

Abstract
The fundamental mechanisms of charge migration in DNA are pertinent for current developments in molecular electronics and electrochemistry-based chip technology. The energetic control of hole (positive ion) multistep hopping transport in DNA proceeds via the guanine, the nucleobase with the lowest oxidation potential. Chemical yield data for the relative reactivity of the guanine cations and of charge trapping by a triple guanine unit in one of the strands quantify the hopping, trapping, and chemical kinetic parameters. The hole-hopping rate for superexchange-mediated interactions via two intervening AT base pairs is estimated to be 109 s−1 at 300 K. We infer that the maximal distance for hole hopping in the duplex with the guanine separated by a single AT base pair is 300 ± 70 Å. Although we encounter constraints for hole transport in DNA emerging from the number of the mediating AT base pairs, electron transport is expected to be nearly sequence independent because of the similarity of the reduction potentials of the thymine and of the cytosine.
The understanding of charge migration phenomena in DNA is central for the development of avenues in DNA-based molecular technologies, in particular, electrochemical sequencing techniques (1–3) and functional nanoscale electronic devices (4–9). Although, in recent years, a large number of experimental studies have evolved (10–21), the mechanism of charge transport in DNA over long distances of the order of 100 Å remained elusive. We advance a kinetic analysis of recent chemical yield data for the relative reactivity of guanine (G) cations G+ and of charge trapping by a distant triple G unit (GGG) within one of the strands of the helix (15, 18, 19). Thereby, we establish the occurrence of hole transport via hopping and define the conditions for the realization of chemistry over large distances (50–300 Å) in DNA.
Energetic Control of Charge Migration in DNA.
The majority of the available experimental information on charge migration, separation, shift, and recombination in DNA pertains to hole (positive ion) transfer and/or transport in solution (10–21). This process involves electron transfer from the hole acceptor to the electronically excited or positively charged hole donor. On the basis of a theoretical analysis (22) of the experimental results, two distinct mechanisms were considered for hole transfer and transport in DNA: (i) Unistep charge transfer between localized donor and acceptor sites involving intervening DNA-nucleobases as superexchange-mediators and (ii) multistep hopping-type charge transport between the redox centers along the nucleobases forming the bridge. The superexchange mechanism (i) is characterized by an exponential donor–acceptor distance (RDA) dependence of the charge transfer rate, kCT ∝ exp(−βRDA) ∝ exp(−βnR1) with β = 0.6–1.4 Å−1 (13–15, 18), where n is the number of base pairs in the bridge and R1 is their nearest-neighbor distance. The hopping mechanism (ii), which heuristically results in small values of β = 0.01–0.1 Å−1 (10–12, 16, 19, 21), can be described in terms of a weak (algebraic) distance dependence of the rate, i.e., kCT ∝ n−η with η = 1–2 (22). There is no dichotomy between the different mechanisms, superexchange-mediated charge transfer and hopping charge transport. Rather, the prevalence of one of the two mechanisms is determined by the energetic constraints (22) given by the specific relative energies of the charge donor and the bridge. This element of energetic control is based on the distinction between off-resonance and resonance donor-bridge coupling (22). It explains the occurrence of superexchange, off-resonance coupling (mechanism i), which is now well documented (13–15, 18, 19), and establishes the conditions for the realization of hopping charge transport in mechanism ii of resonance coupling. The experimental evidence for the latter case was inferred (10, 11, 16–19, 21) from the weak distance dependence of charge migration. A second element of the energetic control involves the relative energies of the nucleobases in the bridge. For the case of hole hopping, the positive charge will be exclusively located on the guanines, i.e., the nucleobase with the lowest oxidation potential separated from the next higher nucleobase, adenine, by 0.4 eV (1 eV = 1.602 × 10−19 J; refs. 23 and 24). This conclusion concurs with recent experiments (18, 19) that explored hole transport from the guanine (G) cation G+ to the hole trap triple G unit (GGG). The chemical yield data for charge migration in the G+(TTG)NGG strand shown in Scheme S1 show a weak distance dependence over a distance scale of r = 10–40 Å (N = 1–4), in accord with the hopping model (18, 19, 22). Here, A, T, and C denote adenine, thymine, and cytosine, respectively. A quantitative analysis of the chemical yield data (18, 19) reported herein provides quantitative information on hopping, trapping, and chemical kinetic parameters.
