig. 3E), emphasizing that the subcellular fraction containing KCNQ2/Q3 heteromeric channels was distinct from that containing PSD-95 and the NMDA receptor.
The above results suggested that the Triton X-100 insolubility of KCNQ2 and KCNQ3 might be caused by interaction with membrane-associated proteins and/or the cytoskeleton. We therefore solubilized brain membranes as before (i.e., Fig. 3A) with Triton X-100, isolated KCNQ2 from nonassociated proteins by immunoaffinity chromatography, and probed for cytoskeletal proteins or proteins implicated in KCNQ channel regulation. Tubulin and actin were present in the KCNQ2 affinity-purified fraction, but PSD-95, NMDA receptor subunits NR1 and NR2, and calcineurin were absent (Fig. 4 A and B). AKAP79 and the RIIB subunit of protein kinase A also were detected within the KCNQ2-associated complexes (Fig. 4B).
Figure 4.

KCNQ3, cytoskeletal proteins, and PKA are associated with KCNQ2 in a Triton X-100 insoluble complex. (A) KCNQ2-associated proteins were affinity-purified using Q2C1 or Q2C2 antibodies, and probed with the indicated antibodies. KCNQ2, KCNQ3, and tubulin were detected, but PSD-95, although abundant in the starting membranes, was absent in the affinity-purified fraction. (B) AKAP79, PKA RIIB, tubulin, and actin are present in the Q2C2 affinity-purified complex, but calcineurin, CaM Kinase, NMDA-R1, and NMDA-R2 are not present.
Discussion
In electrophysiological studies of KCNQ2 and KCNQ3 using Xenopus oocytes, each of the subunits when expressed alone forms functional channels at low levels (8–10). Coexpression may result in 10- to 50-fold increases in current levels. The marked increases in the amount of protein expression resulting from coexpression that we observe (Fig. lA) may partially explain the increase in current density seen in functional studies. We have shown using immunocytochemistry that KCNQ2 and KCNQ3 exhibit overlapping patterns of expression in the human cortex and hippocampus. Both subunits are expressed postsynaptically in a similar, markedly punctate pattern by numerous pyramidal and polymorphic neurons in both brain regions. The somatic immunofluorescent staining for KCNQ2 and KCNQ3 appeared to include components localized internally and at the cell surface, and thus may in part represent one or more biosynthetic pools of channels not contributing directly to somatic excitability, but instead destined for transport to and cell-surface expression at other subcellular domains. In human brain, native KCNQ2 and KCNQ3 proteins are contained within a triton-insoluble subcellular fraction. After treatments that strip cell membranes of cytoskeletal attachments and membrane-associated proteins, both subunits are rendered triton-soluble and can be coimmunoprecipitated. Indeed, actin, tubulin, and components of protein kinase A [shown to regulate KCNQ2 in heterologous expression studies (10)] are copurified with KNCQ2 and KCNQ3 by affinity chromatography (Fig. 4). These data suggest that heteromeric KCNQ2/KCNQ3 channels are expressed postsynaptically, where they may be associated with the cytoskeleton directly, or perhaps more likely, by means of unknown scaffolding proteins. Because KCNQ2 is more detergent resistant than KCNQ3 (Fig. 2B), we hypothesize that it may contain a region essential for such cytoskeletal interaction. This parallels the reliance by heteromeric NMDA, γ-aminobutyric acid, and glycine receptor channels on specific interactions between particular channel subunits and scaffold proteins that link the channels to the cytoskeleton (22–24). Heteromeric KCNQ2/KCNQ3 channels located on proximal dendrites and somata could regulate neuronal excitability by controlling the ability of excitatory postsynaptic potentials received at dendritic branches and spines to spread centrally and generate action potentials. Presynaptic KCNQ2-containing channels may potentially control action potential propagation and neurotransmitter release. Thus, the ability of linopirdine and other selective KCNQ channel blockers to enhance stimulus-evoked acetylcholine and glutamate release in vivo (25) likely reflects a combination of presynaptic and postsynaptic mechanisms. It is unclear whether the presynaptic channels are homotetramers of KCNQ2 subunits or heteromeric channels containing KCNQ2 and additional, as yet unknown, KCNQ subfamily members. Our findings highlight the possibility that reductions in KCNQ2 and KCNQ3 channel activity in BNFC may result from reductions in channel protein level, or impaired channel assembly, trafficking, or localization as well as alteration in intrinsic functional properties. Pharmacological, mRNA expression, and electrophysiological data suggest that heteromeric KCNQ2/KCNQ3 channels form postsynaptic M-channels in sympathetic neurons (9, 11, 12). Inhibition of M-channels also contributes to the excitation of central neurons by muscarinic cholinergic agonists. This cholinergic excitation is important for alertness, arousal, learning, and memory and has been implicated in Alzheimer's disease and temporal lobe epilepsy (reviewed in ref. 3). It therefore will be of great interest to determine whether molecules important for the cholinergic inhibition of M-channels are associated with KCNQ2 and KCNQ3 in a triton-insoluble complex and to analyze channel activity and modulation in those neurons expressing KCNQ2 and KCNQ3 in the human brain.
Acknowledgments
We thank members of the Jan laboratories for comments on this manuscript and Dr. Thomas Jentsch for KCNQ cDNA clones. This work was supported by a National Institute of Neurological Disorders and Stroke Clinical Investigator Development Award and Parke–Davis Young Investigator Award from the American Academy of Neurology (to E.C.C.), and by an National Institute of Mental Health Silvio Conte Center grant at the University of California, San Francisco. L.Y.J. and Y.N.J. are Howard Hughes Investigators.
Abbreviations
- BNFC
benign neonatal familial convulsions
- PKA
cAMP-dependent protein kinase
- NMDA
N-methyl-d-aspartate
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.090092797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.090092797
References
- 1.Ptacek L. Am J Med. 1998;105:58–70. doi: 10.1016/s0002-9343(98)00123-5. [DOI] [PubMed] [Google Scholar]
- 2.Noebels J L. Neuron. 1996;16:241–244. doi: 10.1016/s0896-6273(00)80042-2. [DOI] [PubMed] [Google Scholar]
- 3.Cooper E C, Jan L Y. Proc Natl Acad Sci USA. 1999;96:4759–4766. doi: 10.1073/pnas.96.9.4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leppert M, Anderson V E, Quattlebaum T, Stauffer D, O'Connell P, Nakamura Y, Lalouel J M, White R. Nature (London) 1989;337:647–648. doi: 10.1038/337647a0. [DOI] [PubMed] [Google Scholar]
- 5.Singh N A, Charlier C, Stauffer D, DuPont B R, Leach R J, Melis R, Ronen G M, Bjerre I, Quattlebaum T, Murphy J V, et al. Nat Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- 6.Charlier C, Singh N A, Ryan S G, Lewis T B, Reus B E, Leach R J, Leppert M. Nat Genet. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- 7.Steinlein O, Schuster V, Fischer C, Haussler M. Hum Genet. 1995;95:411–415. doi: 10.1007/BF00208966. [DOI] [PubMed] [Google Scholar]
- 8.Biervert C, Schroeder B C, Kubisch C, Berkovic S F, Propping P, Jentsch T J, Steinlein O K. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- 9.Wang H-S, Pan Z, Shi W, Brown B S, Wymore R S, Cohen I S, Dixon J E, McKinnon D. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- 10.Schroeder B C, Kubisch C, Stein V, Jentsch T J. Nature (London) 1998;396:687–690. doi: 10.1038/25367. [DOI] [PubMed] [Google Scholar]
- 11.Shapiro M S, Roche J P, Kaftan E J, Cruzblanca H, Mackie K, Hille B. J Neurosci. 2000;20:1710–1721. doi: 10.1523/JNEUROSCI.20-05-01710.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Selyanko, A. A., Hadley, J. K., Wood, I. C., Abodagie, F. C., Jentsch, T. J. & Brown, D. A. J. Physiol. (London)522, 349–355. [DOI] [PMC free article] [PubMed]
- 13.Marrion N V. Annu Rev Physiol. 1997;59:483–504. doi: 10.1146/annurev.physiol.59.1.483. [DOI] [PubMed] [Google Scholar]
- 14.Cruzblanca H, Koh D-S, Hille B. Proc Natl Acad Sci USA. 1998;95:7151–7156. doi: 10.1073/pnas.95.12.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheng M, Cummings J, Roldan L A, Jan Y N, Jan L Y. Nature (London) 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- 16.Brown D A, London E. Annu Rev Cell Dev Biol. 1998;14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- 17.Cooper E C, Milroy A, Jan Y N, Jan L Y, Lowenstein D H. J Neurosci. 1998;18:965–974. doi: 10.1523/JNEUROSCI.18-03-00965.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strappe P M, Wang T H, McKenzie C A, Lowrie S, Simmonds P, Bell J E. J Virol Methods. 1997;67:103–112. doi: 10.1016/s0166-0934(97)00083-9. [DOI] [PubMed] [Google Scholar]
- 19.Loup F, Weinmann O, Yonekawa Y, Aguzzi A, Wieser H G, Fritschy J M. J Histochem Cytochem. 1998;46:1129–1139. doi: 10.1177/002215549804601005. [DOI] [PubMed] [Google Scholar]
- 20.Buckmaster P S, Wenzel H J, Kunkel D D, Schwartzkroin P A. J Comp Neurol. 1996;366:271–292. doi: 10.1002/(sici)1096-9861(19960304)366:2<270::aid-cne7>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 21.Müller B M, Kistner U, Kindler S, Chung W J, Kuhlendahl S, Fenster S D, Lau L F, Veh R W, Huganir R L, Gundelfinger E D, Garner C C. Neuron. 1996;17:255–265. doi: 10.1016/s0896-6273(00)80157-9. [DOI] [PubMed] [Google Scholar]
- 22.Kornau H C, Seeberg P H, Kennedy M B. Curr Opin Neurobiol. 1997;7:368–373. doi: 10.1016/s0959-4388(97)80064-5. [DOI] [PubMed] [Google Scholar]
- 23.Meyer G, Kirsch J, Betz H, Langosch D. Neuron. 1995;15:563–572. doi: 10.1016/0896-6273(95)90145-0. [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Bedford F K, Brandon N J, Moss S J, Olsen R W. Nature (London) 1999;397:69–72. doi: 10.1038/16264. [DOI] [PubMed] [Google Scholar]
- 25.Nickolson V J, Tam S W, Myers M J, Cook L. Drug Dev Res. 1990;19:285–300. [Google Scholar]