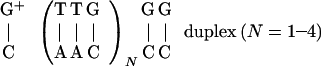
The energetic control (15) of the hole migration mechanism in the G1TTG2TTG3…GNTTGGG strand (18, 19), which contains N G bases, rests on redox potential data (23, 24) in solution, assuming that the energy differences are maintained in DNA. The lowest-energy hole states correspond to Gj+/Gj (j = 1…N), whereas the energies of the A+/A, T+/T, and C+/C states are higher than those of Gj+/Gj by about 0.5–0.7 eV. Accordingly, the coupling between the nearest-neighbor Gj+… Gj±1 bridge bases is resonant, and hole transport will occur between G1+ and GGG via hopping through the Gj bases. On the other hand, the Gj+T intrastrand coupling is off-resonance, as are the interstrand Gj+C and Gj+A couplings. Thus, hole hopping from Gj+ to T, A, and C is precluded, and these bases can mediate the resonant Gj+… Gj±1 interaction via superexchange coupling. Finally, the energy of GGG+/GGG is lower from that of Gj+/Gj by about 0.7 eV according to ab initio calculations of ionization potentials (25); thus, GGG acts as a hole trap.
The Kinetic Scheme for Hole Transport.
Hole hopping in the G+TTGTTGTT…TTGGG strand (18, 19) is shown in Scheme S2. This scheme corresponds to reversible hole hopping with a rate k along the chain between nearest-neighbor G bases, which is terminated by trapping at GGG with a rate kt. Effective charge separation from the initially oxidized site G1+ to the subsequent sites G2…GN should not be constrained by Coulomb attraction within the primary radical pair. This requirement restricts the choice of the exogenous injector. To minimize a Coulomb barrier in the primary radical pair, the injecting species either should be charged suitably or, if neutral, should be a component of an exogenous donor–acceptor system that allows primary charge separation to be followed by a removal of the charge from the injector. In the case of the experiments to be analyzed here, hole injection proceeds via charge shift from an adjacent deoxyribose cation to G1. The initially formed cation G1+ can undergo several side reactions, e.g., deprotonation and a reaction with water (15, 18, 19) with a rate constant kr. The yield for the reaction of G1+ with water is taken to be proportional to the total product yield P1 of all the side reactions. Similar side reactions (15, 18, 19) can take place at Gj+ (j = 2…N), with a global rate kd leading to the products Pj (j = 2…N), with kd being taken to be independent of j. Making contact with quantum mechanical theory of charge transfer (22, 26), the rates k and kt correspond to nearest-neighbor superexchange rates in Gj+TTGj±1 (for k) and in GN+TTGGG (for kt), which are mediated by off-resonance coupling of G+ with the bridging elements TT. Thus, the kinetic Scheme S2 represents a hopping mechanism with the individual rates k and kt being determined by superexchange interactions.
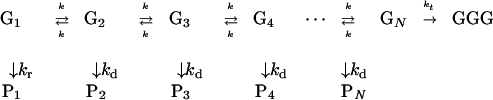
The experimental observables are the time-independent reaction yields of the oxidized guanines with water, i.e., the yields at the initial site Y(P1) and at the intermediate sites Y(Pj) (j = 2…N) as well as the acceptor yield Y(GGG). The experimental procedures for the determination of the chemical yields are described elsewhere (18, 19). The experimental yield data are given in terms of the ratio of the yields of GGG and of the water reaction products Pj (j = 1…N)
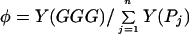 |
1 |
or, alternatively, in terms of the ratio of the yield of GGG and the product P1, i.e.,
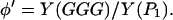 |
2 |
On the assumption that the total yield of side reactions of each oxidized guanine is proportional to the water reaction yield, Eqs. 1 and 2 also determine the ratios of the total yield of side reactions. A complete solution of the kinetic scheme provides general results and numerical data. An exact and simple expression for φ′ in the case kd = 0 is
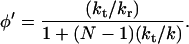 |
3 |
For finite values of kd, the algebraic N dependence of φ′ changes its character with increasing N. There is a crossover that occurs at approximately N ≅ Nc = (k/kd)1/2, between the algebraic behavior given by Eq. 3 and an exponential dependence on N of the form φ ∝ xN = exp(Nlnx), where x = [1 + (kd/2k)] − [(kd/k) + (kd/k)2]1/2, for N ≫ Nc. For (kd/k) ∼< 1, the asymptotic solution is −lnx = (kd/k)1/2; thus, for both φ and φ′,
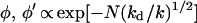 |
4 |
follows. We emphasize that the asymptotic result (for kd/k ∼< 1) provides a weak exponential distance dependence of φ (and of φ′) of the form φ ∝ exp(−β̄RDA) with a small numerical value of the exponent β̄ = (kd/k)1/2/R0, where R0 is the nearest neighbor G…G distance.
Hopping, Trapping, and Chemical Reaction Rates.
Analysis of the experimental data for the sequence G(TTG)NGG with N = 1–4 was provided in terms of the kinetic model, Scheme S2, with the yield ratios being expressed in terms of the three independent parameters kr/k, kd/k, and kt/k, with all the rate constants being measured in units of the hopping rate k. The N = 1 case corresponds to φ and φ′ = kt/kr. The experimental yield data of φ and φ′ are well accounted for in Fig. 1 by the numerical solution of the kinetic Scheme S2 with the parameters 0.08 < kr/k < 0.1, kd/k = 0.08, and 0.6 < kt/k < 1.1. We note that the yield data for N = 1–4 are approximated by the algebraic relation Eq. 3 for φ′, as appropriate for this system, where Nc ≅ 4. From these results, we infer that the rates of the water reactions with G+ are slow on the time scale of charge hopping. More interestingly, the ratio kt/k is close to unity. This result is in accord with the predictions of electron transfer theory (26) for the two superexchange-mediated rates with equal electronic couplings, i.e., Gj+TTGj±1 (with an energy gap ΔGj = 0 for all j) and GN+TTGGG (with ΔG = −0.7 eV). The nearly identical values of the endoergic hopping rate k in the normal region (−ΔG < λ) and of the exoergic rate kt in the inverted region (−ΔG > λ) imply that the low-frequency reorganization energy λ for hole transfer is λ = −ΔG/2, i.e., λ = 0.35 eV, a value that is consistent with the experimental result of Harriman (27). This relatively large value of λ is compatible with the response of a moderately polar environment to charge hopping.
Figure 1.

Bridge-size dependence of hole transport in DNA strands: G+(TTG)nGG DNA with N = 1–4. (a) The ratio φ of the yields of GGG and of all the products Pj (N = 1…N). (b) The ratio φ′ of the yields of GGG and of P1. Experimental data of Giese et al. (ref. 19 and S.W. and B.G., unpublished data) are marked by open squares with error bars. The calculated results from the numerical solution of Scheme S2 are, for the solid lines, kr/k = 0.08, kd/k = 0.08, and kt/k = 0.6 and, for the dotted lines, kr/k = 0.1, kd/k = 0.08, and kt/k = 1.1.
Hole-Hopping Rate Mediated by Two AT Base Pairs.
Because experimental information on the absolute value of the nearest-neighbor GTTG hopping rate is not yet available, it will be instructive to present an estimate of k. Using nonadiabatic charge transfer theory for the symmetric charge exchange reaction (ΔG = 0), we determine that the hopping rate is
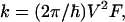 |
5 |
where V is the electronic coupling, F is the thermally averaged Franck–Condon density, incorporating both medium and intramolecular vibrational modes,
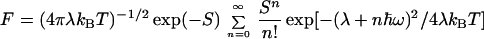 |
6 |
and λ ≅ 0.38 − 0.48 eV (27). The high-frequency intramolecular vibrational modes are taken to be characterized by the mean vibrational frequency ω ≅ 1,500 cm−1 and coupling S ≅ 1. The electronic coupling for the G+TTG superexchange interaction (19) is given by V = [ν2/(ΔG + λ)](ν/λ), where ΔG ≅ 0.7 eV is the G+T − GT+ energy gap inferred from redox potential data (23, 24) and ν is the nearest-neighbor pair G–T and T–T hole transfer integral. From the measurements of Giese et al. (15, 18, 19) on the G+…GGG transfer, the superexchange trapping rate is of the form kt ∝ exp(−βRGG) with β = 0.7 ± 0.1 Å−1, where RGG is the corresponding G…GGG distance, and we take the same functional distance dependence for k. Thus, each pair of AT bases reduces the superexchange hole transfer rate by a numerical factor of 10. Superexchange theory (26) then suggest that this reduction factor is (ν/λ)2 = 1/10, i.e., ν ≅ 0.12 eV. The value of ν is lower than the theoretical calculations of Dee and Bauer (28) for the hole transfer integrals for the GT, TT, GA, AA, and AT pairs. Accordingly, V = 0.006 eV for the G+TTG coupling. Eq. 6 now gives k ∼ 109 s−1 at T = 300 K for hole hopping mediated by the TT bridge. The large value of the hole transfer integrals results in short superexchange-mediated hopping lifetimes in the range of ≈1 ns in this system, providing dynamic rulers for hopping hole transport in DNA. This rough estimate, together with the ratios of the rates for the TT bridge (Fig. 1), result in the approximate values
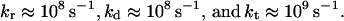 |
Maximal Distance for Hole Hopping.
Interesting additional information emerges from the superexchange mediation of the individual hopping and trapping rates. The dependence of the hole-hopping rate k and of the trapping rate kt on the number of AT base pairs between the sites G…G and G…GGG, as inferred from the experimental data (18, 19), gives a reduction factor of 1/10 for each extra AT base pair in the bridge. This information, together with the kinetic data for the TT bridge (Fig. 1), can be used for the semiquantitative interpretation of additional experimental results. In the sequence G+ACGTCTGACTCGACTGGG (Fig. 2), the guanine bases are separated by a single AT base pair (18). This system can be modeled as a chain of N = 7 in which both the hopping rate k and the trapping rate kt are larger by a numerical factor of 10 than those for the TT bridge. The simulations of Fig. 2 give φ ≅ 3.7 for N = 7, in reasonable agreement with the experimental result φ = 2.3 ± 0.7 (18). This moderately high value of φ for charge hopping over the G1… GGG distance of 54 Å reflects the efficiency of individual hopping and trapping rates for a single mediating AT base pair. From this analysis, we can infer the maximal distance for hole hopping between G bases separated by single AT base pairs, which is limited by the chemical side reactions. Making use of Eqs. 1 and 4, together with the experimental value kd/k = 0.08, we set φ = Y(GGG)/[1 − Y(GGG)] ≅ Y(GGG), whereas, from numerical data, Y(GGG) = 132 exp(−0.28 N) for large values of N. Accordingly, Y(GGG) assumes the values of 10−1 and of 10−2 for N = 35 (a distance scale of 240 Å) and for N = 55 (a distance scale of 375 Å), respectively. In this system, the maximum distance over which a measurable (1–10%) charge transport via hopping can occur is 300 ± 70 Å. This estimate specifies the initiation of chemistry over large distances of a few hundreds of angstroms in DNA.
Figure 2.

Numerical simulations of the dependence of the chemical yield ratios φ and φ′ for hole trapping and reactivity on the hopping and trapping rates in the duplex G+ACGTCTGACTCGACTGGG and C TGCAGACTGAGCTGACCC (N ≅ 7), where the G1…GGG spacing is 54 Å; φ (solid line) and φ′ (dotted line) denote the ratios of yields for k = 10 with kr/k = 0.008, kd/k = 0.008, and kt/k = 0.6.
Time-Resolved Information on Hole Hopping.
Finally, we advance time-resolved information from kinetic Scheme S2. In the limit kd = 0, the time τ for reducing the total charge population f(t) on the chain to 1/e of its initial value is extremely well represented by τ = (N/kt) + (N2/2k), which, for Nkt ≫ k, results in τ ∝ N2, manifesting diffusion to an absorbing sink (22, 29). Model calculations for our kinetic Scheme S2 with realistic kinetic parameters reveal that the time-resolved population f(t) of the acceptor GGG+ (Fig. 3) exhibits the rise time τR for the increase of this population to the value of (1 − e−1) of its final value given by τR ∝ N. This behavior could serve as a fingerprint for the diffusive-reactive process underlying the reaction scheme for hole hopping in DNA.
Figure 3.

Model calculations of the time-dependence of the population of the acceptor GGG+. Kinetic parameters are as described for the solid line in Fig. 1.
Epilogue.
This analysis of chemical yield data provides quantitative information on the transport of positive charges in DNA over distances as long as 50–300 Å. The hopping mechanism involves the guanines exclusively and is simplified in the sense that it does not account for the potential role of a coupled proton transfer in the G…C pair (30). In contrast to long-distance hole transport, the corresponding transport of negative charge is expected to proceed via the reduction of both thymine and cytosine, because their reduction potentials are similar (23, 24). Because one of these pyrimidine bases is present in each base pair, long-distance electron transport might turn out to be highly efficient if large electronic couplings (28) between adjacent base pairs of the order of 0.1 eV indeed prevail. Consequently, long-range transport of electrons along arbitrary duplexes is expected to be favored over holes, because it should not—or hardly—depend on the sequence, provided that protonic equilibria are not interfering (30). This feature might help to explain electrical conduction through DNA (8, 9) and therefore might be relevant for DNA nanoelectronics (4–9) and a diversity of electrochemical DNA-based technologies. The latter ones address primarily the fields of biosensors in which DNA is used as a conducting spacer (2) and of molecular recognition and sequencing methods that rely on interfacing of DNA to macroscopic electrodes (1, 2).
Biological implications of long-range charge transport in DNA pertain to the generation of damage rather than to its repair. Damage of DNA can be induced by radiation and by chemical reactions (31). In both cases, specific radical reactions with nucleobases occur, which finally lead to oxidized guanine, which may be followed by cation migration. For such systems, our analysis of hole transport is relevant. In the context of the repair of UV-induced damage of DNA by long-range charge transport, we are not aware of any convincing argument supporting such a mechanism. This biological system prefers a strictly short-range (≈10 Å) electron-transfer process from the enzyme photolyase onto thymine dimers to initiate photorepair (32–34). Short-range charge transfer rests on the availability of a highly specialized enzymatic apparatus, including an appropriate set of cofactors and a specific binding site. Although expensive in terms of evolution, this strategy avoids a set of destructive shortcomings associated with long-range charge transport in DNA, e.g., the extreme redox capability for charge injection, the obvious chemical instability of the charged DNA, and the problem of back donation of the charge after repair over long distances.
Acknowledgments
The support of this research by the Volkswagen Foundation, the Swiss National Science Foundation, and the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 377) is gratefully acknowledged.
References
- 1.Marshall A, Hodgson J. Nat Biotechnol. 1998;16:27–31. doi: 10.1038/nbt0198-27. [DOI] [PubMed] [Google Scholar]
- 2.Kelley S O, Jackson N M, Hill M G, Barton J K. Angew Chem Int Ed Engl. 1999;38:941–945. doi: 10.1002/(SICI)1521-3773(19990401)38:7<941::AID-ANIE941>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 3.Lisdat F, Ge B, Scheller F W. Electrochem Commun. 1999;1:65–68. [Google Scholar]
- 4.Mirkin C A, Letsinger R L, Mucic R C, Stofhoff J J. Nature (London) 1996;382:607–609. doi: 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]
- 5.Alivisatos A P, Johnsson K P, Wilson T E, Loveth C J, Bruchez M P, Schulz P G. Nature (London) 1996;382:609–611. doi: 10.1038/382609a0. [DOI] [PubMed] [Google Scholar]
- 6.Winfree E, Liu F, Wenzler L A, Seeman N C. Nature (London) 1998;394:539–544. doi: 10.1038/28998. [DOI] [PubMed] [Google Scholar]
- 7.Braun E, Eichen Y, Sivan U, Ben-Joseph G. Nature (London) 1998;391:775–778. doi: 10.1038/35826. [DOI] [PubMed] [Google Scholar]
- 8.Fink H-W, Schönenberger C. Nature (London) 1999;398:407–410. doi: 10.1038/18855. [DOI] [PubMed] [Google Scholar]
- 9.Porath, D., Bezryadin, A., de Vries, S. & Dekker, C. (1999) Nature (London), in press. [DOI] [PubMed]
- 10.Dandliker P J, Holmlin R E, Barton J K. Science. 1997;275:1465–1468. doi: 10.1126/science.275.5305.1465. [DOI] [PubMed] [Google Scholar]
- 11.Holmlin R E, Dandliker P J, Barton J K. Angew Chem Int Ed Engl. 1997;36:2715–2730. [Google Scholar]
- 12.Dandliker P J, Nunez M E, Barton J K. Biochemistry. 1998;37:6491–6502. doi: 10.1021/bi980041w. [DOI] [PubMed] [Google Scholar]
- 13.Lewis F D, Wu T, Zhang Y, Letsinger R L, Greenfield S R, Wasielewski M R. Science. 1997;277:673–676. doi: 10.1126/science.277.5326.673. [DOI] [PubMed] [Google Scholar]
- 14.Fukui K, Tanaka K. Angew Chem Int Ed Engl. 1998;37:158–161. [Google Scholar]
- 15.Meggers E, Kusch D, Spichty M, Wille U, Giese B. Angew Chem Int Ed Engl. 1998;37:460–462. doi: 10.1002/(SICI)1521-3773(19980302)37:4<460::AID-ANIE460>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 16.Breslin D T, Schuster G B. J Am Chem Soc. 1996;118:2311–2319. [Google Scholar]
- 17.Gasper S M, Schuster G B. J Am Chem Soc. 1997;119:12762–12771. [Google Scholar]
- 18.Meggers E, Michel-Beyerle M E, Giese B. J Am Chem Soc. 1998;120:12950–12955. [Google Scholar]
- 19.Giese B, Wessely S, Spormann M, Lindemann U, Meggers E, Michel-Beyerle M E. Angew Chem Int Ed Engl. 1999;38:996–998. doi: 10.1002/(SICI)1521-3773(19990401)38:7<996::AID-ANIE996>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 20.Wan C, Fiebig T, Kelly S O, Treadway C R, Barton J K, Zewail A H. Proc Natl Acad Sci USA. 1999;96:6014–6019. doi: 10.1073/pnas.96.11.6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henderson P T, Jones D, Hampikian G, Kan Y, Schuster G B. Proc Natl Acad Aci USA. 1999;96:8353–8358. doi: 10.1073/pnas.96.15.8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jortner J, Bixon M, Langenbacher T, Michel-Beyerle M E. Proc Natl Acad Sci USA. 1998;95:12759–12765. doi: 10.1073/pnas.95.22.12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seidel C A M, Schultz A, Sauer M H M. J Phys Chem. 1996;100:5541–5553. [Google Scholar]
- 24.Steenken S, Jovanovic S V. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 25.Saito I, Takayama M, Nakatami K, Tsuchida A, Yamamoto M. J Am Chem Soc. 1995;117:6406–6407. [Google Scholar]
- 26.Bixon M, Jortner J. Adv Chem Phys. 1999;106:35–208. [Google Scholar]
- 27.Harriman A. Angew Chem Int Ed Engl. 1999;38:945–949. doi: 10.1002/(SICI)1521-3773(19990401)38:7<945::AID-ANIE945>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 28.Dee D, Bauer M E. J Chem Phys. 1974;60:541–550. [Google Scholar]
- 29.Bar-Haim A, Klafter J. J Chem Phys. 1998;109:5187–5193. [Google Scholar]
- 30.Steenken S. Biol Chem. 1997;378:1293–1297. [PubMed] [Google Scholar]
- 31.Demple B, Harrison L. Annu Rev Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 32.Sancar A. Biochemistry. 1994;33:2–9. doi: 10.1021/bi00167a001. [DOI] [PubMed] [Google Scholar]
- 33.Heelis P F, Hartman R F, Rose S D. Chem. Soc. Rev. 1995. 289–297. [Google Scholar]
- 34.Langenbacher T, Zhao X, Bieser G, Heelis P F, Sancar A, Michel-Beyerle M E. J Am Chem Soc. 1997;118:10532–10536. [Google Scholar]